

Abstract
A photochemical organocatalytic strategy for the direct enantioselective β‐benzylation of α,β‐unsaturated aldehydes is reported. The chemistry capitalizes upon the light‐triggered enolization of 2‐alkyl‐benzophenones to afford hydroxy‐o‐quinodinomethanes. These fleeting intermediates are stereoselectively intercepted by chiral iminium ions, transiently formed upon condensation of a secondary amine catalyst with enals. Density functional theory (DFT) studies provided an explanation for why the reaction proceeds through an unconventional Michael‐type addition manifold, instead of a classical cycloaddition mechanism and subsequent ring‐opening.
Keywords: asymmetric catalysis, benzylation, organocatalysis, photochemistry, synthetic methods
The photoenolization/Diels–Alder sequence1 is an established synthetic strategy (Figure 1 a) which has found widespread application for the one‐step construction of stereochemically dense benzannulated carbocyclic molecules (2).2 The approach exploits the light‐triggered enolization of 2‐alkyl‐benzophenones (1) affording transient hydroxy‐o‐quinodinomethanes (A),3 which are highly reactive intermediates that can serve as dienes in [4+2] cycloadditions with electron‐poor alkenes of type B. Herein, we document that the reactivity of the photoenol A is not restricted to cycloaddition mechanisms, but can also participate in conjugate addition manifolds.4 More specifically, we have found that a chiral iminium ion intermediate (C),5 formed upon condensation of a secondary amine catalyst (4) and α,β‐unsaturated aldehydes (3), can stereoselectively intercept A and lead to the exclusive formation of the Michael addition product 5 (Figure 1 b). No traces of the expected Diels–Alder adduct of type 2 were detected.
Figure 1.
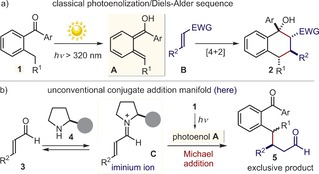
a) The classical photoenolization/Diels–Alder strategy for the one‐pot synthesis of chiral cyclic compounds (2). b) The discovered Michael‐type addition of the photoenol A to the chiral iminium ion C leading to the enantioenriched β‐benzylated aldehyde 5. Filled grey circle represents a bulky substituent on the chiral amine catalyst. EWG=electron‐withdrawing group.
To account for this unexpected reactivity, we have conducted theoretical studies which indicate that the reaction proceeds through an unconventional direct Michael‐type addition path instead of a classical cycloaddition manifold, followed by the retro‐aldol‐mediated ring‐opening of 2. From a synthetic perspective, the chemistry described in Figure 1 b is relevant since it provides a straightforward method for the direct β‐benzylation of enals (3), a transformation for which there are few effective catalytic enantioselective precedents.6 Metal‐catalyzed conjugate additions to enals are generally plagued7 by the intrinsic instability of the benzyl‐metallic reagents and the competing 1,2‐addition manifold, while organocatalytic iminium‐ion‐based strategies have been successful only for a specific class of highly activated nitro‐toluene substrates.8
The initial motivations for this study stemmed from our recent discovery that a chiral bifunctional organic catalyst, by activation of a dienophilic maleimide of type B, could allow the stereoselective trap of the photoenol A, thus leading to enantioenriched cycloaddition products of type 2.9 This strategy provided the first effective enantioselective catalytic variant of the photoenolization/Diels–Alder sequence.10 Building on this precedent, we wondered whether the electrophilic iminium ion intermediate C 5 could provide another organocatalytic tool useful for successfully intercepting A (Figure 1 b). The feasibility of our plan was tested by reacting the commercially available 2‐methylbenzophenone (1 a) and trans‐2‐pentenal (3 a) in the presence of commercially available diphenylprolinol trimethylsilylether (4 a)11 as the chiral amine catalyst (Table 1). The experiments were conducted over 20 hours, in toluene, and under irradiation by an ordinary 15 W black light bulb (BLB; λ max=365 nm). Initial experiments confirmed the possibility of trapping the fleeting A, generated from 1 a. This observation came about with an unanticipated reactivity, since the light‐triggered process led to the exclusive formation of the conjugate addition product 5 a with high stereocontrol, albeit with poor chemical yield (entry 1). A screening of the reaction media identified 1,2‐dichlorobenzene as the most appropriate solvent (entry 2). The addition of an acidic additive (diphenylphosphoric acid, DPP, 20 mol %) had a beneficial effect on the reactivity (entry 3), while modification of the catalyst scaffold, by introducing a bulkier silyl protecting group, resulted in a better enantioselectivity (entry 4).
Table 1.
Exploratory studies.
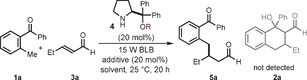
| Entry | Catalyst (R) | Solvent | Additive | Yield [%][a] | ee [%][b] |
|---|---|---|---|---|---|
| 1 | 4 a (TMS) | toluene | – | 38 | 90 |
| 2 | 4 a (TMS) | o‐Cl2C6H4 | – | 52 | 83 |
| 3 | 4 a (TMS) | o‐Cl2C6H4 | DPP | 60 | 86 |
| 4 | 4 b (TBS) | o‐Cl2C6H4 | DPP | 61 | 90 |
| 5[c] | 4 b (TBS) | o‐Cl2C6H4 | DPP | 0 | – |
| 6 | none | o‐Cl2C6H4 | DPP | 18 | – |
[a] Yield of isolated 5 a. [b] Determined by HPLC analysis on a chiral stationary phase. [c] Reaction in the dark. BLB=black light bulb (λ max=365 nm), DPP=diphenylphosphoric acid, o‐Cl2C6H4=1,2‐dichlorobenzene, TMS=trimethylsilyl, TBS=tert‐butyldimethylsilyl.
We then conducted control experiments to better clarify the nature of the photochemical organocatalytic process. As expected, the reaction was completely inhibited in the dark (Table 1, entry 5). When the process was performed in the absence of the aminocatalyst 4, the racemic adduct 5 a was isolated in 18 % yield after 20 hours (entry 6). Thus, the high stereocontrol inferred by the chiral amine 4 b indicated that the rate acceleration afforded by the catalyst must be large enough to overcome a racemic background process.12 In all of these experiments, we could not detect any traces of the Diels–Alder adduct 2 a.13
Adopting the optimized reaction conditions described in Table 1, entry 4, we then demonstrated the generality of the asymmetric organocatalytic photoenolization/conjugate addition sequence (Figure 2 a) by evaluating a variety of enals (3). Figure 2 b shows that different aliphatic groups at the β‐position are viable substituents. Short (adducts 5 a and 5 b) and long alkyl chains, containing either aromatic or alkene functionalities (5 e and 5 f), as well as a more sterically hindered isopropyl group (5 c), are all well tolerated. The reaction of the carboxybenzyl (Cbz)‐protected 5‐amino‐2‐pentenal affords the cyclic compound 5 g, originating from a spontaneous intramolecular condensation between the aldehyde and the amino group within the β‐benzylated product precursor. The method is synthetically useful, with a good efficiency maintained when running the reaction on a 1 mmol scale (5 a). Cinnamaldehyde derivatives are also competent substrates (Figure 2 c) since different substitution patterns at the aromatic moiety of 3 are well tolerated, regardless of their electronic properties (5 h–j). A heteroaryl framework can also be included in the product, as shown by the furyl‐substituted adduct 5 k.
Figure 2.
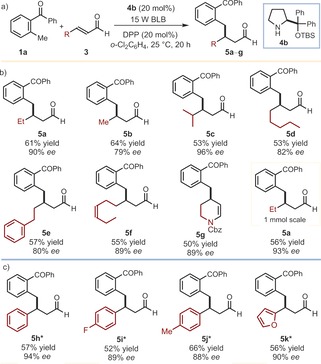
a) Survey of the enals 3 which can participate in the photochemical β‐benzylation process. Reactions of b) aliphatic and c) aromatic β‐substituted enals. Reactions performed on a 0.2 mmol scale. Yields of the isolated products 5 are given. The enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. *Performed in toluene using the catalyst 4 a.
We then sought to explore the scope of the benzophenone substrate 1 (Figure 3 a). Different substituents on both the enolizable (products 5 l–o) and the non‐enolizable aromatic ring of 1 (5 p–s) are well tolerated (Figure 3 b). The presence of substituents at the benzylic position (R2) of 1 brings about the formation of two contiguous stereogenic centers (5 t–w). When R2=Ph, a valuable diarylmethine stereocenter is forged with very high fidelity (5 t). Finally, as a demonstration of the synthetic versatility of the β‐benzylated products, 5 p, obtained from a 1 mmol scale reaction, was easily converted into the cyclic adduct 6, whose absolute configuration was unambiguously inferred by single‐crystal X‐ray analysis (Figure 3 c).14
Figure 3.
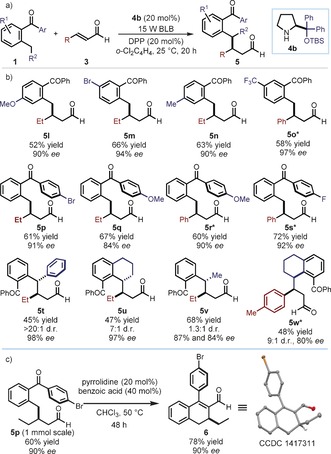
Substrate scope for the photoenolization/β‐benzylation sequence. a) The developed photochemical organocatalytic reaction. b) Survey of the benzophenones 1 which can participate in the reaction. c) Product manipulation and stereochemical assignment. Reactions performed on a 0.2 mmol scale. Yields of the isolated products 5 are given. The enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. *Performed in toluene using catalyst 4 a.
After delineating its synthetic potential, we sought to shed light on the mechanism of the reported process, for the uncommon ability of the photoenol A to undergo a conjugate addition was mechanistically intriguing. Since we could not collect any experimental evidence supporting a [4+2] cycloaddition pathway followed by the opening of the cyclic intermediate 2,13 the question remained as to why this generally preferred pathway was disabled in the present system. We investigated the mechanism using a density functional theory (DFT) computational study15 at the M06‐2X level in a toluene solvent. DFT calculations are established tools for mechanistically elucidating organocatalytic16 and light‐triggered processes.17 They have recently been used to support a hetero‐Diels–Alder/ring‐opening sequence as the underlying mechanism of the light‐driven carboxylation of benzophenones (1) proceeding by the trapping of A with CO2.17b
The reaction of crotonaldehyde and 1 a, yielding 5 b (Figure 2 b), catalyzed by 4 a was chosen as the model system. Systematic conformational analyses were carried out, and we discuss here only the results concerning the most stable conformations.18 We computed the reaction between the transiently generated A and the chiral iminium ion C. The results are summarized in Figure 4. We could locate two transition states, one leading to the intermediate D (the precursor of the cycloaddition adduct 2 b) and the other to E (which provided the Michael addition product 5 b upon hydrolysis). The cycloaddition transition state (TS1) leads to the very stable D (−47.4 kcal mol−1) through the formation of two carbon–carbon (C−C) bonds. The alternative transition state (TS2), which involves only one C−C bond‐forming event, leads to the intermediate F upon proton transfer from A to the iminium ion nitrogen atom. Then F, which is also quite stable (−30.7 kcal mol−1), undergoes an intramolecular proton transfer through TS3, ultimately leading to E (−44.8 kcal mol−1).19 The computed reaction pathways in Figure 4 are not surprising, as one would expect both the cycloaddition and the Michael addition to be mechanistically viable. But this data is in sharp disagreement with the experimental results. The cycloaddition transition state TS1 (7.0 kcal mol−1) is 1.1 kcal mol−1 lower in energy than the Michael addition transition state TS2 (8.1 kcal mol−1), meaning that the Diels–Alder adduct 2 b should be the preferred kinetic product. In addition, since 2 b is the more stable product, there is no chance that it could evolve towards 5 b through a retro‐aldol/ring‐opening process.13
Figure 4.
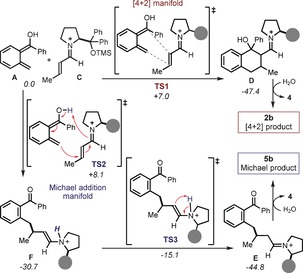
Reaction mechanism in the absence of a proton shuttle. Free energies in kcal mol−1. Filled grey circle represents the trimethylsiloxy group of the chiral amine catalyst 4 a.
Since we could not computationally reproduce the experimental observation, we considered the potential role of additional species in the reaction media, and how they could affect the relative energy of the transition states reported in Figure 4. The condensation of 4 and 3 to afford C, assisted by DPP as an acidic additive, releases water into the system.20 Both water and the resulting diphenylphosphate can act as a proton shuttle. Proton shuttles have been shown to lower the barrier of processes involving proton transfer,21 a key step underlying the Michael addition mechanism (see TS2 in Figure 4). Since the DPP anion has no acidic protons and cannot serve as hydrogen‐bonding donor, we repeated the calculations including an explicit water molecule as the most likely proton shuttle.22 The results are summarized in Figure 5.
Figure 5.
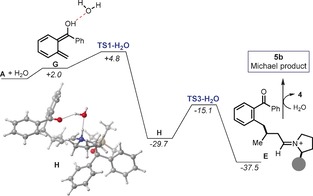
Free‐energy profile of the reaction assisted by a proton shuttle. The optimized structure of intermediate H (at −29.7 kcal mol−1) is highlighted. Free energies in kcal mol−1.
A water molecule can engage in a hydrogen‐bonding network with A to form the adduct G, which is only 2.0 kcal mol−1 above the separate species. The approach of G to C has only one transition state (TS1‐H2O), which leads to the Michael addition product E. No transition state could be located for the cycloaddition manifold. Remarkably, the structure of TS1‐H2O is conformationally similar to that of TS1 (Figure 4), which affords the cycloaddition adduct 2 b instead. We confirmed, by an intrinsic reaction coordinate (IRC) calculation, that TS1‐H2O exclusively relaxes to the intermediate H with the proton being transferred from the photoenol to the iminium ion nitrogen atom by a water‐assisted proton shuttle mechanism. The structure of H shows how the water molecule participates in multiple hydrogen‐bonding interactions with both the H‐N+ moiety within the enammonium ion and the O=C group of the benzophenone. From this intermediate, an intramolecular proton transfer readily forms the Michael adduct precursor E.19 The key feature of the water‐assisted mechanism is the free energy of TS1‐H2O, which is 2.2 kcal mol−1 lower than the free energy for TS1 reported in Figure 4, thus explaining why the Michael addition manifold dominates over the classical cycloaddition mechanism.
In conclusion, we have demonstrated that chiral iminium ions can stereoselectively intercept photochemically generated hydroxy‐o‐quinodinomethanes. The chemistry, which provides a rare example of enantioselective catalytic β‐benzylation of electron‐poor olefins, proceeds through an unconventional Michael‐type addition path instead of a classical cycloaddition manifold. Computational studies revealed that the intrinsic preference for the cycloaddition is offset in this system by a network of proton‐transfer mechanisms facilitated by the presence of proton shuttles. Similar behaviors can be expected in related systems where such key structural elements are conserved.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the CERCA Programme (Generalitat de Catalunya), MINECO (projects CTQ2013‐45938‐P and CTQ2014‐57661‐R, Severo Ochoa Excellence Accreditation 2014–2018, SEV‐2013‐0319), the AGAUR (2014 SGR 1059 and 2014 SGR 0409), and the European Research Council (ERC 681840‐CATA‐LUX to P.M.). L.D. thanks the Marie Curie COFUND action (2014‐1‐ICIQ‐IPMP) for a postdoctoral fellowship. V.F. is grateful to MINECO for an FPI fellowship (ref BES‐2012–057655).
L. Dell'Amico, V. M. Fernández-Alvarez, F. Maseras, P. Melchiorre, Angew. Chem. Int. Ed. 2017, 56, 3304.
Contributor Information
Dr. Luca Dell'Amico, http://www.iciq.org/research/research group/prof‐paolo‐melchiorre/.
Prof. Dr. Feliu Maseras, Email: fmaseras@iciq.es
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Sammes P. G., Tetrahedron 1976, 32, 405–422; [Google Scholar]
- 1b.“Photoenolization and its applications”: Klán P., Wirz J., Gudmundsdottir A. in CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed. (Ed.: A. Griesbeck), CRC, Boca Raton, 2012, Chap. 26, pp. 627–651. [Google Scholar]
- 2.For selected examples, see:
- 2a. Nicolaou K. C., Gray D., Tae J., Angew. Chem. Int. Ed. 2001, 40, 3675–3678; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 3787–3790; [Google Scholar]
- 2b. Nicolaou K. C., Gray D. L. F., Tae J. S., J. Am. Chem. Soc. 2004, 126, 613–627. [DOI] [PubMed] [Google Scholar]
- 3.For the original report, see:
- 3a. Yang N. C., Rivas C., J. Am. Chem. Soc. 1961, 83, 2213 For the mechanism of formation of the photoenol A, see: [Google Scholar]
- 3b. Scaiano J. C., Acc. Chem. Res. 1982, 15, 252–258. [Google Scholar]
- 4.Generally, the formation of conjugate-addition-type adducts is rationalized on the basis of a [4+2]/ring-opening sequence. For a nonstereoselective precedent that invokes a direct Michael-type addition mechanism, see: Wilson R. M., Hannemann K., Heineman W. R., Kirchhoff J. R., J. Am. Chem. Soc. 1987, 109, 4743–4745. [Google Scholar]
- 5. Lelais G., MacMillan D. W. C., Aldrichimica Acta 2006, 39, 79–87. [Google Scholar]
- 6.For a recent example of enantioselective catalytic direct addition of benzyl radicals to electron-poor olefins, see: Huo H., Harms K., Meggers E., J. Am. Chem. Soc. 2016, 138, 6936–6939. [DOI] [PubMed] [Google Scholar]
- 7.To our knowledge, no catalytic asymmetric conjugate additions of benzyl metal reagents to enals have been reported. For a nonstereoselective process, see: Van Heerden P. S., Bezuidenhoudt B. C. B., Steenkamp J. A., Ferreira D., Tetrahedron Lett. 1992, 33, 2383–2386. [Google Scholar]
- 8.
- 8a. Li T., Zhu J., Wu D., Li X., Wang S., Li H., Li J., Wang W., Chem. Eur. J. 2013, 19, 9147–9150; [DOI] [PubMed] [Google Scholar]
- 8b. Dell'Amico L., Companyó X., Naicker T., Bräuer T. M., Jørgensen K. A., Eur. J. Org. Chem. 2013, 5262–5265. [Google Scholar]
- 9. Dell'Amico L., Vega-Peñaloza A., Cuadros S., Melchiorre P., Angew. Chem. Int. Ed. 2016, 55, 3313–3317; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3374–3378. [Google Scholar]
- 10.Asymmetric catalytic approaches are complicated by the fleeting nature A For an asymmetric method using a stoichiometric amount of a chiral complexing agent, see: Grosch B., Orlebar C. N., Herdtweck E., Massa W., Bach T., Angew. Chem. Int. Ed. 2003, 42, 3693–3696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3822–3824. [Google Scholar]
- 11. Donslund B. S., Johansen T. K., Poulsen P. H., Halskov K. S., Jørgensen K. A., Angew. Chem. Int. Ed. 2015, 54, 13860–13874; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14066–14081. [Google Scholar]
- 12.In analogy with our preceding study (Ref. [9]), we evaluated the possibility that the chiral amine 4 a attenuates the rate of the racemic background process by reducing the concentration of A in solution by means of an electron-transfer mechanism. Flash photolysis quenching studies of A at the millisecond resolution established that 4 a does not significantly influence its formation. We also established that the presence of pentenal 3 a or cinnamaldehyde did not affect the formation of A to any extent.
- 13.In all of the experiments conducted in this study, we could not detect any trace of the Diels–Alder adducts of type 2 In addition, when an authentic sample of the cyclic adduct 2 a (synthesized according to: E. Block, R. Stevenson, J. Chem. Soc. Perkin Trans. 1 1973, 308–313) was subjected to the optimized photo-organocatalytic conditions, we did not detect the open product 5 a Along with the unreacted adduct 2 a, a minor amount (ca. 20 %) of the dehydration product of type 6 (see Figure 3 c) was recovered. This experiment indicates that the retro-aldol-mediated ring-opening of 2 is not a viable pathway for the formation of 5 a.
- 14.Crystallographic data for 6 have been deposited with the Cambridge Crystallographic Data Centre, accession number CCDC 1417311 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 15.See the Supporting Information for full computational details. A data set collection of computational results is available in the IoChem-BD repository and can be accessed via htpp://dx.doi.org/10.19061/iochem-bd-1-28. See: Álvarez-Moreno M., de Graaf C., López N., Maseras F., Poblet J. M., Bo C., J. Chem. Inf. Model. 2015, 55, 95–103. [DOI] [PubMed] [Google Scholar]
- 16.For selected reviews, see:
- 16a. Kerske E. H., Houk K. N., Acc. Chem. Res. 2013, 46, 979–989; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Sameera W. M. C., Maseras F., WIREs Comput. Mol. Sci. 2012, 2, 375–385. [Google Scholar]
- 17.
- 17a. Fernández-Alvarez V. M., Nappi M., Melchiorre P., Maseras F., Org. Lett. 2015, 17, 2676–2679; [DOI] [PubMed] [Google Scholar]
- 17b. Masuda Y., Ishida N., Murakami M., J. Am. Chem. Soc. 2015, 137, 14063–14066. For a review on enantioselective catalytic photochemical processes, see: [DOI] [PubMed] [Google Scholar]
- 17c. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. [Google Scholar]
- 18. Besora M., Braga A. A. C., Ujaque G., Maseras F., Lledós A., Theor. Chem. Acc. 2011, 128, 639–646. [Google Scholar]
- 19. Sorgi K. L., Maryanoff C. A., McComsey D. F., Graden D. W., Maryanoff B. E., J. Am. Chem. Soc. 1990, 112, 3567–3579. [Google Scholar]
- 20.Both water and DPP are beneficial for reactivity. The reactions in Figures 2 and 3 were performed using reagent-grade solvents. When using rigorously dry solvents, an appreciable decrease in reactivity was observed, while the absence of DPP severely reduced the reaction rate. For explicative results concerning the reaction of 1 a with cinnamaldehyde to give 5 h (reaction over 24 hours, conditions as in Figure 2 c): reagent-grade toluene: 57 % yield, 94 % ee; dry toluene: 41 % yield, 94 % ee; reagent-grade toluene in the absence of DPP: 30 % yield, 92 % ee.
- 21.
- 21a. Bellarosa L., Díez J., Gimeno J., Lledós A., Suárez F. J., Ujaque G., Vicent C., Chem. Eur. J. 2012, 18, 7749–7765; [DOI] [PubMed] [Google Scholar]
- 21b. Monot J., Brunel P., Kefalidis C. E., Espinosa-Jalapa N. A., Maron L., Martín-Vaca B., Bourissou D., Chem. Sci. 2016, 7, 2179–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Water-assisted proton shuttle mechanisms have been extensively discussed in quantum mechanical investigations of organocatalytic processes. For a review, see: Cheong P. H.-Y., Legault C. Y., Um J. M., Çelebi-Ölcüm N., Houk K. N., Chem. Rev. 2011, 111, 5042–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary