

Abstract
Background
Vascular endothelial growth factor (VEGF) and its receptors (VEGFR-1 and VEGFR-2) regulate vascular permeability and endothelial cell survival. We hypothesized that hemorrhagic shock (HS) and chronic stress (CS) would increase expression of lung VEGF and its receptors, potentiating pulmonary edema in lung tissue.
Materials and Methods
8-9 week old male Sprague-Dawley rats were randomized: naïve control, lung contusion (LC), LC followed by HS (LCHS), and LCHS with CS in a restraint cylinder for two hours per day (LCHS/CS). Animals were sacrificed on day one and day seven. Expression of lung VEGF, VEGFR-1, VEGFR-2 was determined by PCR. Lung Injury Score (LIS) was graded on light microscopy by inflammatory cell counts, interstitial edema, pulmonary edema, and alveolar integrity (range: 0=normal; 8=severe injury).
Results
Seven days following LC, lung VEGF and VEGFR-1 were increased, and lung tissue healed (LIS 0.8±0.8). However, seven days after LCHS and LCHS/CS, lung VEGF and VEGFR-1 expression was decreased. VEGFR-2 was also decreased following LCHS/CS. LIS was elevated seven days after LCHS and LCHS/CS (6.5±1.0 and 8.2±0.8). Increased LIS following LCHS and LCHS/CS was due to higher inflammatory cell counts, increased interstitial edema, and loss of alveolar integrity, whereas pulmonary edema was unchanged.
Conclusions
Elevation of lung VEGF and VEGFR-1 expression following LC alone was associated with healing of injured lung tissue. Expression of VEGF, VEGFR-1, and VEGFR-2 was reduced following LCHS and LCHS/CS, and injured lung tissue did not heal. Persistent lung injury following severe trauma was due to inflammation rather than pulmonary edema.
Keywords: pulmonary contusion, lung injury, inflammation, pulmonary edema, vascular endothelial growth factor, tissue repair
Introduction
Vascular endothelial growth factor (VEGF) contributes to lung homeostasis by several mechanisms: modulating growth and apoptosis of vascular endothelial cells, increasing vascular permeability, potentiating pulmonary endothelial cell growth and survival, and stimulating type II pneumocyte surfactant production.1, 2 VEGF binds to two receptor tyrosine kinases: VEGFR-1 (also known as flt-1) and VEGFR-2 (also known as flk-1) 1. The physiology of VEGFR-1 remains controversial, in part because its signaling properties depend on the animal and cell type.1, 3 VEGFR-2 is the major mediator of VEGF-induced angiogenesis and vascular permeability.1 Lung VEGF is primarily produced by epithelial cells, and also produced in smaller quantities by endothelial cells, macrophages, and neutrophils.1, 4-7 Its major targets are VEGFR-1 and VEGFR-2 on endothelial cells, but also it targets type I and type II pneumocytes.8-10
VEGF is an essential growth and survival factor for pulmonary epithelial and endothelial cells, but also promotes vascular permeability in the lung, which may cause detrimental pulmonary edema.7, 11, 12 Therefore, both abnormally low and abnormally high lung VEGF activity may be harmful by different mechanisms. Several studies have investigated VEGF and VEGF receptor function following non-traumatic lung injury, producing inconsistent results, which may be attributable to differences in experimental models, the timing of interventions, and different approaches to measuring the potentially beneficial and adverse effects of lung VEGF.8, 12-16 However, the effects of traumatic lung injury on VEGF are unknown. Pulmonary contusion followed by hemorrhagic shock and chronic stress from the intensive care unit environment is a relatively common clinical scenario. Understanding of the role of VEGF in lung tissue repair under these conditions may elucidate therapeutic strategies. Based on evidence that hypoxia and hypercatecholaminemia increase VEGF and VEGF receptor expression and function,8, 17-21 we hypothesized that lung contusion (LC) and hemorrhagic shock (HS) would increase expression of lung VEGF and VEGF receptors, the addition of daily chronic restraint stress (CS) would exacerbate this effect, and that VEGF overexpression would be associated with pulmonary edema and impaired lung tissue healing.
Materials and Methods
Male Sprague-Dawley rats (Charles River, Raleigh, NC) age 8 weeks and weighing 300-400g were housed in pairs and fed ad lib with Teklad Diet #7912 (Harlan Laboratories Inc., Tampa, FL) and water during a one week acclimation period. Light and dark cycles were 12 hours each throughout acclamation and experimental periods. All animal care was conducted in accordance with University of Florida Institutional Animal Care and Use Committee standards. Animals were randomly allocated to one of four groups: naïve (n=8), LC (1 day model: n=5, 7 day model: n=7), LC followed immediately by HS (LCHS) (1 day model: n=5, 7 day model: n=7), and LCHS with daily chronic restraint stress (LCHS/CS) (1 day model: n=5, 7 day model: n=7). These injury models were chosen to recapitulate common clinical scenarios: isolated blunt chest trauma (LC), blunt chest trauma accompanied by hemorrhage with early recovery (LCHS), and blunt chest trauma accompanied by hemorrhage followed by chronic stressors associated with the intensive care unit environment.
Prior to LC and HS, animals were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg). Pentobarbital was used in order to avoid confounding effects on the neuroendocrine response to injury and stress. LC was performed by applying a percussive staple gun (PowerShot Model 5700M, Saddle Brook, NJ) to a 12 mm metal plate applied to the right lateral chest wall 1 cm below the axillary crease. This model has previously been shown to produce a clinically significant and reproducible pulmonary contusion based on histologic findings.22-24
Rats that would also undergo HS were then placed on a heating pad and PE-50 tubing was inserted into the right internal jugular vein and right femoral artery under direct visualization. The arterial catheter was used for continuous blood pressure monitoring with the BP-2 Digital Blood Pressure Monitor (Columbus Instruments, Columbus, OH). Blood was then withdrawn through the venous catheter into a heparinized syringe (10 units/ml) until a mean arterial pressure of 30-35 mm Hg was obtained. This blood pressure was maintained for a 45 minute period by withdrawing or reinfusing blood as necessary. After 45 minutes, shed blood was reinfused at a rate of 1 ml/min.
CS was performed by placing animals in a restraint cylinder (Kent Scientific Corporation, Torrington, CT) for two hours a day. CS began one day after LCHS in the LCHS/CS group. In order to prevent acclimation during the two hour period, the cylinders were rotated 180 degrees every 30 minutes, and alarms were transmitted by speakers placed immediately adjacent to the cylinders for a two minutes period each time the cylinders were rotated. Because animals undergoing CS had no access to food or water while in the restraint cylinder, all other groups were also subjected to a two hour daily fast. There were no deaths associated with LC alone, and no late deaths attributable to daily CS. HS was associated with approximately 15-20% mortality within 3 hours.
Animals were sacrificed by cardiac puncture following intraperitoneal injection of ketamine (80-100 mg/kg) and xylazine (5-10 mg/kg) on day 1 and day 7. Day 1 LC and LCHS rats were sacrificed one day after interventions were performed. Day 1 LCHS/CS rats had LCHS, then a single episode of CS occurring one day later, and were sacrificed one day after CS. Right lung, left lung, and plasma specimens were collected. Lung specimens were initially placed in phosphate buffered saline (PBS). Portions of the contused right lung and the non-contused left lung were placed in formalin for hematoxylin and eosin staining and histologic analysis by light microscopy, and a non-contused portion of the right lung was placed immediately in dry ice and then stored at -80°C. Plasma samples were obtained during cardiac puncture by withdrawing 7-10 ml of blood into a heparinized syringe (10 units/ml). Samples were stored at -80°C.
Lung VEGF, VEGFR-1, and VEGFR-2 expression were assessed by endpoint polymerase chain reaction. The following primers were selected: VEGF forward 5′ gtggacttgagttgggagga and reverse 5′ caaacagacttcggcctctc (product region: 2135-2228, product size: 147 bp), VEGFR-1 forward 5′ agtggctccacgaccttaga and reverse 5′ gaagaccgcttcagttttcgt (product region: 2258-2575, product size: 317 bp), VEGFR-2: forward 5′ acagcatcaccagcagtcag and reverse 5′ ccaagaactccatgccctta (product region: 3127-3274, product size: 147 bp). Amplifications were performed using a SimpliAmp thermal cycler (Applied Biosystems, Carlsbad, CA) with an initial 4 minute denaturation phase at 95°C, followed by 32 cycles with denaturation at 95°C, annealing at 60°C, and extension at 72°C for 45 seconds each. Products were separated on 1.5% agarose gel stained with Ethidium Bromide (Invitrogen, Carlsbad, CA). VEGF, VEGFR-1, and VEGFR-2 were assessed in the contused right lung and the non-contused left lung to determine if effects were consistent with local tissue trauma (seen in the contused right lung only), or systemic changes in VEGF physiology.
Plasma VEGF was measured by enzyme linked immunosorbent assay (Quantikine ELISA Rat VEGF, R&D Systems, Minneapolis, MN) according to manufacturer instructions. All samples were run in duplicate. 50 μl of assay diluent were added to each well followed by 50 μl of the standard solution, control solution, or rat plasma. The plate was incubated at room temperature for two hours on a microplate shaker at 500 rpm. Each well was then aspirated and washed with 400 μl of wash buffer. This aspiration and wash sequence was repeated four more times. 100 μl of rat VEGF conjugate were then added to each well. The plate was then incubated for one hour at room temperature on a horizontal microplate shaker at 500 rpm. The aspiration and wash sequence was repeated five more times. 100 μl of stop solution was then added to each well. Optical density of each well was determined with a microplate reader set at 450 nm with wavelength correction set at 540 nm.
Lung injury was assessed by a board certified pathologist using light microscopy to visualize hematoxylin and eosin stained portions of the contused right lung. The pathologist was blinded to the study group and day of tissue procurement. The degree of lung injury was reported according to the LIS scoring system described in Table 1, adapted from Claridge et al.25 and Matute-Bello et al.26
Table 1.
Lung Injury Score calculator (HPF: high-power field).
| Points | |
|---|---|
| Inflammatory cells/HPF | |
| <5 | 0 |
| 6-10 | 1 |
| 11-15 | 2 |
| 16-20 | 3 |
| >20 | 4 |
| Interstitial edema | |
| None | 0 |
| Minimal | 1 |
| Moderate | 2 |
| Severe | 3 |
| Pulmonary edema | |
| <5% | 0 |
| 5-25% | 1 |
| >25% | 2 |
| Alveolar integrity | |
| Normal | 0 |
| Moderately abnormal | 1 |
| Severely abnormal | 2 |
Statistical analysis was performed using GraphPad Prism version 6.05 (GraphPad Software, La Jolla, CA) to calculate one-way analysis of variance. Significance was set at α = 0.05 and data were reported as mean ±standard deviation.
Results
Day 1 and day 7: VEGF, VEGFR-1, and VEGFR-2
In the contused right lung, VEGF expression remained similar to naïve expression on day 1 for all injury models (Figure 1). Following LC alone, VEGF expression increased over the course of seven days. However, seven days following LCHS and LCHS/CS, VEGF expression decreased 22% and 29%, respectively (Figure 1). On day 7, lung VEGF expression was significantly lower following LCHS and LCHS/CS compared to naïve animals (Figure 1). VEGF was undetectable in the plasma in all four groups: naïve, LC, LCHS, and LCHS/CS (data not shown).
Figure 1.
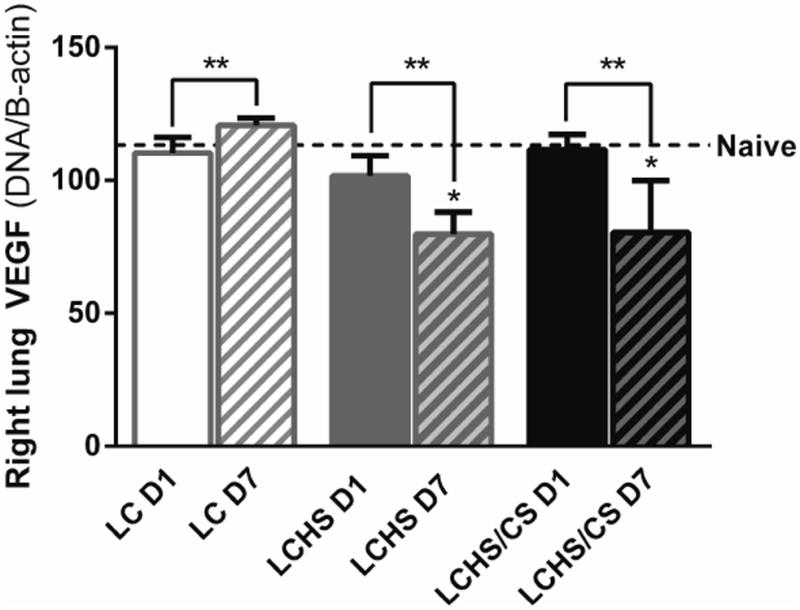
VEGF expression in the contused right lung increased over time following LC alone but was suppressed seven days after LCHS and LCHS/CS. (LC: lung contusion; D1: day 1; D7: day 7; HS: hemorrhagic shock; CS: chronic stress; *p < 0.05 vs. Naïve; **p < 0.05 within the group)
Lung VEGFR-1 expression followed a nearly identical pattern to VEGF expression (Figure 2A). On day 1, LC and LCHS/CS VEGFR-1 expression was similar to that of naïve animals. LCHS VEGFR-1 expression did significantly increase on day 1 compared to naïve (LC: 95±11 vs. naïve: 80±9). Following LC alone, VEGFR-1 expression increased over the course of seven days (Figure 2A). In contrast, seven days following LCHS and LCHS/CS, VEGFR-1 expression decreased 45% and 61%, respectively (Figure 2A). On day 7, lung VEGFR-1 expression was significantly lower following LCHS and LCHS/CS compared to naïve animals (Figure 2A).
Figure 2.
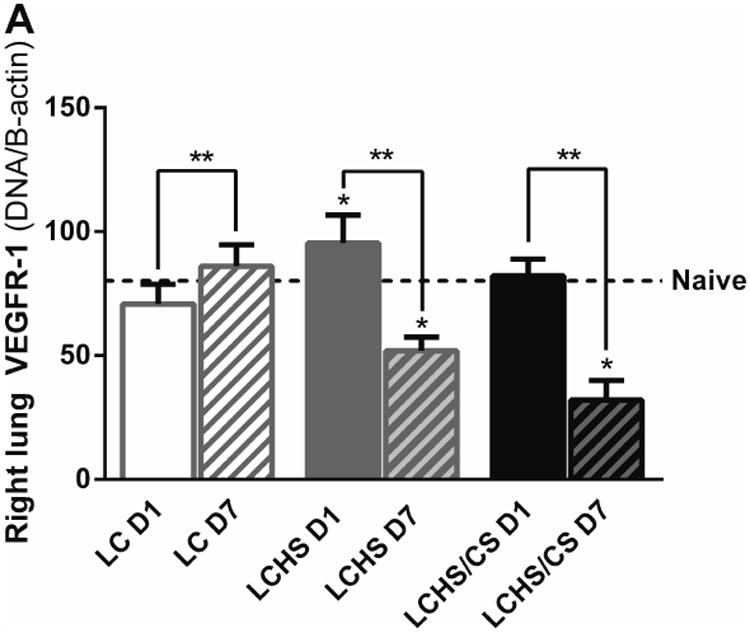
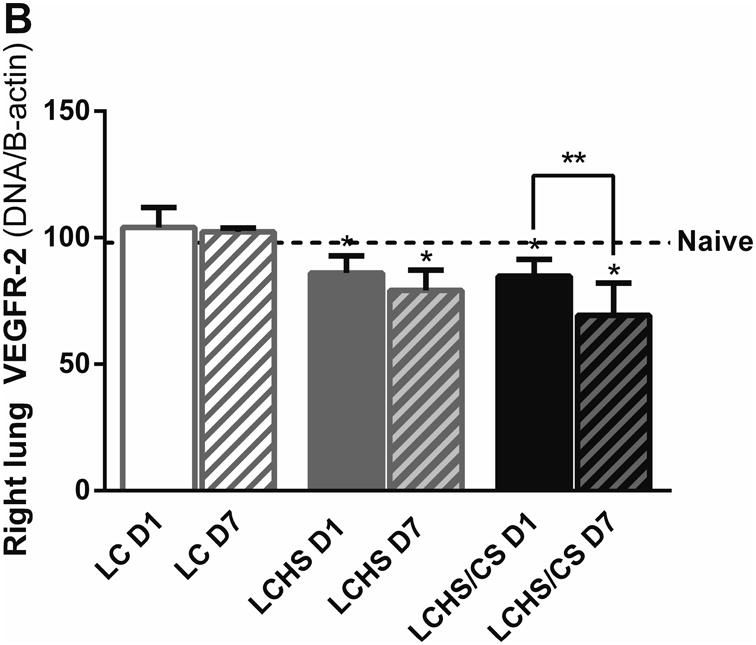
A: VEGFR-1 expression in the contused right lung increased seven days after LC alone but was decreased following LCHS and LCHS/CS. B: VEGFR-2 expression in the contused right lung was decreased after LCHS and LCHS/CS. (LC: lung contusion; D1: day 1; D7: day 7; HS: hemorrhagic shock; CS: chronic stress; *p < 0.05 vs. Naïve; **p < 0.05 within the group)
There was no change in lung VEGFR-2 expression from day 1 to day 7 following LC alone (Figure 2B). Day 1 VEGFR-2 expression was significantly decreased in LCHS and LCHS/CS groups compared to naïve (Figure 2B). Following LCHS, VEGFR-2 expression remained low, but did not significantly change from day 1 to day 7 (Figure 2B). Following LCHS/CS, VEGFR-2 expression decreased 19% from day 1 to day 7 (Figure 2B). For VEGF, VEGFR-1, and VEGFR-2, the greatest magnitude of suppression uniformly occurred in the LCHS/CS group.
Assessment of the non-contused left lung
VEGF, VEGFR-1, and VEGFR-2 were also assessed in the non-contused left lung at 7 days to determine if the observed effects in the right lung were due to local tissue injury or systemic effects. In the non-contused left lung, VEGF expression was unchanged compared to naïve seven days following LC, LCHS, and LCHS/CS (Figure 3), suggesting that the increase in VEGF expression following LC and the decrease in VEGF expression following LCHS and LCHS/CS was related to local tissue injury.
Figure 3.
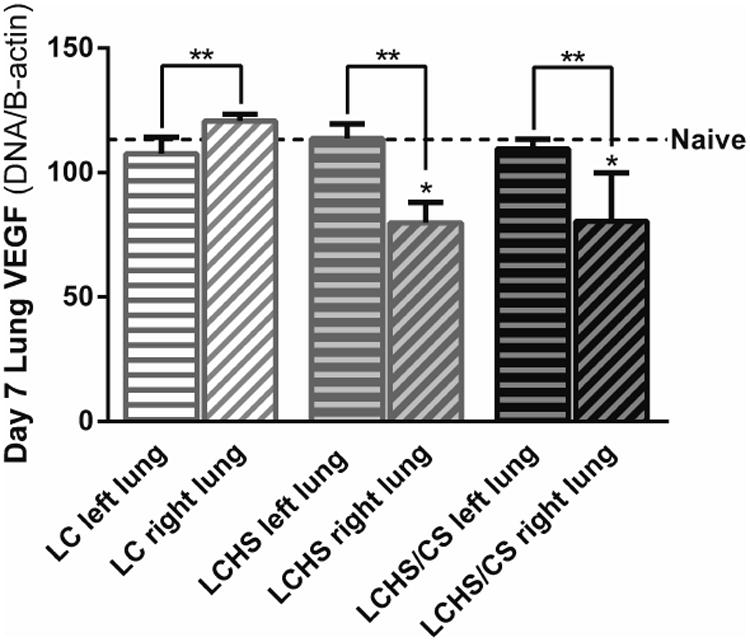
VEGF expression in the non-contused left lung was similar to naïve expression seven days following all three injury models. (LC: lung contusion; HS: hemorrhagic shock; CS: chronic stress; *p < 0.05 vs. Naïve; **p < 0.05 within the group)
Similarly, VEGFR-1 expression was increased seven days following LC, LCHS, and LCHS/CS in the non-contused left lung compared to naïve (Figure 4A). Compared to the non-contused left lung, VEGFR-1 expression in the contused right lung was decreased 18% following LC, 55% following LCHS, and 71% following LCHS/CS (Figure 4A).
Figure 4.
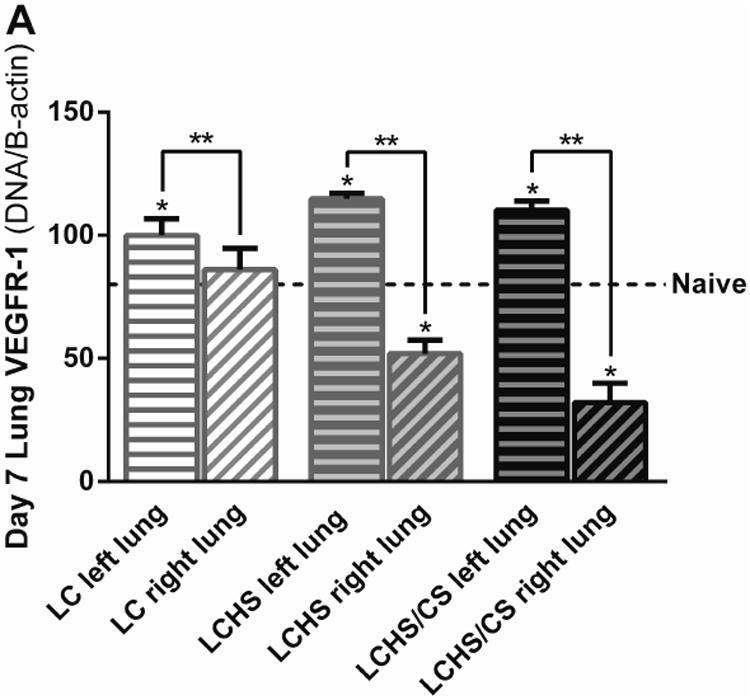
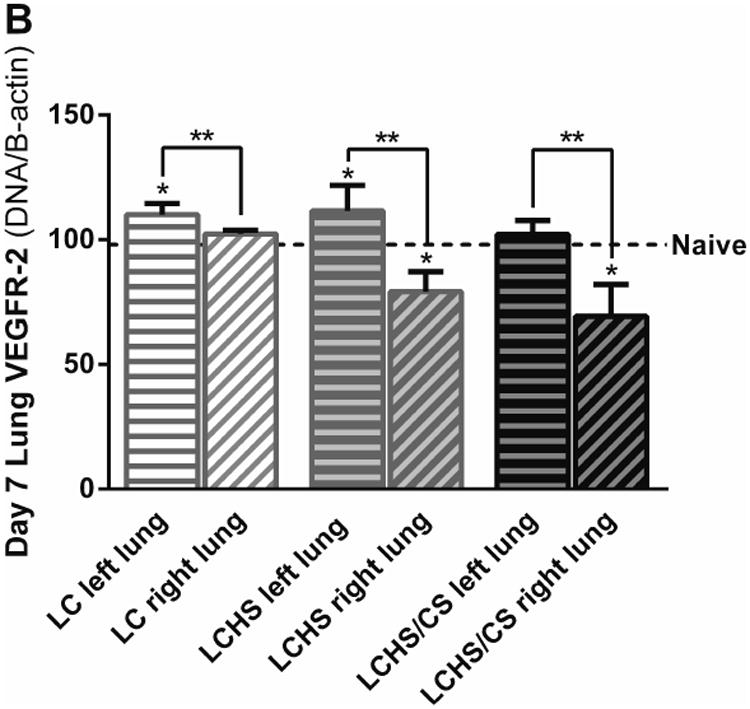
A: VEGFR-1 expression in the non-contused left lung was increased seven days following LCHS and LCHS/CS. B: VEGFR-2 expression in the non-contused left lung was increased seven days following LC and LCHS. (LC: lung contusion; HS: hemorrhagic shock; CS: chronic stress; *p < 0.05 vs. Naïve; **p < 0.05 within the group)
Figure 4B illustrates VEGFR-2 expression in the non-contused left lung and the contused right lung seven days after injury. VEGFR-2 expression was increased seven days following LC and LCHS in the non-contused left lung compared to naïve (Figure 4B). Compared to the non-contused left lung, VEGFR-1 expression in the contused right lung was decreased 3% following LC, 32% following LCHS, and 43% following LCHS/CS (Figure 4B).
Histologic characterization of lung injury
Histologic evidence of lung injury correlated with VEGF and VEGFR-1 expression. Seven days following LC, lung tissue healed completely with a LIS score of 0.8±0.4 (Figure 5A and 5B). Seven days after LCHS, lung tissue has not healed, with a LIS score of 6.5±1.0. With the addition of CS following LCHS, LIS score remained elevated at 8.2±0.8 (Figure 5A and 5B). When further examining the differences in the LIS between groups, significant changes were primarily due to inflammatory cell counts, interstitial edema, and alveolar integrity (Table 2). There were no significant differences in pulmonary edema among naïve, LC, LCHS, and LCHS/CS animals.
Figure 5.
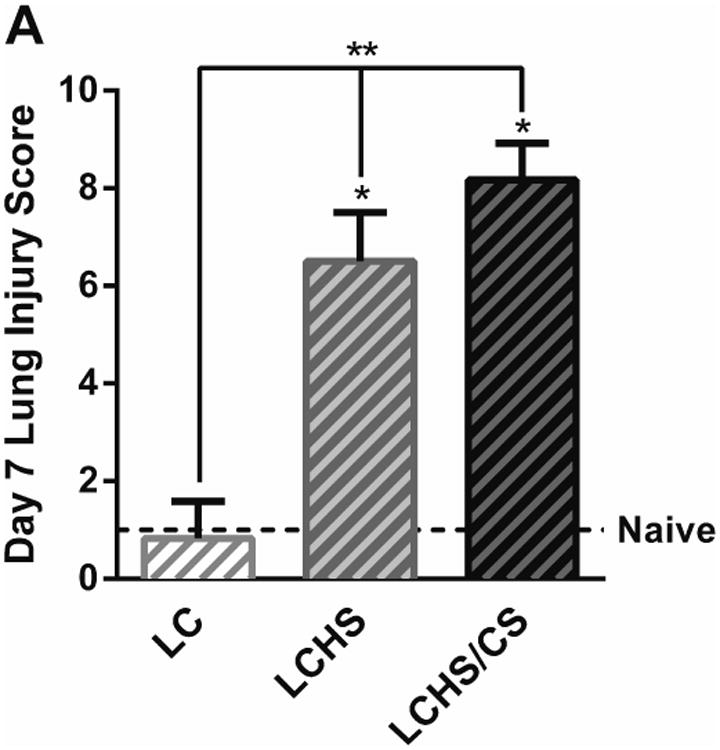
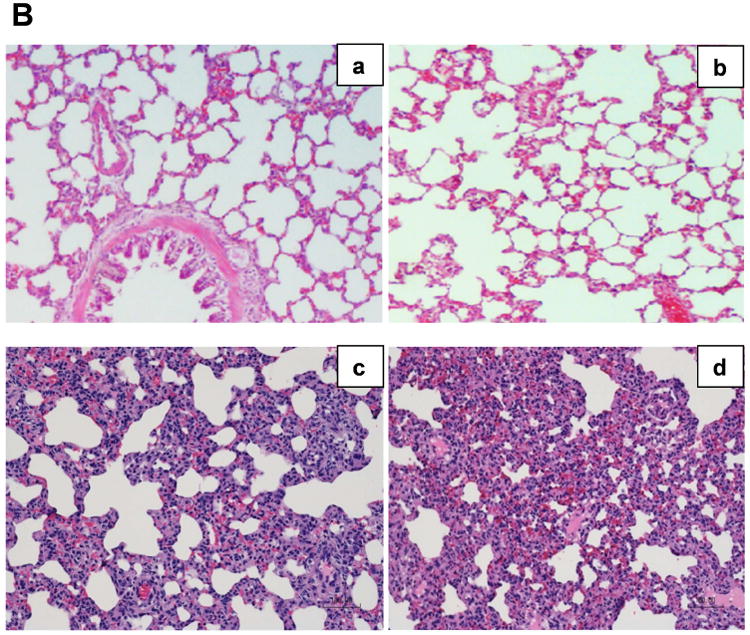
A: Contused lung tissue healed completely seven days following LC alone, but healing was impaired following LCHS and more severely impaired by the addition of CS. B: Hematoxylin and eosin stained rat lungs for the following groups: a=Naïve, b=LC, c=LCHS, d=LCHS/CS (LC: lung contusion; HS: hemorrhagic shock; CS: chronic stress; *p < 0.05 vs. Naïve; **p < 0.05 between injury models)
Table 2. Composition of the Lung Injury Score for each model.
| Naïve | LC | LCHS | LCHS/CS | |
|---|---|---|---|---|
| Inflammatory cells/HPF | 0.3 ±0.5 | 0.0 ±0.0 | 3.3 ±0.5* | 3.8 ±0.4* |
| Interstitial edema | 0.0 ±0.0 | 0.8 ±0.4 | 2.0 ±0.0* | 2.2 ±0.4* |
| Pulmonary edema | 0.7 ±0.5 | 0± 0.0 | 0.0 ±0.0 | 0.3 ±0.5 |
| Alveolar integrity | 0.0 ±0.0 | 0± 0.0 | 1.3 ±1.0* | 1.8 ±0.4* |
| Lung Injury Score | 1.0 ±0.9 | 0.8 ±0.4 | 6.5 ±1.0* | 8.2 ±0.8*# |
LC: lung contusion; HS: hemorrhagic shock; CS: chronic stress; HPF: high-power field;
p < 0.05 vs. LC;
p < 0.05 vs. LCHS.
Data are reported as mean ±standard deviation.
Discussion
In this study, VEGF, VEGFR-1 and VEGFR-2 expression was suppressed by the combination of traumatic injury and hemorrhagic shock, and daily chronic stress exacerbated these effects. In addition, decreased VEGF and VEGF receptor expression correlated with impaired tissue healing. Although VEGFR-1 and VEGFR-2 both appeared to be associated with this process, VEGFR-1 played a more dominant role and correlated more closely with histologic findings, especially when adding daily CS to LCHS. Unlike lung VEGF, VEGFR-1 and VEGFR-2 expression were elevated in the non-contused left lung compared to naïve animals, which may indicate the presence of a systemic upregulation of VEGF receptor expression following injury. Interestingly, pulmonary edema was the only factor that did not contribute to higher LIS among the LCHS and LCHS/CS groups, despite the propensity of VEGF to increase vascular permeability.7 The fact that alveolar integrity was adversely affected by LCHS and LCHS/CS models is also consistent with the ability of VEGF to promote growth and survival for pulmonary epithelial and endothelial cells.11, 12 Finally, these effects appeared to be related to direct tissue injury, as there was no evidence of VEGF or VEGF receptor suppression in the non-contused left lung. Previous experiments have shown that LIS one day following LC is 8.5, suggesting that differences in day seven LIS among different injury models represent the presence of absence of tissue repair, rather than a delayed measure of the original injury severity.27
Our data did not support our hypothesis, which was based on previous work in non-traumatic lung injury models, hemorrhagic shock models without tissue injury, and cell line studies which may explain our current findings. Acute hypoxia has been shown to increase lung tissue mRNA expression of lung VEGF and its receptors.8 After hemorrhagic shock with resuscitation, lung VEGF and VEGFR-1 levels were moderately increased, and VEGFR-2 levels were similar to controls.17 Hemorrhagic shock without resuscitation resulted in high levels of VEGF and VEGFR-1 and moderately increased VEGFR-2.17 Overexpression of lung VEGF has been associated with pulmonary edema in an animal model utilizing intratracheal delivery of VEGF complementary protein by an adenovirus vector.7 The effects of tissue injury followed by hemorrhagic shock and the effects of chronic stress on lung VEGF and its receptors have not previously been investigated. We presumed that the effects of catecholamines on VEGF would serve as an appropriate surrogate. In cancer cells, VEGF expression is dependent on upon hypoxia-inducible factor (HIF)-1 induction. 18-21 HIF-1 is stimulated by norepinephrine (NE) in a dose-dependent fashion, and the non-selective beta blocker propranolol significantly inhibits NE-induced HIF-1 protein production and VEGF expression.18-21 In our rat model, the addition of chronic stress to LCHS increased urine NE levels.28
Considering each of these factors, it seemed reasonable that hemorrhagic shock and chronic stress would each increase VEGF expression. However, observed increases in VEGF and VEGF receptor expression following lung contusion alone correlated with lung tissue healing in this study, and is consistent with previous work demonstrating that persistently low levels of alveolar neutrophil VEGF (6.3 pg/ml) are associated with prolonged illness in human subjects with Acute Respiratory Distress Syndrome (ARDS), whereas higher levels (13.0 pg/ml) correlate with recovery four days after the onset of symptoms.29 In our study, decreased expression of VEGF, VEGFR-1, and VEGFR-2 following LCHS and LCHS/CS were local effects seen only in the injured lung tissue.
Despite these findings in the literature, the effects of VEGF on lung tissue healing remain largely unclear.8, 12-16 In addition, the relationship between traumatic lung injury and VEGF function has not been previously reported. Therefore, this study was largely exploratory, and had several limitations. The mechanisms by which VEGF and VEGF receptors are suppressed remain unknown. Hematopoietic progenitor cell mobilization and T regulatory cell function appear to play a role in lung tissue repair in our model,23, 24, 30 but the pathophysiology of lung tissue repair following LC, LCHS, and LCHS/CS is not fully understood. Also, accurate interpretation of our data depends upon assumptions that animals subjected to LC, LCHS, and LCHS/CS had changes in NE levels similar to those previously described,28 and that our hemorrhagic shock model resulted in acute and global hypoxia. Future research should investigate lung VEGF and VEGF receptor physiology following LC followed by CS with no component of HS, and assess the impact of resuscitation with crystalloid intravenous fluid rather than blood.
Conclusions
Lung VEGF and VEGFR-1 were increased seven days following lung contusion alone, and lung tissue healed completely under these conditions. VEGF and VEGFR-1 were decreased seven days following LCHS and LCHS/CS, and the injured lung tissue did not heal. VEGFR-2 expression was decreased seven days following LCHS/CS. The addition of chronic stress to LCHS resulted in further suppression of VEGFR-1, VEGFR-2, and higher LIS, implicating both VEGF receptors in the pathophysiology of chronic stress-induced impairment of lung tissue healing. VEGF and VEGF receptor expression were not suppressed in the non-contused lung, suggesting that these phenomena are dependent upon direct tissue injury. Future research should seek to clarify the mechanisms of VEGF and VEGF receptor suppression following traumatic injury, hemorrhagic shock, and chronic stress.
Acknowledgments
Disclosure: This work was supported in part by grants P30 AG028740 (PAE) awarded by the National Institute on Aging and by R01 GM113945-01 (PAE), R01 GM105893-01A1 (AMM), P50 GM111152–01 (PAE, AMM) awarded by the National Institute of General Medical Sciences (NIGMS). TJL was supported by a post-graduate training grant (T32 GM-08721) in burns, trauma and perioperative injury by NIGMS.
Footnotes
AJT, KBK, IGA, and AMM contributed to the literature review and study design. TJL, AJT, KBK, IGA, HNR, and EEW contributed to data collection. TJL, AJT, KBK, IGA, and AAM contributed to data analysis and interpretation. PAE and AAM provided critical revisions.
The authors have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 3.Gille H, Kowalski J, Li B, LeCouter J, Moffat B, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 4.Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- 5.Hoshino M, Nakamura Y, Hamid QA. Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. J Allergy Clin Immunol. 2001;107:1034–1038. doi: 10.1067/mai.2001.115626. [DOI] [PubMed] [Google Scholar]
- 6.Skurk C, Maatz H, Rocnik E, Bialik A, Force T, et al. Glycogen-Synthase Kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res. 2005;96:308–318. doi: 10.1161/01.RES.0000156273.30274.f7. [DOI] [PubMed] [Google Scholar]
- 7.Kaner RJ, Ladetto JV, Singh R, Fukuda N, Matthay MA, et al. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am J Respir Cell Mol Biol. 2000;22:657–664. doi: 10.1165/ajrcmb.22.6.3779. [DOI] [PubMed] [Google Scholar]
- 8.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klekamp JG, Jarzecka K, Perkett EA. Exposure to hyperoxia decreases the expression of vascular endothelial growth factor and its receptors in adult rat lungs. Am J Pathol. 1999;154:823–831. doi: 10.1016/S0002-9440(10)65329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaner RJ, Crystal RG. Compartmentalization of vascular endothelial growth factor to the epithelial surface of the human lung. Mol Med. 2001;7:240–246. [PMC free article] [PubMed] [Google Scholar]
- 11.Lahm T, Crisostomo PR, Markel TA, Wang M, Lillemoe KD, et al. The critical role of vascular endothelial growth factor in pulmonary vascular remodeling after lung injury. Shock. 2007;28:4–14. doi: 10.1097/shk.0b013e31804d1998. [DOI] [PubMed] [Google Scholar]
- 12.Song J, Lu H, Zheng X, Huang X. Effects of Vascular Endothelial Growth Factor in Recovery Phase of Acute Lung Injury in Mice. Lung. 2015;193:1029–1036. doi: 10.1007/s00408-015-9803-x. [DOI] [PubMed] [Google Scholar]
- 13.Koh H, Tasaka S, Hasegawa N, Yamada W, Shimizu M, et al. Protective role of vascular endothelial growth factor in endotoxin-induced acute lung injury in mice. Respir Res. 2007;8:60. doi: 10.1186/1465-9921-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamada N, Kuwano K, Yamada M, Hagimoto N, Hiasa K, et al. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol. 2005;175:1224–1231. doi: 10.4049/jimmunol.175.2.1224. [DOI] [PubMed] [Google Scholar]
- 15.Karmpaliotis D, Kosmidou I, Ingenito EP, Hong K, Malhotra A, et al. Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2002;283:L585–595. doi: 10.1152/ajplung.00048.2002. [DOI] [PubMed] [Google Scholar]
- 16.Nin N, Lorente JA, de Paula M, El Assar M, Vallejo S, et al. Rats surviving injurious mechanical ventilation show reversible pulmonary, vascular and inflammatory changes. Intensive Care Med. 2008;34:948–956. doi: 10.1007/s00134-007-0959-6. [DOI] [PubMed] [Google Scholar]
- 17.Ekerbicer N, Tarakci F, Barut T, Inan S. Immunolocalization of VEGF, VEGFR-1 and VEGFR-2 in lung tissues after acute hemorrhage in rats. Acta Histochem. 2008;110:285–293. doi: 10.1016/j.acthis.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Park SY, Kang JH, Jeong KJ, Lee J, Han JW, et al. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer. 2011;128:2306–2316. doi: 10.1002/ijc.25589. [DOI] [PubMed] [Google Scholar]
- 19.Lutgendorf SK, Cole S, Costanzo E, Bradley S, Coffin J, et al. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin Cancer Res. 2003;9:4514–4521. [PubMed] [Google Scholar]
- 20.Yang EV, Sood AK, Chen M, Li Y, Eubank TD, et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006;66:10357–10364. doi: 10.1158/0008-5472.CAN-06-2496. [DOI] [PubMed] [Google Scholar]
- 21.Deng GH, Liu J, Zhang J, Wang Y, Peng XC, et al. Exogenous norepinephrine attenuates the efficacy of sunitinib in a mouse cancer model. J Exp Clin Cancer Res. 2014;33:21. doi: 10.1186/1756-9966-33-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bible LE, Pasupuleti LV, Gore AV, Sifri ZC, Kannan KB, et al. Daily propranolol prevents prolonged mobilization of hematopoietic progenitor cells in a rat model of lung contusion, hemorrhagic shock, and chronic stress. Surgery. 2015;158:595–601. doi: 10.1016/j.surg.2015.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gore AV, Bible LE, Livingston DH, Mohr AM, Sifri ZC. Can mesenchymal stem cells reverse chronic stress-induced impairment of lung healing following traumatic injury? J Trauma Acute Care Surg. 2015;78:767–772. doi: 10.1097/TA.0000000000000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah S, Ulm J, Sifri ZC, Mohr AM, Livingston DH. Mobilization of bone marrow cells to the site of injury is necessary for wound healing. J Trauma. 2009;67:315–321. doi: 10.1097/TA.0b013e3181a5c9c7. discussion 321-312. [DOI] [PubMed] [Google Scholar]
- 25.Claridge JA, Enelow RI, Young JS. Hemorrhage and resuscitation induce delayed inflammation and pulmonary dysfunction in mice. J Surg Res. 2000;92:206–213. doi: 10.1006/jsre.2000.5899. [DOI] [PubMed] [Google Scholar]
- 26.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannoush EJ, Sifri ZC, Elhassan IO, Mohr AM, Alzate WD, et al. Impact of enhanced mobilization of bone marrow derived cells to site of injury. J Trauma. 2011;71:283–289. doi: 10.1097/TA.0b013e318222f380. discussion 289-291. [DOI] [PubMed] [Google Scholar]
- 28.Bible LE, Pasupuleti LV, Gore AV, Sifri ZC, Kannan KB, et al. Chronic restraint stress after injury and shock is associated with persistent anemia despite prolonged elevation in erythropoietin levels. J Trauma Acute Care Surg. 2015;79:91–96. doi: 10.1097/TA.0000000000000686. discussion 96-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thickett DR, Armstrong L, Millar AB. A role for vascular endothelial growth factor in acute and resolving lung injury. Am J Respir Crit Care Med. 2002;166:1332–1337. doi: 10.1164/rccm.2105057. [DOI] [PubMed] [Google Scholar]
- 30.Badami CD, Livingston DH, Sifri ZC, Caputo FJ, Bonilla L, et al. Hematopoietic progenitor cells mobilize to the site of injury after trauma and hemorrhagic shock in rats. J Trauma. 2007;63:596–600. doi: 10.1097/TA.0b013e318142d231. discussion 600-592. [DOI] [PubMed] [Google Scholar]