

Abstract
The X-linked KDM5C gene plays an important role in brain development and behavior. It encodes a histone demethylase that is involved in gene regulation in neuronal differentiation and morphogenesis. When mutated, it causes neuropsychiatric symptoms, such as intellectual disability, delayed language development, epilepsy, and impulsivity. To better understand how the patient mutations affect neuronal development, we expressed KDM5C mutations in Neuro2a cells, a mouse neuroblastoma cell line. Retinoic acid (RA) induced-neurite growth was suppressed by the mutation KDM5CY751C, KDM5CH514A, and KDM5CF642L, but not KDM5CD87G or KDM5CA388P. RNA-seq analysis indicated an up-regulation of genes important for neuronal development, such as Ntng2, Enah, Gas1, Slit2, and Dscam, in response to the RA treatment in control Neuro2a cells transfected with GFP or wild type KDM5C. In contrast, in cells transfected with KDM5CY751C, these genes were not up-regulated by RA. Ntng2 was down-regulated in cells with KDM5C mutations, concordant with the lower levels of H3K4 methylation at its promoter. Moreover, knocking down Ntng2 in control Neuro2a cells led to the phenotype of short neurites similar to that of cells with KDM5CY751C, whereas Ntng2 overexpression in the mutant cells rescued the morphological phenotype. These findings provide new insight into the pathogenesis of phenotypes associated with KDM5C mutations.
Keywords: Memory, Autism, Aggression, Sex difference, Synapse, X chromosome
Introduction
X-linked mutations are one major cause of intellectual disability (ID), accounting for ∼10% cases in male individuals with this condition (Ropers and Hamel, 2005, des Portes, 2013). Among the X-linkedID cases, lysine (K)-specific demethylase 5C (KDM5C) has been frequently identified to be mutated, resulting in non-syndromic as well as syndromic ID with additional clinical features such as developmental delay, epilepsy, impulsivity, autism-like behavior, short adult stature, and facial dysmorphism (Jensen et al., 2005, Tzschach et al., 2006, Abidi et al., 2008, Adegbola et al., 2008, Rujirabanjerd et al., 2010, Santos-Reboucas et al., 2011). Mutations and genetic variations of KDM5C have also been implicated in neuropsychiatric disorders such as Huntington's disease and drug addiction (Vashishtha et al., 2013, Aguilar-Valles et al., 2014). Therefore a better understanding of KDM5C in brain development has direct relevance to basic and clinical research.
The KDM5C protein targets the methylation modifications of histone protein H3 at lysine 4 (methyl-H3K4), which are often formed at active gene promoters and likely involved in the assembly of the transcription initiation complex (Christensen et al., 2007, Iwase et al., 2007, Tahiliani et al., 2007). KDM5C-mediated demethylation, on the other hand, leads to gene repression. In consistence with the neurological symptoms of individuals with KDM5C mutations, KDM5C has been found to be crucial to neuronal development, affecting events such as neuronal differentiation, cell death, and dendritic growth (Iwase et al., 2007, Tahiliani et al., 2007, Wynder et al., 2010, Shen et al., 2014). However, it remains to be determined which genes are targeted by KDM5C and misregulated in individuals with KDM5C mutations, and which among these KDM5C-regulated genes are involved in neuronal development. We set out to test specific KDM5C patient mutations for their effects on gene expression and neuronal development, using Neuro2a (N2a) cells as a model which are a mouse neuroblastoma cell line (Olmsted et al., 1970).
N2a cells are normally kept in a neural progenitor-like stage; upon retinoic acid (RA) treatment, they undergo morphogenesis with neurite growth that closely resembles the differentiation and dendritic growth of a developing neuron (Olmsted et al., 1970). These cells have served as a convenient yet informative model for functionality studies of genes: mutations can be readily introduced into N2a cells by plasmid or viral vectors, and a stably transfected cell line can be established by chemical or genetic selection (for e.g. (Goshima et al., 1993, Kojima et al., 1994)). We adopted this strategy in our study of KDM5C and generated N2a cells stably transfected with KDM5C patient mutations, among which some, but not all, mutations caused a reduction in neurite growth.
We performed genomic analysis to determine which genes and pathways might be affected in the mutant N2a cells, and identified Netrin G2 (Ntng2) as one of the down-regulated genes. The protein product of Ntng2 has in fact been known to be important for brain development and specifically in neurite growth: the Ntng2 protein is secreted at an axon terminal; it traverses the synaptic cleft, binds to its postsynaptic receptor NGL-2 (netrin-G2 ligand), and in turn activates signal transduction that leads to the postsynaptic growth of both dendrites and exons (Nakashiba et al., 2002, Soto et al., 2013). In addition, it plays a key role in circuit formation by guiding the axon to its correct postsynaptic target – in the hippocampus, for instance, the CA3 neurons make synaptic connections with the CA1 neurons at the proximal, not distal, dendritic segments, a precision largely dependent on an interaction between Ntng2 and NGL-2 (Nishimura-Akiyoshi et al., 2007, DeNardo et al., 2012). Consistent with these findings, Ntng2 down-regulation in N2a cells with KDM5C mutations was accompanied by reduced neurite lengths. Using chromatin immunoprecipitation (ChIP), we detected higher levels of mutant Kdm5c proteins at the Ntng2 promoter, a possible explanation for Ntng2 down-regulation. We were able to rescue the phenotype of short neurites by overexpressing Ntng2 in mutant N2a cells, suggesting that Kdm5c's effects on neurite growth are mediated, at least in part, by Ntng2. Together, these results shed new light on the etiology of neuropsychiatric symptoms associated with KDM5C mutations.
Materials and Methods
Mice and primary neuronal culture
Kdm5c knockout (KO) and wild type (WT) mouse pups were generated by breeding a heterozygous female on the C57BL6 background with a WT 129 male. The genotypes were determined by PCR analysis of tail clip DNAs (PCR primer sequence available upon request). Hippocampi were extracted from newborn male pups for primary neuronal culture, following the protocol described in Beaudoin et al. 2012 (Beaudoin et al., 2012). Briefly, the hippocampal tissue was digested in trypsin (0.25%, wt/vol) for 15 min at 37°C, stopped by an equal volume of Dulbecco's Modified Eagle Medium (DMEM supplemented with 10% FBS, Life Technologies, Carlsbad, CA). The lysate was spun down and the pellet resuspended by trituration in the Neurobasal medium supplemented with B-27 and glutamine (Life Technologies). The dissociated cells were seeded in poly-lysine coated wells at the density of ∼100×103 cells per well in the 12-well plate (Nunc, Rochester, NY), and grown in an incubator following the standard conditions. All procedures involving animals were approved by the IACUC of the Washington State University (Pullman, WA), in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Neuro2a cell culture
N2a cells were cultured in DMEM/F12 supplemented with FBS (10%) and PenStrep, split (1:10) every 3 days. Plasmid transfection was carried out using Lipofectamine 2000, followed by selection with puromycin (WT or mutant KDM5C plasmid) or zeocin (Ntng2 cDNA plasmid). The exogenous KDM5C protein can be identified in subsequent analyses by their hemagglutinin (HA) tag. Neurite growth was initiated by adding RA in the medium (20 μM) and reducing the concentration of FBS (0.5%). All reagents mentioned above are from Life Technologies unless specified otherwise.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed following the protocol described in Nelson et al., 2006 (Nelson et al., 2006). Briefly, chromatin was cross-linked with formaldehyde (1.42%) for 15 min at room temperature, followed by a glycine quenching (125 mM, 5 min). Cells were lysed in RIPA buffer using a hand-held homogenizer (Kimble-Chase, Vineland, NJ); the chromatins were pelleted and then sheared in an ultrasonicater (10 min, 30 s on/off, 4°C; Misonix, Newtown, CT), resulting in fragments between 200 and 500bp. The sheared chromatins were sequentially incubated with an anti-trimethyl-H3K4 or anti-HA antibody (1:100 dilution; Millipore, Billerica, MA) at 4°C overnight, and the protein A/G agarose beads (1:10 dilution; Millipore) at 4°C for 1 hr. Some chromatin samples were incubated with IgG as reference. The recovered chromatins were washed repeatedly, reverse cross-linked by Chelax-100 (10% wt/vol; Bio-Rad, Hercules, CA), and purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA). An aliquot of fixed, sonicated chromatins was reverse cross-linked and processed in parallel, serving as DNA input. Enrichment of trimethyl-H3K4 or HA-conjugated KDM5C at a DNA sequence was quantified using an ABI7500 qPCR machine (Life Technologies). A gene promoter was executively defined as the 1000bp sequence upstream of the transcription start site, retrieved from the UCSC genome browser (GRCm38/mm10). qPCR primers were designed using Primer Express software (ABI-Life Technologies) and specificity verified by the alignment of melting curves at the same temperature across independent chromatin samples. Relative enrichment of a histone modification or DNA binding protein at the promoter of interest was calculated according to the formula below:
where CtAntibody, CtIgG, and CtInput are respectively the qPCR cycle threshold (Ct) numbers of the antibody-processed chromatin sample, the IgG sample, and DNA input (at a 1:10 dilution).
RNA-seq analysis
RNA was extracted using the RNeasy Mini kit (Qiagen) and RNA integrity verified on a Bioanalyzer to be adequate (>0.8; Agilent Technologies, Santa Clara, CA). Libraries were constructed following the manufacturer's instructions (Illumina, San Diego, CA) and sequencing was performed at the Genomics Core, University of Washington. The RNA-seq reads were aligned to the reference genome using TopHat, and differentially expressed genes (adjusted p < 0.05) determined by Cuffdiff. GO ontology and pathway analysis were performed in the differentially expressed genes using DAVID Bioinformatics Resources (Huang da et al., 2009b, a).
Reverse transcription – quantitative PCR (RT-qPCR)
cDNA was synthesized using the Maxima First Strand kit (ThermoFisher, Waltham, MA), and gene expression quantified on the ABI qPCR machine (Life Technologies). Gene-specific PCR primers were designed with the PrimerExpress software (Life Technologies), and primer pairs were screened for specificity, indicated by single-peaked dissociation curves across independent cDNA samples, and amplification efficiency which was assessed with serial dilution samples. The chosen PCR primer sequences for Ntng2, for e.g., are as follows.
Forward: 5′-GGCAGTCAGACCAAGCCAC-3′
Reverse: 5′-CAATGGGTATCACCTGAGGAGA-3′
All PCR reactions were run in duplicates (highly expressed genes) or triplicates (lowly expressed genes). Gapdh mRNA was quantified in sample as reference for the cDNA quantity. Relative expression of a gene of interest (GOI) was calculated as follows.
where CtGOI and CtGapdh are the Ct numbers for amplification of the GOI or Gapdh cDNA, is the average value of ΔCtGOI across the WT or control samples while n is the number of these samples.
Fluorescent immunocytochemistry
For immunocytochemistry, cells were grown on glass coverslips and transfected with viral and/or plasmid vectors as described above. At the specified time points, cells were fixed with paraformaldehyde (4% in PBS) for 15 min at room temperature, and then incubated sequentially in normal serum (3%, 1 hr, room temperature), the primary antibody (overnight, 4°C), and finally a fluorescent secondary antibody (Life Technologies; 1 hr, room temperature). Both primary and secondary antibodies were diluted in PBS with 3% normal serum. The coverslips were mounted on slides and images were captured on a fluorescence microscope (DM4000, Leica, Wetzlar, Germany) equipped with a camera and the imaging software Leica Application Suite.
Western blotting
Cells were lysed in RIPA buffer (Cell Signaling, Danvers, MA) and pelleted by centrifugation (13,000 rpm) for 10 min at 4°C. Supernatant was collected and the protein concentrations were quantified using Pierce BCA Protein Assay Kit (ThermoFisher). Aliquots of proteins (25 μg per sample) were electrophoresed on a NuPAGE Bis-Tris gel (TheomaFisher) and transferred to a polyvinylidene fluoride (PVDF) membrane (Life Technologies). The blot was incubated with the primary and secondary antibody sequentially (primary: 4°C, overnight; secondary: room temperature, 1 hr), and developed using the ECL kit (PerkinElmer, Waltham, MA). The protein signal was digitized using the ChemiDoc MP system (BioRad, Hercules, CA) and quantified with ImageJ (Schneider et al., 2012).
Antibodies and plasmids
The anti-HA and anti-MAP2 antibody were purchased from Sigma (St. Louis, MO), anti-His tag antibody from Abcam (Cambridge, MA), anti-tri-methyl H3K4 antibody from Millipore (Billerica, MA), anti-KDM5C antibody from Active Motif (Carlsbad, CA), and anti-actin antibody from Sigma. cDNA of wild type or mutated (D87G, A388P, H514A, F642L, and Y751C) human KDM5C was cloned in the pHAGE-HA backbone, and the transfected cells can be selected with puromycin. A second set of KDM5C plasmids were made which co-express EYFP (pLenti-KDM5C-IRES-EYFP), making it possible to directly observe the neurite growth of the transfected cells in real time. The Myc-His-tagged Ntng2 plasmid was in the pcDNA4/myc-his backbone which carries in addition the zeocin resistance gene. The Ntng2 plasmid was a generous gift from Dr. Shigeyoshi Itohara (RIKEN Brain Science Institute, Wako, Japan), in whose lab Ntng2 was initially cloned and characterized (Nakashiba et al., 2002, Aoki-Suzuki et al., 2005, Meerabux et al., 2005). The shRNA lentiviruses against Ntng2 or Kdm5c mRNA were purchased from Sigma.
Statistical analysis
When the data met the normality and equal variance assumptions, the effect of genotype was tested with Student's t-tests between any two experimental groups; otherwise, Mann-Whitney U tests were used to assess the effects. The significance level was set in all tests at p < 0.05.
Results
KDM5C patient mutations H514A, F642L and Y751C suppressed neurite growth in Neuro2a cells
To investigate the etiology of KDM5C patient mutations, we made Neuro2a (N2a) cell lines stably transfected with plasmids encoding the wild type (WT) or mutant copy of human KDM5C, including four KDM5C patient mutations (D87G, A388P, F642L, and Y751C; Fig.1A) as well as one synthetic mutation in the Jumonji domain important for the histone demethylase activity of KDM5C (H514A; Fig.1A). As a control, some N2a cells were transfected with GFP plasmids. Stable transfection was confirmed by immunocytochemical detection of the HA tag, genetically fused to the exogenous human KDM5C protein, in all cells after multiple passages (Fig.1B). This exogenous fusion protein was localized in the nucleus consistent with it being a histone modification enzyme (Fib.1B). We designated these cell lines as KDM5CWT, KDM5CD87G, KDM5CA388P, KDM5CH514A, KDM5CF642L, KDM5CY751C, and GFP N2a cell, respectively. In addition, using PCR primers specific for human or mouse KDM5C, RT-qPCR analysis indicated comparable expression between the exogenous WT and mutant KDM5C, which was higher than that of the endogenous mouse Kdm5c gene (data not shown).
Figure 1. Establishment of Neuro2a cell lines stably expression wild type or patient mutant KDM5C.
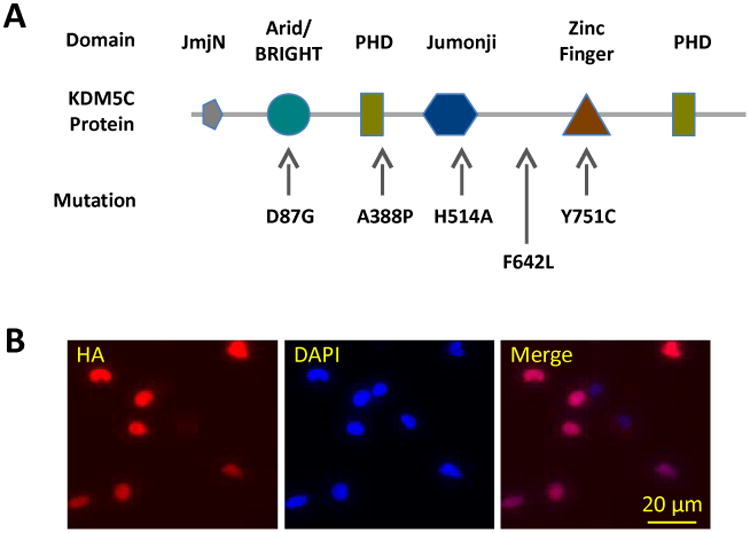
(A) Schematic representation of the functional motifs of the KDM5C protein and selective mutations relevant to the current study. (B) Stable expression of the exogenous copies of KDM5C was confirmed by immunocytochemical detection of HA, which was localized to the nuclei of the transfected cells.
We then tested retinoic acid (RA)-induced neurite growth in these cells. Relative to that of the KDM5CWT cells, the percentage of cells with neurites was significantly lower in the KDM5CF642L and KDM5CY751C cells after a 12 hr RA treatment, and the total neurite length per cell was reduced in the KDM5CH514A and KDM5CY751C cells (Table 1, Fig.2A & -B). Neither measure was affected in the KDM5CD87G and KDM5CA388P cells, suggesting distinct effects between these patient mutations on neuronal development (Table 1, Fig.2A & -B). There was also a significant difference in percentage of cells with neurites between the KDM5CWT and GFP cells, possibly due to an effect of the increased amount of KDM5C proteins in the former (Table 1, Fig.2B). Moreover, there was likely a similar effect of KDM5C mutations on hippocampal neurons: they were transfected at DIV 2 with KDM5CWT or KDM5CY751C using the EYFP expressing plasmid as vector (pLenti-KDM5C-IRES-EYFP); when examined four days post transfection, neurons with KDM5CY751C were found to have fewer and shorter dendrites compared to that with KDM5CWT (Fig. 2C).
Table 1. Reduced neurite growth of N2a cells with KDM5C patient mutations.
Percentage of cells with neurites and total neurite length per cell were measured after 24 hrs of retinoic acid treatment, shown below as mean ± standard error. Six independent samples per cell line were examined.
| Cell Line | % Cells with Neurites | Total Length of Neurites (μm) |
|---|---|---|
| GFP | 68.17 ± 2.6* | 54.9 ± 6.2 |
| WT | 58.5 ± 3.3 | 55.1 ± 3.4 |
| D87G | 51.1 ± 2.6 | 59.7 ± 5.4 |
| A388P | 49.8 ± 3.6 | 57.9 ± 3.4 |
| H514A | 48.1 ± 3.3* | 37.1 ± 4.1* |
| F642L | 36.6 ± 1.5** | 47.3 ± 3.6 |
| Y751C | 27.9 ± 3.9** | 35.2 ± 5.5* |
p< 0.05,
p <0.0001, t-test relative to cells transfected with wild type KDM5C (WT).
Figure 2. The effect of KDM5C mutations on neuronal development.
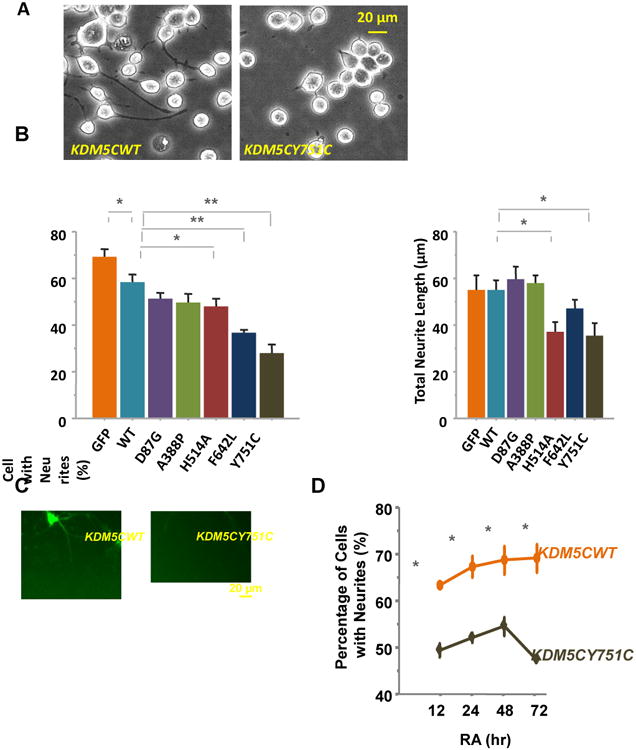
(A) Neurite growth was elicited by RA in N2a cells stably transfected with KDM5CWT or mutant KDM5C and morphological analysis was performed after a 12 hr incubation. Images were taken using an inverted phase contrast microscope (Leica DMIL). (b) Specific patient mutations caused lower percentages of cells with neurites (KDM5CH514A, KDM5CF642L, and KDM5CY751C) and short neurite lengths (KDM5CH514A and KDM5CY751C) relative to that of N2a cells expressing wild type KDM5C in response to the RA treatments (*: p < 0.05, **: p < 0.01). In addition, there was a difference in percentage of neurited cells between the KDM5CWT and GFP N2a cells. At least 6 independent samples were examined per cell line and four images per sample were quantified representing the four quadrants of a well. (c) Cultured hippocampal neurons were shown to grow shorter and fewer dendrites when transfected with KDM5CY751C compared to that of KDM5CWT. The transfected neurons were identified by EYFP that is encoded by the plasmid (i.e. pLenti-KDM5C-IRES-EYFP) and not fused with KDM5C. (D) Extended RA incubation to 72 hrs did not increased the percentage of cells with neurites in KDM5CY751C N2a cells relative to that of the KDM5CWT N2a cells.
We tested the possibility that reduced neurite growth might be due to a delayed response of the mutant cells to RA, which might take longer periods to reach the percentage of neurited cells comparable to that of the KDM5CWT cells. At 12, 24, 48, and 72 hrs of RA treatment, we detected consistently lower percentages of KDM5CY751C cells with neurites than that of the KDM5CWT cells, arguing against a slower response of the mutant cells (p < .01 at each of the four time points, Mann-Whitney U; Fig.2D). In KDM5CY751C cells, the percentage in fact peaked at 48 hrs before it started to taper off, suggesting the possibility of less stabilized neurites in these cells.
KDM5C regulation of gene expression in RA-induced neurite growth
To determine the KDM5C-regulated genes in RA-induced neurite growth of N2a cells, we employed RNA-seq and identified genes which were differentially expressed between the KDM5CY751C, KDM5CWT, and GFP N2a cells before and after a 12 hr RA treatment, with two samples in each genotype X treatment group and each sample being pooled from three biologically independent wells. We reasoned that (a) the genes up- or down-regulated by RA are likely ones that are involved in N2a neurite growth, and (b) the genes responsive to RA in the KDM5CWT and GFP cells, but not KDM5CY751C cells, likely include the candidates for the short neurite phenotype of the latter. Setting the significance level at adjusted p < 0.05, 40 genes were identified to be up-regulated and 38 genes down-regulated by RA in the KDM5CWT and GFP cells but not KDM5CY751C cells (Table. 2). A functional annotation of the 78 genes (DAVID Bioinformatics Resources) led to three clusters of biological processes (p < .1), namely neuron project morphogenesis (Dscam, Ntng2, Enah, Gas1, Slit2; p = .0052), chordate embryonic development (G2e3, Enah, Gas1, Tcf7l2, Tgfb3, Zeb2; p = .014), and regulation of apoptosis (Dcun1d3, G2e3, Aifm2, Dedd2, Gas1, Tgfb3; p = .068). Between the five genes implicated in neuron project morphogenesis, Ntng2 expression exhibited the largest differences between the KDM5CY751C cells and the other two cell types before and after RA (Table 3). We therefore decided to focus on this gene with additional analyses.
Table 2. Differential expression of RA-responsive genes between GFP, KDM5CWT, and KDM5CY751C N2a cells identified in RNA-seq analysis.
These genes were found to be up- or down-regulated by RA in GFP and KDM5CWT (adjusted p < .05) but not KDM5CY751C N2a cells. FC: fold change in the number of reads between the cells with and without RA. Biological Processes are shown below only one identified by functional annotation (David Bioinformatics Resources, https://david.ncifcrf.gov), and genes involved in these processes are highlighted.
| Symbol | Gene Name | FC (GFP) | FC (KDM5CWT) | Biological Process |
|---|---|---|---|---|
| RA-Induced Gene Up-Regulation | ||||
| Ccnjl | cyclin J-like | 7.77 | 11.83 | |
| Gas1 | growth arrest specific 1 | 3.58 | 3.70 | Neuron Project Morphogenesis / Chordate Embryonic Development / Regulation of Apoptosis |
| Rasl10b | RAS-like, family 10, member B | 3.09 | 3.91 | |
| Ntng2 | netrin G2 | 2.75 | 2.99 | Neuron Project Morphogenesis |
| Gnb3 | guanine nucleotide binding protein (G protein), beta 3 | 2.93 | 2.72 | |
| F630110N24Rik | RIKEN cDNA F630110N24 gene | 2.96 | 1.70 | |
| Aifm2 | apoptosis-inducing factor, mitochondrion-associated 2 | 2.04 | 2.51 | Regulation of Apoptosis |
| Frmpd1 | FERM and PDZ domain containing 1 | 2.31 | 2.17 | |
| Tcp11l2 | t-complex 11 (mouse) like 2 | 2.42 | 1.84 | |
| Dscam | Down syndrome cell adhesion molecule | 1.76 | 2.49 | Neuron Project Morphogenesis |
| Rec8 | REC8 homolog (yeast) | 2.32 | 1.91 | |
| Gfi1 | growth factor independent 1 | 2.24 | 1.99 | |
| Procr | protein C receptor, endothelial | 1.77 | 2.35 | |
| 5730409E04Rik | RIKEN cDNA 5730409E04Rik gene | 1.92 | 2.12 | |
| Rnd2 | Rho family GTPase 2; predicted gene 4768 | 1.90 | 1.93 | |
| Isoc1 | isochorismatase domain containing 1 | 1.52 | 2.30 | |
| Gm8909 | predicted gene 8909 | 1.77 | 1.94 | |
| Zfp809 | zinc finger protein 809 | 1.70 | 2.00 | |
| 8430427H17Rik | RIKEN cDNA 8430427H17 gene | 2.21 | 1.43 | |
| Zfp438 | zinc finger protein 438 | 1.71 | 1.93 | |
| Ttc8 | tetratricopeptide repeat domain 8 | 2.22 | 1.38 | |
| Ttc28 | tetratricopeptide repeat domain 28 | 1.63 | 1.97 | |
| Gpr124 | G protein-coupled receptor 124 | 1.89 | 1.56 | |
| Bicd1 | bicaudal D homolog 1 (Drosophila) | 1.53 | 1.81 | |
| Dedd2 | death effector domain-containing DNA binding protein 2 | 1.59 | 1.65 | Regulation of Apoptosis |
| Snhg12 | hypothetical protein LOC100039864 | 1.54 | 1.63 | |
| Phyh | phytanoyl-CoA hydroxylase | 1.52 | 1.63 | |
| Dcun1d3 | DCN1, defective in cullin neddylation 1, domain containing 3 (S. cerevisiae) | 1.42 | 1.71 | Regulation of Apoptosis |
| Zfp445 | zinc finger protein 445 | 1.47 | 1.62 | |
| 1700017B05Rik | septin 14; RIKEN cDNA 1700017B05 gene | 1.65 | 1.41 | |
| Slc25a20 | solute carrier family 25 (mitochondrial carnitine/acylcarnitine translocase), member 20 | 1.52 | 1.53 | |
| Slit2 | slit homolog 2 (Drosophila) | 1.54 | 1.45 | Neuron Project Morphogenesis |
| Ncoa3 | nuclear receptor coactivator 3 | 1.54 | 1.43 | |
| Eml5 | echinoderm microtubule associated protein like 5 | 1.52 | 1.44 | |
| Uck1 | uridine-cytidine kinase 1; predicted gene 4482 | 1.44 | 1.51 | |
| Ofd1 | oral-facial-digital syndrome 1 gene homolog (human) | 1.53 | 1.41 | |
| Ptpn21 | protein tyrosine phosphatase, non-receptor type 21 | 1.53 | 1.40 | |
| Arhgef10l | Rho guanine nucleotide exchange factor (GEF) 10-like | 1.40 | 1.49 | |
| Enah | enabled homolog (Drosophila) | 1.40 | 1.41 | Neuron Project Morphogenesis / Chordate Embryonic Development |
| Ucp2 | uncoupling protein 2 (mitochondrial, proton carrier) | 1.33 | 1.40 | |
| RA-Induced Gene Down-Regulation | ||||
| Pde9a | phosphodiesterase 9A | 0.27 | 0.30 | |
| Cdh22 | similar to cadherin 22; cadherin 22 | 0.33 | 0.31 | |
| Tgfb3 | transforming growth factor, beta 3 | 0.43 | 0.43 | Chordate Embryonic Development / Regulation of Apoptosis |
| Sertad1 | SERTA domain containing 1 | 0.28 | 0.59 | |
| Tcf7l2 | transcription factor 7-like 2, T-cell specific, HMG-box | 0.40 | 0.47 | Chordate Embryonic Development |
| Tmtc1 | transmembrane and tetratricopeptide repeat containing 1 | 0.40 | 0.49 | |
| Ctla2b | cytotoxic T lymphocyte-associated protein 2 beta | 0.55 | 0.40 | |
| Igfbp7 | insulin-like growth factor binding protein 7 | 0.50 | 0.49 | |
| Mapk10 | mitogen-activated protein kinase 10 | 0.59 | 0.54 | |
| Adck4 | aarF domain containing kinase 4 | 0.59 | 0.53 | |
| Plod2 | procollagen lysine, 2-oxoglutarate 5-dioxygenase 2 | 0.58 | 0.58 | |
| Pmp22 | peripheral myelin protein 22 | 0.69 | 0.49 | |
| Mical2 | microtubule associated monoxygenase, calponin and LIM domain containing 2 | 0.57 | 0.62 | |
| Plekha2 | pleckstrin homology domain-containing, family A (phosphoinositide binding specific) member 2 | 0.67 | 0.53 | |
| Ets1 | E26 avian leukemia oncogene 1, 5′ domain | 0.62 | 0.59 | |
| Rgs9 | regulator of G-protein signaling 9 | 0.64 | 0.58 | |
| Slc44a1 | solute carrier family 44, member 1 | 0.72 | 0.55 | |
| Capn6 | calpain 6 | 0.63 | 0.66 | |
| Brp44l | similar to brain protein 44-like protein; brain protein 44-like; predicted gene 3452; predicted gene 8219 | 0.71 | 0.59 | |
| Cxxc4 | CXXC finger 4; similar to Dvl-binding protein Idax | 0.63 | 0.68 | |
| Zeb2 | zinc finger E-box binding homeobox 2 | 0.67 | 0.66 | Chordate Embryonic Development |
| Rab3c | similar to RAB3C, member RAS oncogene family; RAB3C, member RAS oncogene family | 0.72 | 0.60 | |
| Uqcrh | predicted gene 14088; ubiquinol-cytochrome c reductase hinge protein | 0.70 | 0.65 | |
| Slc6a8 | solute carrier family 6 (neurotransmitter transporter, creatine), member 8 | 0.66 | 0.69 | |
| Abat | 4-aminobutyrate aminotransferase | 0.67 | 0.68 | |
| Wipi1 | WD repeat domain, phosphoinositide interacting 1 | 0.74 | 0.63 | |
| Scrn1 | secernin 1 | 0.69 | 0.71 | |
| Wbp5 | WW domain binding protein 5 | 0.73 | 0.69 | |
| G2e3 | G2/M-phase specific E3 ubiquitin ligase | 0.69 | 0.75 | Chordate Embryonic Development / Regulation of Apoptosis |
| Fn1 | fibronectin 1 | 0.74 | 0.72 | |
| Spock2 | sparc/osteonectin, cwcv and kazal-like domains proteoglycan 2 | 0.73 | 0.74 | |
| Plcxd3 | phosphatidylinositol-specific phospholipase C, X domain containing 3 | 0.75 | 0.72 | |
| 1700025G04Rik | RIKEN cDNA 1700025G04 gene | 0.72 | 0.75 | |
| Slc4a7 | solute carrier family 4, sodium bicarbonate cotransporter, member 7 | 0.73 | 0.75 | |
| Me2 | malic enzyme 2, NAD(+)-dependent, mitochondrial | 0.74 | 0.76 | |
Table 3. Effect of KDM5CY751C on Ntng2 expression.
Among the genes implicated in neuron project morphogenesis, Ntng2 exhibited the largest differences in expression between the KDM5CY751C cell line and the other two cell lines, namely GFP and KDM5CWT without or with RA treatment. Normalized numbers of reads are shown below averaged between the two independent samples.
| Gene | No RA | RA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GFP | KDM5CWT | KDM5CYC | K5CYC vs GFP (%) | K5CYC vs K5CWT (%) | GFP | KDM5CWT | KDM5CYC | K5CYC vs GFP (%) | K5CYC vs K5CWT (%) | |
| Ntng2 | 6.76 | 5.16 | 1.23 | 18.2 | 23.8 | 18.6 | 15.44 | 3.41 | 18.3 | 22.1 |
| Dscam | 2.01 | 1.12 | 0.51 | 25.4 | 45.5 | 3.54 | 2.78 | 1.18 | 33.3 | 42.4 |
| Gas1 | 0.93 | 1.12 | 1.01 | 108.6 | 90.2 | 3.31 | 4.16 | 2.6 | 78.5 | 62.5 |
| Enah | 43.98 | 51.55 | 44.08 | 100.2 | 85.5 | 61.53 | 72.41 | 54.37 | 88.4 | 75.1 |
| Slit2 | 11.79 | 12.33 | 14.77 | 125.3 | 119.8 | 18.18 | 17.88 | 17.88 | 98.3 | 100.0 |
KDM5C mutations caused Ntng2 down-regulation
Ntng2 down-regulation in the KDM5CY751C cells relative to that of KDM5CWT cells was confirmed in independent samples using RT-qPCR (p < .05, Mann-Whitney U tests; Fig.3A). It was also down-regulated in the KDM5CH514A and KDM5CF642L cells (p < .05 in both cases, Mann-Whitney U tests; Fig.3A), but not in the KDM5CD87G, KDM5CA388P, or GFP cells, concordant to the neurite phenotypes of these cell lines (Fig. 2B).
Figure 3. Ntng2 down-regulation by KDM5C mutations.
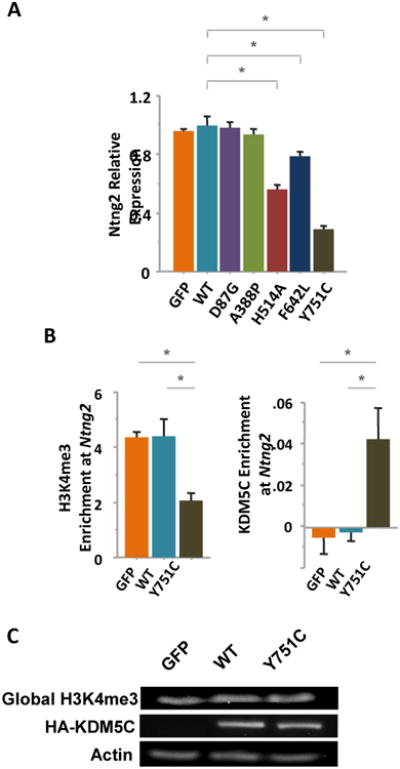
Ntng2 expression was quantified with RT-qPCR assays and found to be down-regulated in the KDM5CH514A, KDM5CF642L, and KDM5CY751C N2a cells relative to that of the KDM5CWT cells. Three independent samples per cell line were included in the analysis and relative expression was calculated using the AACt formula with Gapdh expression as reference. *: p < .05, Mann-Whitney U test. (b) ChIP assays indicated that, at the Ntng2 promoter (-65bp to -142bp), the H3K4me3 levels were decreased while that of exogenous KDM5C proteins were increased in the KDM5CY751C N2a cells compared to that of KDM5CWT or GFP N2a cells (*: p < .05, Mann-Whitney). The KDM5C levels were estimated by ChIP analysis of the HA tag. Three independent chromatin samples per cell line were processed. (c) Western blotting suggested that the global levels of H3K4me3 and HA-KDM5C were comparable between the GFP, KDM5CWT, and KDM5CY751C N2a cells.
In line with Ntng2 down-regulation, trimethylation of histone H3 at lysine 4 (H3K4me3), an active chromatin mark and the enzymatic substrate of Kdm5c, was found to be reduced at the Ntng2 promoter in KDM5CY751C cells relative to that of the KDM5CWT and GFP cells, quantified with ChIP assays at a sequence upstream from the transcription start site between -65bp and -142bp; meanwhile, there was a higher level of KDM5C demethylase at this sequence, measured with ChIP assays against HA (in both cases, U = 0, Z = 1.96, p = 0.0495, Mann-Whitney U tests; Fig.3B). There was no difference between the GFP and KDM5CWT cells in either the H3K4me3 or KDM5C levels (p > .05 in both cases; Fig.3B), suggesting that the differences between these cell lines in Ntng2 expression are likely attributed to the KDM5CY751C mutation, not a higher level of exogenous KDM5C protein. Moreover, the KDM5CY751C mutant protein seemed to target specific sequences without causing a global change in the H3K4me3 levels (Fig.3C).
We also quantified Ntng2 expression and neurite growth in primary hippocampal cultures from neonatal Kdm5c KO mice as well as N2a cells with Kdm5c knocked down by lentivirus-derived shRNAs. In cultured neurons the dendritic morphology was examined by immunolabeling the dendritic protein Map2 at DIV3, which revealed significantly shorter dendrites in the Kdm5c KO than WT neurons (WT neuron: 189.2 ± 9.8 μm, Kdm5c KO neuron: 155.9 ± 8.8 μm; U = 2, Z = 1.96, p = .050, Mann-Whitney U; Fig.4). In N2a cells treated with shRNAs, Kdm5c knockdown (KD) was confirmed to be ∼37% that of the control cells treated with non-targeting shRNA (p < .05, Mann-Whitney), which led to a reduced percentage of KD cells with neurites compared to that of control cells (Control: 65.7 ± 4.0 %, Kdm5c KD cell: 46.7 ± 2.7 %; U = 0, Z = 1.96, p < .05, Mann-Whitney U; Fig.4). However, despite of the similar morphological phenotypes between these and cells with KDM5C patient mutations, Ntng2 was not down-regulated in either Kdm5c KO neurons or Kdm5c KD N2a cells (p > .05 in both cases, Mann-Whitney U; Fig.4), suggesting that Ntng2 down-regulation might be a result of the gain-of-function KDM5C mutations.
Figure 4. Kdm5c deficiency caused reduced neurite growth but not Ntng2 down-regulation.
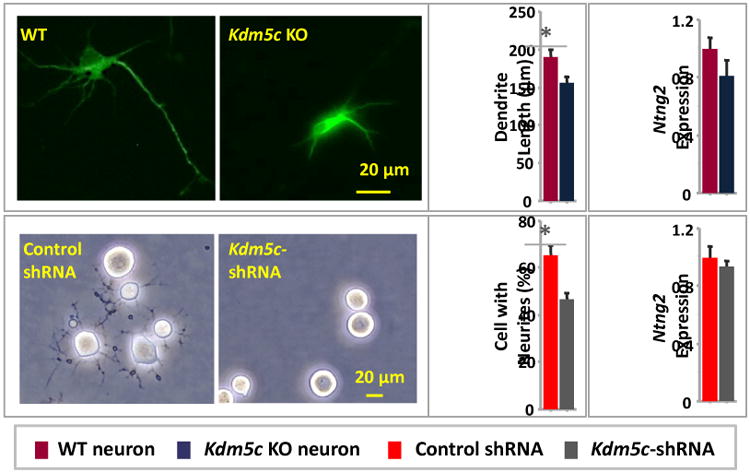
Dendritic lengths were shorter in cultured Kdm5c KO hippocampal neurons relative to that of WT neurons, and percentages of cells with neurites were lower in Kdm5c knockdown N2a cells than that treated with control shRNA particles. However, no difference in Ntng2 expression was detected in either case.
Ntng2 overexpression rescued the neurite phenotype of KDM5CY751C cells
We tested whether Ntng2 down-regulation could by itself cause reduced neurite growth of N2a cells. Ntng2 KD cells were generated using lentiviral particles expressing gene-specific shRNAs, leading to a down-regulation of Ntng2 relative to that of cells treated with control viruses (p < .05, Mann-Whitney; Fig.5A). In these Ntng2 KD cells, the percentages of cells with neurites was significantly decreased, indicating an implication of Ntng2 in neurite growth of N2a cells (Ntng2 KD cells with neurites: 24.8 ± 1.5%; control cells with neurites: 61.7 ± 5.7%; U = 0, Z = 2.31, p = .021, Mann-Whitney; Fig.5A).
Figure 5. Ntng2 down-regulation mediated the effect of KDM5C mutations on neurites.
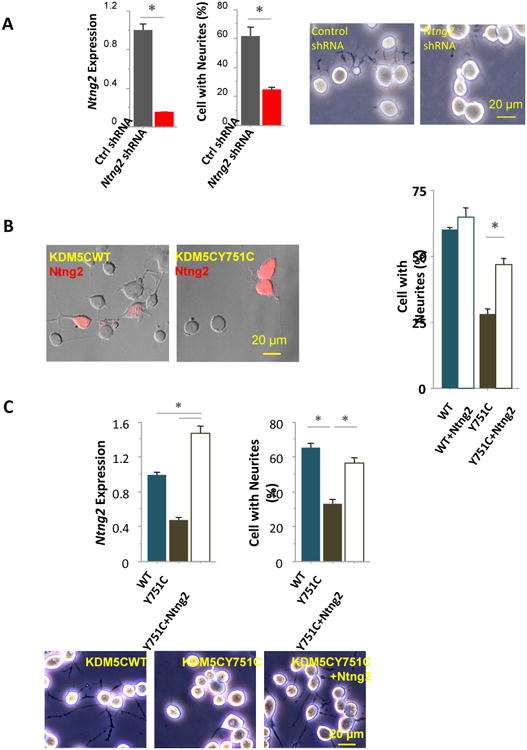
(A) Expression of Ntng2 was suppressed in N2a cells treated with lentiviral particles encoding gene-specific shRNAs (Ntng2 shRNA), leading to significantly lower percentages of cells with neurites, compared to that of control cells treated with non-gene targeting viral particles (Ctrl shRNA). (B) The percentages of cells with neurites were significantly higher in KDM5CY751C cells transiently transfected with Ntng2 plasmids than control KDM5CY751C cells, although no difference was found between the KDM5CWT cells with and without transient Ntng2 transfection. Cells transfected with Ntng2 were identified by the plasmid-derived His tag, immunolabeled and visualized with Alexa fluor 594. (c) In KDM5CY751C cells stably transfected with Ntng2 plasmids (Y751C + Ntng2), Ntng2 expression was significantly increased compared to that of KDM5CY751C or KDM5CWT cells; the percentages of cells with neurites were comparable to that of KDM5CWT cells and significantly higher than that of KDM5CY751C cells.
We then tested whether Ntng2 over-expression would rescue the phenotype of short neurites in KDM5CY751C cells. The morphological effects of both transient and stable transfection of Ntng2 plasmids were examined in these cells. A transient transfection was sufficient to cause a significant increase in the percentage of KDM5CY751C cells with neurites, rising from 28.2 ± 1.9% to 46.7 ± 2.5% (U = 0, Z = 1.96, p = 0.0495, Mann-Whitney; Fig. 5B), without affecting this measure in KDM5CWT cells (p > .05, Mann-Whitney; Fig.5B). Once the KDM5CY751C cells were stably transfected following antibiotic selection, designated as KDM5CY751C + Ntng2 cell, the Ntng2 expression reached a significantly higher level than that of the control KDM5CY751C and KDM5CWT cells (U = 0, Z = 1.96, p = 0.0495 in both cases, Mann-Whitney; Fig.5C).
Accompanying Ntng2 up-regulation, the percentage of cells with neurites was also increased in the KDM5CY751C + Ntng2 cells, which became comparable to that of the KDM5CWT cells and significantly higher than that of the control KDM5CY751C cells (KDM5CWT: 65.4 ± 2.2 %; control KDM5CY751C: 33.5 ± 2.5 %; KDM5CY751C + Ntng2: 56.8 ± 2.8 %; U = 0, Z = 2.3, p = 0.021, between control KDM5CY751C and KDM5CY751C + Ntng2 or KDM5CWT cells; p > 0.05 between KDM5CY751C + Ntng2 and KDM5CWT cells; Mann-Whitney, Fig.5C). Therefore, the phenotype of short neurites in KDM5CY751C cells can be rescued by up-regulating Ntng2, suggesting that this gene indeed plays a role in the effect of KDM5CY751C mutation on neuronal development.
Discussion
An increasing number of mutations have been detected in the KDM5C gene among individuals with intellectual disability (Jensen et al., 2005, Tzschach et al., 2006, Adegbola et al., 2008, Rujirabanjerd et al., 2010, Santos-Reboucas et al., 2011). A majority of these individuals have additional symptoms, such as epilepsy, delayed language development, and impulsivity, which are more variable, exhibited by some individuals but not others. No explanation is currently available for the high degree of individual differences in symptoms. It is also not clear how KDM5C is implicated in discrete neurodevelopmental events, including neuronal differentiation, dendritic growth, and regulation of apoptosis, possibly involving different protein interaction partners and gene targets of KDM5C (Iwase et al., 2007, Tahiliani et al., 2007). Our recent study of the Kdm5c deficient mice suggests that the dendritic abnormality seem to be particularly relevant to the behavioral phenotypes of these animals (Iwase et al., 2016), although it remains to be tested whether a Kdm5c deletion mutation affects neuronal development similarly to that of KDM5C patient mutations. Moreover, enzymatic activity analysis of KDM5C patient mutations indicates that they compromise the histone demethylase activity, i.e. they are loss-of-function mutations. However, no similar test has been done in neurons or animals.
To investigate the effects of KDM5C patient mutations on neuronal development, dendrite growth in particular, we expressed these mutations in N2a cells. These are mouse neuroblastoma cells that have gained increasing popularity in recent years for functionality studies, including analysis of guidance cues (e.g. Netrins, Slits, Ephrins, and Semaphorins) and cell adhesion molecules as well as intracellular signaling pathways, determining the contributions of these genes to neuronal differentiation, axon/dendrite growth, and cell migration (Goshima et al., 1993, Nakata and Troy, 2005, Jankowski et al., 2006, Dasgupta and Milbrandt, 2007, Shin et al., 2007, Yoong and Too, 2007, Schwaibold and Brandt, 2008, Chen et al., 2011, Hagiyama et al., 2011, Li et al., 2011, Takahashi et al., 2012, Ito et al., 2014). In these studies, the gene of interest is typically mutated, knocked down, or over-expressed in N2a cells, taking advantage of the high efficiency of plasmid transfection in these cells. Analytical procedures are then performed in these genetically modified cells, in particular the ones that require a large number of homogeneous cells, such as chromatin immunoprecipitation, gene expression, and protein pull-down assays. It is common in these and many other studies using N2a cells that findings obtained in N2a cells are subsequently reproduced or collaborated by analyses of primary neurons and/or animal models, either in the same or separate studies, lending credibility to the approach that we adopted in this project. Additional concerns for validity of this system include that, as neuroblastoma cells, N2a cells likely have a gene expression profile different from that of neural progenitor cells; the concentrations of the exogenous, mutant or wild type, KDM5C proteins in the stably transfected N2a cells are likely much higher than that found in a normal human cell, and subsequently genes not normally regulated by KDM5C are now subjected to its regulation; ectopic regulation could also be due to a mix-match between a human KDM5C protein and the mouse genome of N2a cells. The concern of interspecific incompatibility is somewhat ameliorated by the high similarity between humans and mice in the KDM5C amino acid sequences (Agulnik et al., 1994), and the comparable enzymatic activities between the human and mouse KDM5C protein – a human KDM5C protein is capable of rescuing the morphological phenotype in rat neurons with Kdm5c deficiency (Iwase et al., 2007). Moreover, a crucial confirmation for us came from the RNA-seq analysis between the three cell lines, GFP, KDM5CWT, and KDM5CY751C, which led to a cluster of five genes important for neuron projection morphogenesis. This group of genes was in fact top-ranked among three clusters identified by functional annotation, and all five genes were up-regulated by RA in GFP and KDM5CWT cells at adjusted p < .05; however, in line with the reduced neurite growth of KDM5CY751C cells, the five genes were not up-regulated by RA in these cells. Therefore, these N2a cells are indeed an informative model which provides clues to the candidate genes and pathways relevant to the phenotypes elicited by KDM5C mutations.
It's particularly exciting to identify Ntng2 as a target for KDM5C patient mutations which was down-regulated in the KDM5CY751C cells and likely contributed to the short neurite phenotype. Netrin G2, the protein product of Ntng2, is a glycosyl phosphatidyl-inositol-linked membrane protein and a member of the Netrin family of axon guidance molecules (Rajasekharan and Kennedy, 2009). It is highly expressed in the developing brain, where it is typically anchored on the cell membrane and promotes the outgrowth of both axons and dendrites (Nakashiba et al., 2002). It has a close homologue Netrin G1, which is distributed in the brain in a complementary fashion. These two homologues bind to different postsynaptic receptor proteins – Netrin G1 with NGL1 while G2 with NGL2 (Nakashiba et al., 2002). Together they ensure the separation of afferent axons into segments along a dendrite based on the presence of Netrin G1 or G2 at the axonal terminal, creating dendritic segments that consist of primarily exhibitory or inhibitory synapses (Nishimura-Akiyoshi et al., 2007, DeNardo et al., 2012). In N2a cells, Ntng2 is up-regulated by RA, consistent with its role in neurite growth, as shown in the GFP and KDM5CWT cells. In contrast, in KDM5CY751C N2a cells, the KDM5CY751C protein was detected at the Ntng2 promoter using ChIP assays, concordant with the low levels of H3K4me3 at this DNA sequence and down-regulated Ntng2 expression. This mutation is within a Zinc finger domain, possibly affecting protein-protein and/or protein-DNA interaction. It is a gain-of-function mutation, at least in N2a cells, in contrast to the results of a prior analysis where all KDM5C mutations examined were found to compromise the H3K4 demethylase activity and thus categorized as loss-of-function mutations (Iwase et al., 2007). The Ntng2 down-regulation is likely related to the neurite growth deficits of KDM5CY751C N2a cells, since an over-expression of Ntng2 successfully rescued this phenotype (Fig. 5). If Ntng2 is similarly down-regulated in individuals with this KDM5C mutation, neural circuits might be affected as a result, which in turn bring about neuropsychiatric symptoms. It should point out that, although we focused on Ntng2 as the target gene for KDM5C patient mutations, the other genes identified by RNA-seq, e.g. Dscam, Gas1, Enah, and Slit2, could also play a role in the neurite phenotype. In fact, a great deal could be learned about neurite growth of N2a cells from an ongoing bioinformatics analysis of the RNA-seq datasets.
It's not clear why neurite growth of N2a cells is affected by some KDM5C patient mutations but not other ones. No clear pattern can be readily made between the two types of mutations, not by the clinical features or by locations in the KDM5C protein. One interesting possibility is that different neurodevelopmental processes might be affected by different mutations, with some mutations causing changes in neuronal differentiation or apoptosis instead of dendritic growth. More information will likely become available soon about the functional relevance of its protein motifs from the large scale, genome-wide association studies, which will identify a large number of KDM5C sequence variations and the associated physiological features. At the same time, biochemical analysis could be informative by determining the KDM5C protein interaction partners and its genomic binding sites. One excitement is to perform this type of analysis in iPSCs derived from patients.
On the other hand, a similar phenotype of reduced neurite growth was noticed in various model systems, including N2a cells with KDM5C patient mutations, Kdm5c knockdown N2a cells, and cultured neurons deficient for Kdm5c. However, different genes seem to be affected by a mutant KDM5C protein and a deficiency of this protein. For one, the mutant KDM5C could target genes different from that of the wild type KDM5C protein. It is also possible that KDM5C deficiency might be compensated for by its homologues (e.g. KDM5A, KDM5B, and KDM5D), which is less likely if the mutant KDM5C proteins continue their occupation at the target genes. Both Kdm5c KD and KO cells grow shorter neurites without Ntng2 down-regulation; there are therefore other genes in these cells that are misregulated and responsible for the neurite reduction. It remains a possibility that these genes and Ntng2 are implicated in the same genetic pathways.
In sum, we established N2a cell lines stably transfected with KDM5C mutations, and demonstrated the merits of this model system for identification of genes targeted by these mutations. We verified specifically that Ntng2 is down-regulated by KDM5CY751C, and it mediates the effects of this mutation on neurite growth. These results shed new light on the pathogenesis of KDM5C mutations.
Acknowledgments
We thank Dr. Shigeyoshi Itohara for kindly providing us with the Ntng2 plasmid. This study was supported by NIH grants MH096066 (JX), NS089896 (SI), GM113943 (CMD), and GM046883 (CMD), as well as an award from the Farrehi Family Foundation (SI).
References
- Abidi FE, Holloway L, Moore CA, Weaver DD, Simensen RJ, Stevenson RE, Rogers RC, Schwartz CE. Mutations in JARID1C are associated with X-linked mental retardation, short stature and hyperreflexia. J Med Genet. 2008;45:787–793. doi: 10.1136/jmg.2008.058990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD) Am J Med Genet A. 2008;146A:505–511. doi: 10.1002/ajmg.a.32142. [DOI] [PubMed] [Google Scholar]
- Aguilar-Valles A, Vaissiere T, Griggs EM, Mikaelsson MA, Takacs IF, Young EJ, Rumbaugh G, Miller CA. Methamphetamine-associated memory is regulated by a writer and an eraser of permissive histone methylation. Biol Psychiatry. 2014;76:57–65. doi: 10.1016/j.biopsych.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulnik AI, Mitchell MJ, Mattei MG, Borsani G, Avner PA, Lerner JL, Bishop CE. A novel X gene with a widely transcribed Y-linked homologue escapes X-inactivation in mouse and human. Human molecular genetics. 1994;3:879–884. doi: 10.1093/hmg/3.6.879. [DOI] [PubMed] [Google Scholar]
- Aoki-Suzuki M, Yamada K, Meerabux J, Iwayama-Shigeno Y, Ohba H, Iwamoto K, Takao H, Toyota T, Suto Y, Nakatani N, Dean B, Nishimura S, Seki K, Kato T, Itohara S, Nishikawa T, Yoshikawa T. A family-based association study and gene expression analyses of netrin-G1 and -G2 genes in schizophrenia. Biol Psychiatry. 2005;57:382–393. doi: 10.1016/j.biopsych.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Beaudoin GM, 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nature protocols. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Chen K, Mi YJ, Ma Y, Fu HL, Jin WL. The mental retardation associated protein, srGAP3 negatively regulates VPA-induced neuronal differentiation of Neuro2A cells. Cellular and molecular neurobiology. 2011;31:675–686. doi: 10.1007/s10571-011-9664-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128:1063–1076. doi: 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo LA, de Wit J, Otto-Hitt S, Ghosh A. NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron. 2012;76:762–775. doi: 10.1016/j.neuron.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V. X-linked mental deficiency. Handbook of clinical neurology. 2013;111:297–306. doi: 10.1016/B978-0-444-52891-9.00035-X. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Ohsako S, Yamauchi T. Overexpression of Ca2+/calmodulin-dependent protein kinase II in Neuro2a and NG108-15 neuroblastoma cell lines promotes neurite outgrowth and growth cone motility. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1993;13:559–567. doi: 10.1523/JNEUROSCI.13-02-00559.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiyama M, Furuno T, Hosokawa Y, Iino T, Ito T, Inoue T, Nakanishi M, Murakami Y, Ito A. Enhanced nerve-mast cell interaction by a neuronal short isoform of cell adhesion molecule-1. Journal of immunology (Baltimore, Md : 1950) 2011;186:5983–5992. doi: 10.4049/jimmunol.1002244. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ito Y, Asada A, Kobayashi H, Takano T, Sharma G, Saito T, Ohta Y, Amano M, Kaibuchi K, Hisanaga S. Preferential targeting of p39-activated Cdk5 to Rac1-induced lamellipodia. Molecular and cellular neurosciences. 2014;61:34–45. doi: 10.1016/j.mcn.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Iwase S, Brookes E, Agarwal S, Badeaux AI, Ito H, Vallianatos CN, Tomassy GS, Kasza T, Lin G, Thompson A, Gu L, Kwan KY, Chen C, Sartor MA, Egan B, Xu J, Shi Y. A Mouse Model of X-linked Intellectual Disability Associated with Impaired Removal of Histone Methylation. Cell reports. 2016;14:1000–1009. doi: 10.1016/j.celrep.2015.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jankowski MP, Cornuet PK, McIlwrath S, Koerber HR, Albers KM. SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience. 2006;143:501–514. doi: 10.1016/j.neuroscience.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, Van Esch H, Kleefstra T, Hamel B, Moraine C, Gecz J, Turner G, Reinhardt R, Kalscheuer VM, Ropers HH, Lenzner S. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. American journal of human genetics. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Kurosawa N, Nishi T, Hanai N, Tsuji S. Induction of cholinergic differentiation with neurite sprouting by de novo biosynthesis and expression of GD3 and b-series gangliosides in Neuro2a cells. The Journal of biological chemistry. 1994;269:30451–30456. [PubMed] [Google Scholar]
- Li M, Armelloni S, Ikehata M, Corbelli A, Pesaresi M, Calvaresi N, Giardino L, Mattinzoli D, Nistico F, Andreoni S, Puliti A, Ravazzolo R, Forloni G, Messa P, Rastaldi MP. Nephrin expression in adult rodent central nervous system and its interaction with glutamate receptors. The Journal of pathology. 2011;225:118–128. doi: 10.1002/path.2923. [DOI] [PubMed] [Google Scholar]
- Meerabux JM, Ohba H, Fukasawa M, Suto Y, Aoki-Suzuki M, Nakashiba T, Nishimura S, Itohara S, Yoshikawa T. Human netrin-G1 isoforms show evidence of differential expression. Genomics. 2005;86:112–116. doi: 10.1016/j.ygeno.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Nishimura S, Ikeda T, Itohara S. Complementary expression and neurite outgrowth activity of netrin-G subfamily members. Mechanisms of development. 2002;111:47–60. doi: 10.1016/s0925-4773(01)00600-1. [DOI] [PubMed] [Google Scholar]
- Nakata D, Troy FA., 2nd Degree of polymerization (DP) of polysialic acid (polySia) on neural cell adhesion molecules (N-CAMS): development and application of a new strategy to accurately determine the DP of polySia chains on N-CAMS. The Journal of biological chemistry. 2005;280:38305–38316. doi: 10.1074/jbc.M508762200. [DOI] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nature protocols. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- Nishimura-Akiyoshi S, Niimi K, Nakashiba T, Itohara S. Axonal netrin-Gs transneuronally determine lamina-specific subdendritic segments. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14801–14806. doi: 10.1073/pnas.0706919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted JB, Carlson K, Klebe R, Ruddle F, Rosenbaum J. Isolation of microtubule protein from cultured mouse neuroblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1970;65:129–136. doi: 10.1073/pnas.65.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekharan S, Kennedy TE. The netrin protein family. Genome Biol. 2009;10:239. doi: 10.1186/gb-2009-10-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH, Hamel BC. X-linked mental retardation. Nature reviews Genetics. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- Rujirabanjerd S, Nelson J, Tarpey PS, Hackett A, Edkins S, Raymond FL, Schwartz CE, Turner G, Iwase S, Shi Y, Futreal PA, Stratton MR, Gecz J. Identification and characterization of two novel JARID1C mutations: suggestion of an emerging genotype-phenotype correlation. Eur J Hum Genet. 2010;18:330–335. doi: 10.1038/ejhg.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Reboucas CB, Fintelman-Rodrigues N, Jensen LR, Kuss AW, Ribeiro MG, Campos M, Jr, Santos JM, Pimentel MM. A novel nonsense mutation in KDM5C/JARID1C gene causing intellectual disability, short stature and speech delay. Neuroscience letters. 2011;498:67–71. doi: 10.1016/j.neulet.2011.04.065. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaibold EM, Brandt DT. Identification of Neurochondrin as a new interaction partner of the FH3 domain of the Diaphanous-related formin Dia1. Biochemical and biophysical research communications. 2008;373:366–372. doi: 10.1016/j.bbrc.2008.06.042. [DOI] [PubMed] [Google Scholar]
- Shen E, Shulha H, Weng Z, Akbarian S. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Gu C, Park E, Park S. Identification of phosphotyrosine binding domain-containing proteins as novel downstream targets of the EphA8 signaling function. Molecular and cellular biology. 2007;27:8113–8126. doi: 10.1128/MCB.00794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, Watkins KL, Johnson RE, Schottler F, Kerschensteiner D. NGL-2 regulates pathway-specific neurite growth and lamination, synapse formation, and signal transmission in the retina. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:11949–11959. doi: 10.1523/JNEUROSCI.1521-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Mitoma J, Hosono M, Shiozaki K, Sato C, Yamaguchi K, Kitajima K, Higashi H, Nitta K, Shima H, Miyagi T. Sialidase NEU4 hydrolyzes polysialic acids of neural cell adhesion molecules and negatively regulates neurite formation by hippocampal neurons. The Journal of biological chemistry. 2012;287:14816–14826. doi: 10.1074/jbc.M111.324186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschach A, Lenzner S, Moser B, Reinhardt R, Chelly J, Fryns JP, Kleefstra T, Raynaud M, Turner G, Ropers HH, Kuss A, Jensen LR. Novel JARID1C/SMCX mutations in patients with X-linked mental retardation. Hum Mutat. 2006;27:389. doi: 10.1002/humu.9420. [DOI] [PubMed] [Google Scholar]
- Vashishtha M, Ng CW, Yildirim F, Gipson TA, Kratter IH, Bodai L, Song W, Lau A, Labadorf A, Vogel-Ciernia A, Troncosco J, Ross CA, Bates GP, Krainc D, Sadri-Vakili G, Finkbeiner S, Marsh JL, Housman DE, Fraenkel E, Thompson LM. Targeting H3K4 trimethylation in Huntington disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E3027–3036. doi: 10.1073/pnas.1311323110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynder C, Stalker L, Doughty ML. Role of H3K4 demethylases in complex neurodevelopmental diseases. Epigenomics. 2010;2:407–418. doi: 10.2217/epi.10.12. [DOI] [PubMed] [Google Scholar]
- Yoong LF, Too HP. Glial cell line-derived neurotrophic factor and neurturin inhibit neurite outgrowth and activate RhoA through GFR alpha 2b, an alternatively spliced isoform of GFR alpha 2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:5603–5614. doi: 10.1523/JNEUROSCI.4552-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]