

Abstract
Predicting and controlling infectious disease epidemics is a major challenge facing the management of agriculture, human and wildlife health. Co‐evolutionarily derived patterns of local adaptation among pathogen populations have the potential to generate variation in disease epidemiology; however, studies of local adaptation in disease systems have mostly focused on interactions between competing pathogens or pathogens and their hosts. In nature, parasites and pathogens are also subject to attack by hyperparasitic natural enemies that can severely impact upon their infection dynamics. However, few studies have investigated whether this interaction varies across combinations of pathogen–hyperparasite strains, and whether this influences hyperparasite incidence in natural pathogen populations. Here, we test whether the association between a hyperparasitic fungus, Ampelomyces, and a single powdery mildew host, Podosphaera plantaginis, varies among genotype combinations, and whether this drives hyperparasite incidence in nature. Laboratory inoculation studies reveal that genotype, genotype × genotype interactions and local adaptation affect hyperparasite infection. However, observations of a natural pathogen metapopulation reveal that spatial rather than genetic factors predict the risk of hyperparasite presence. Our results highlight how sensitive the outcome of biocontrol using hyperparasites is to selection of hyperparasite strains.
Keywords: co‐evolution, disease, host–parasite interactions, hyperparasite, local adaptation
Introduction
Parasites and pathogens pose a major risk to the stability of both natural and agricultural systems (Anderson & May 1991). Thus, determining the factors that influence the risk and severity of infection is vital to predict and control outbreaks of disease (Gilligan 2002; Shaw 2002; Zhan et al. 2014). Genetic diversity in both pathogen infectivity and host susceptibility traits can drive variation in disease outcome (Jackson & Tinsley 2005; Bangham et al. 2008), and infection success can vary among combinations of host–pathogen genotypes (Wolinska & King 2009). Selection can act on the variation among such G×G interactions to drive co‐evolutionary dynamics that further promote pathogen–host diversity and therefore influence the risks of infection. To quantify co‐evolutionary process in nature, many studies look for signals of local adaptation (LA), wherein fitness of either organism is higher when facing sympatric rather than allopatric antagonists (Blanquart et al. 2013). Local adaptation has been demonstrated for several host–pathogen systems (Thrall et al. 2002; Laine 2007; Schulte et al. 2011; Koskella 2014) and has been linked with disease dynamics and infection risk (Springer 2007; Tack et al. 2012; Kyle et al. 2014).
To date, almost all studies of G×G and LA in disease systems focus on the pathogen's interaction with its host or competing pathogen strains. However, pathogen dynamics can also be strongly influenced by hyperparasitism, wherein the pathogen itself becomes infected with a parasite. Hyperparasites are likely to be a common in nature, particularly among microorganisms (Parratt & Laine 2016), and theory has shown that they can fundamentally affect the ecological stability and evolutionary trajectory of the host–pathogen system in which they reside (Holt & Hochberg 1998; Taylor et al. 1998). Indeed, the limited studies of natural hyperparasites have shown that they can influence key epidemiological processes and thus greatly impact upon pathogen dynamics (Andersen et al. 2012; Springer et al. 2013; Tollenaere et al. 2014). However, despite being used as biological control measures in human medicine (Nobrega et al. 2015) and agriculture (Swinton & Gilligan 1999), the ecological, spatial and genetic factors governing hyperparasite infection success and epidemiology are poorly understood. In particular, whether or not the infection dynamic between hyperparasites and pathogens is governed by G×G interactions, and whether hyperparasites become locally adapted to their host is mostly unknown.
Hyperparasites are likely to be limited by the population size and distribution of the pathogens that they infect, which are in‐turn limited by the availability of their own host. This has the potential to select for a generalist lifestyle for many hyperparasites, or the ability to disperse much farther than their host in order to maintain an effective population size. Generalism is expected to prevent local adaptation and specialization between interacting species (Kawecki & Ebert 2004; Gómez et al. 2009); however, the impact of high dispersal levels on local adaptation is less clear. Multiple studies have shown that parasites with relatively higher gene flow than their host are most likely to become locally adapted (Greischar & Koskella 2007; Hoeksema & Forde 2008), but if gene flow becomes too strong then it can swamp local selection regimes, preventing local adaptation (Slatkin 1985; Lively 1999; Storfer 1999; Gandon & Michalakis 2002). Evidence for these effects is rare, but studies have found weaker genetic structure in hyperparasitoids compared to their parasite hosts, which alludes to a lack of divergence and specialization at the host‐deme level (Nair et al. 2016). However, environmentally dependent G×G interactions have been found in the interaction between a fungal pathogen and strains of its hyperparasitic virus (Bryner & Rigling 2011). If specific G×G interactions do occur between pathogens and hyperparasites in nature, then we may expect hyperparasites to become rare across fragmented pathogen populations and for their prevalence patterns to be aggregated around specific host genotypes. However, for most hyperparasite systems, simple descriptions of their prevalence, spatial aggregation and host associations are lacking particularly among pathogen individuals or demes of the same species.
Here, we investigate the influence of genetic and ecological factors on the prevalence and infection dynamics of the hyperparasitic fungus Ampelomyces spp, an antagonist of powdery mildew pathogens. Ampelomyces is considered to be a group of generalist hyperparasites (Kiss & Nakasone 1998) as several cross‐inoculation studies have found that isolates are able to infect multiple mildew species (Szentivanyi et al. 2005; Liang et al. 2007; Kiss et al. 2011), and single mildews can become infected with phylogenetically distinct Ampelomyces strains (Pintye et al. 2012). However, molecular evidence suggests that Ampelomyces lineages are broadly associated with specific mildew host species in nature (Liang et al. 2007; Park et al. 2010; Kiss et al. 2011). Microsatellite analysis has demonstrated that this divergence may be driven by host phenology in some instances (Kiss et al. 2011; Pintye et al. 2015). In addition, Ampelomyces isolated from different host species have been shown to vary in their growth rate, morphology, spore germination response in vitro and in their infection severity against different powdery mildew species (Legler et al. 2011, 2016; Angeli et al. 2012a). This evidence suggests that some degree of host association and co‐evolution may occur within Ampelomyces, although strict host‐species fidelity has not evolved. To date, there is little demonstration of any G×G interactions or local adaptation between Ampelomyces and genetically distinct isolates of a single mildew species. The existence of such dynamics could have profound impact for both the hyperparasite's epidemiology and the disease dynamics of the mildew itself.
In this study, we test whether Ampelomyces exhibits G×G infection dynamics and local adaptation across a metapopulation of a single host species: Podosphaera plantaginis. We use cross‐inoculation laboratory studies to expose variation in infection dynamics among hyperparasite and pathogen combinations, and to contrast infection success between sympatric and allopatric pairings. We also test whether hyperparasite infection in the field is clustered by pathogen genotypes, as would be expected if G×G effects are prevalent, or if ecological properties of the mildew host best describe patterns of Ampelomyces. Finally, we contrast hyperparasite prevalence at the pathogen population and metapopulation scales. Our results demonstrate that pathogen and hyperparasite genotypes, as well as their interaction, determine the outcome of infection in the laboratory. We also observed signals of local adaptation that vary in strength among hyperparasite strains. However, these genotypic effects were not readily apparent in the field, primarily due to a strong spatial aggregation of the hyperparasite at the local level.
Methods
Study system
Podosphaera plantaginis (Castagne; U. Braun & Takamatsu) is a specific and obligate fungal pathogen of ribwort plantain (Plantago lanceolata) that causes powdery mildew disease on above ground tissues of the host. Po. plantaginis is aerially dispersed as haploid, asexually produced spores (conidia) during the spring and summer. Po. plantaginis typically undergoes between six to eight clonal cycles, which lead to local epidemics of powdery mildew symptoms. The mildew overwinters as sexually produced [through outcrossing or selfing (Tollenaere & Laine 2013)] resting structures called chasmothecia [formerly cleistothecia (Braun 1995)] that are produced towards the end of the growing season (August–September). Pl. lanceolata is a perennial herb, capable of reproducing clonally through vegetative growth and sexually via wind and animal dispersed seeds. In the Åland archipelago (SW Finland), Pl. lanceolata's preferred dry meadow habitat is fragmented into well‐defined, discrete populations (Ojanen et al. 2013), which support a classical metapopulation of the powdery mildew pathogen (Laine 2004; Laine & Hanski 2006). Annual surveys of the metapopulation since 2001 have revealed that a balance of local extinction and colonization events maintain pathogen prevalence at 1.1–16.9% of the c. 4000 host populations among years (Jousimo et al. 2014).
Ampelomyces spp are obligate mycoparasites of multiple powdery mildew species belonging to the Erysiphaceae, including Po. plantaginis (Kiss et al. 2004). Ampelomyces hyphae invade the tissues and cells of established powdery mildew lesions where they absorb nutrients and degenerate the host cytoplasm. Ampelomyces hijacks the mildew's reproductive structures in which it established pycnidia; sacks of hyperparasite spores that are then released and dispersed aerially and through rain splash to neighbouring mildew hosts (Sullivan & White 2000). The infection process (spore germination–pycnidia formation) takes between 5 and 8 days under laboratory conditions; thus, Ampelomcyes is likely to have a shorter clonal generation time than its host in nature. However, the true within‐host rate of reproduction and the existence or timing of any sexual recombination remains unknown (Kiss et al. 2004).
In the Åland Po. plantaginis metapopulation, Ampelomyces infection has been shown to significantly reduce the probability that the mildew will successfully overwinter (Tollenaere et al. 2014). This is most likely due to the hyperparasite's ability to suppress the formation of powdery mildew chasmothecia (Caffi et al. 2013). Furthermore, in other mildew hosts, Ampelomyces has been shown to have negative effects on within‐season mildew growth, reducing the number and viability of the mildew's asexual conidia (Falk et al. 1995; Abo‐Foul et al. 1996; Verhaar et al. 1996; Shishkoff & McGrath 2002; Romero et al. 2003). This can lead to the collapse of powdery mildew lesions and curtail its spread among plant populations. Given that within‐season transmission is likely to be a major component of mildew fitness [infection level at the end of the epidemic is also an important determinant of mildew overwintering success (Tack et al. 2014a,b)], Ampelomyces spread among conidia is likely to represent a major fitness cost to the mildew. Thus, Ampelomyces infection represents a threat to the mildew's fitness by limiting both within‐season spread and between‐season survival.
Ampelomyces strains originating from different host mildew species have been noted to vary in their infection severity (Angeli et al. 2012b; Legler et al. 2016). However, phenotypic variation among Ampelomyces isolates from the same host mildew at local spatial scales (e.g. within metapopulations) has not been explored in a natural pathosystem. Nor have PathogenG × HyperparasiteG interactions been shown to determine the outcome of infection. Previous work has found genetic variation among Ampelomyces isolates infecting multiple mildew host species (Kiss 1997; Liang et al. 2007). This variation has been linked with a broad host association using conserved rDNA ITS markers in some studies (Park et al. 2010), but multiple Ampelomyces genotypes have been found within single mildew species by others (Pintye et al. 2012). Moreover, microsatellite markers have revealed some divergence among Ampelomyces strains that infect phenologically distinct hosts (Kiss et al. 2011; Pintye et al. 2015). Ultimately, the degree to which Ampelomyces spp co‐evolve with specific mildew species and strains remains an open question. As Ampelomyces is dependent upon its mildew host for survival, it can complete its life cycle (infection to sporulation) within a single clonal cycle of its mildew host. However, the existence or timing of sexual reproduction by Ampelomyces is currently unknown, and thus, the true relative generation times of both organisms are difficult to calculate.
Fungal and plant material
Pl. lanceolata clones were kept in greenhouse conditions at 20 ± 2 °C in 1:1 mixture of potting soil and sand. In the experiments described below, a single clonal plant genotype was used to control for any plant–host genotypic effects on the pathogen–hyperparasite interaction. This plant genotype had been in culture since 2012 and had shown a broad susceptibility to multiple pathogen isolates. It is also allopatric to all fungal isolates used in these experiments.
Po. plantaginis strains were collected from Åland in 2013 and 2015 (see Table S1, Supporting information) as infected leaf lesions. Isolates went through at least three rounds of purification with single‐colony transfers following isolation from the field and were subsequently maintained on detached Plantago leaves in a growth chamber at 20 ± 2 °C with 16:8 L:D photoperiod. Mildews were inoculated onto new maintenance leaves every 14 days.
Ampelomyces isolates used here were collected from Po. plantaginis infected leaves in Åland in 2014 and 2015 (see Table S1, Supporting information). The hyperparasite was maintained in pure culture on custom agar media (3 g NaNO3, 1 g K2HPO4, 0.5 g KCl, 0.5 g MgSO4, 30 g C12H22O11, 115 g Agar & Barley malt, litre−1) in darkness at ambient temperature. Isolates were turned over onto new media every 8 weeks.
Laboratory tests for G×G interactions and local adaptation
We conducted two laboratory inoculation studies to determine whether hyperparasite infection outcome is determined by either organism's genotype and to investigate whether there is evidence for local adaptation between the organisms.
Both experiments were carried out on detached Pl. lanceolata leaves placed onto moist filter paper in a Ø 9‐cm petri dish. Po. plantaginis conidial spores were taken from 14‐day‐old c. 1 cm2 lesions and evenly spread across the recipient leaf surface with a fine, sterile paintbrush. Previous work on this system has demonstrated that this method produces repeatable infection outcomes (Laine 2004; Susi & Laine 2013). Growth of the pathogen was tracked every 2 days starting at 6 days post‐mildew inoculation (DPM) until 16 DPM. Ampelomyces isolates were inoculated onto mildew‐infected leaves at 8 DPM as a 70 ± 2 μL spray of Ampelomyces spore suspension in filter‐sterilized H2O (1 × 106 spore/mL). Spore suspensions were obtained by scraping the surface of four 6‐week‐old Ampelomyces agar into filter‐sterilized H2O. Spore concentrations were assessed with a haemocytometer, and the suspensions were then diluted to the required concentration.
Po. plantaginis development was recorded on a scale modified from Bevan et al. (1993): 0: no growth, 1: spore germination and hyphae visible under a dissecting microscope, 1.5: mycelia with very few conidia visible only under a dissecting microscope, 2: mycelia visible to naked eye and sparse sporulation visible under a dissecting microscope, 3: abundant sporulation and lesion size <0.5 cm2, 4: abundant sporulation and lesion size >0.5 cm2. Established Ampelomyces infections produce distinctive brown spore structures within their host's conidia called pycnidia. Leaves were observed every 48 h with a dissecting microscope for the appearance of pycnidia starting at 10 DPM until the 16 DPM. Ampelomyces infection was scored with a modified version of the scale reported in Falk et al. (1995): 0: no pycnidia observed, 1: 1–20 pycnidia in each Ampelomyces cluster appearing, 2: 20–50 pycnidia in each powdery mildew lesion or between 30 and 50% of powdery mildew covered, 3: 50+ pycnidia in each powdery mildew lesion or >50% of powdery mildew covered. Levels 2 and 3 on this scale can reflect either a set number of Ampelomyces pycnidia or an estimate of pycnidia coverage of the mildew lesion. In this way, the scale controls for the different amounts of powdery mildew tissue available for the hyperparasite to infect; that is, small mildew lesions can still support a level‐3 hyperparasite infection even if there is not enough tissue to produce 50 + pycnidia. Ampelomyces‐negative control leaves infected with just Po. plantaginis were sprayed with H2O only. Negative controls were observed to confirm the absence of Ampelomyces contamination and to follow the growth of the powdery mildew without Ampelomyces infection. Both experiments were run in two blocks, 24 h apart.
Experiment 1: Allopatric variation
In the first experiment, we investigate how much Ampelomyces and Po. plantaginis genotypes, and their potential interaction, determine hyperparasite infectivity and infection severity. We used a fully reciprocal cross‐inoculation design to challenge five distinct genotypes of Po. plantagins that were collected in 2013 with three Ampelomyces isolates collected in 2014. All five mildews and three Ampelomyces were collected from different populations, and all pathogen–hyperparasite combinations were also allopatric. Each combination was replicated 36 times at the outset of the experiment, although this was eroded in some treatments if host leaves died or the mildew became contaminated during the course of the experiment (see Table S2, Supporting information). The spatial origins of the strains used are given in Table S1 (Supporting information). Mildew genotypes [typed at 19 SNP loci detailed in Table S3 (Supporting information) (Tollenaere et al. 2012)] are given in Table S4 (Supporting information).
Experiment 2: Local adaptation
The second inoculation experiment was designed to test for local adaptation between mildew and Ampelomyces. We cross‐inoculated six Ampelomyces isolates onto 12 powdery mildew strains, representing seven mildew populations (see Table S1, Supporting information). All Ampelomyces isolates were exposed to at least one sympatric mildew isolate (from the same population) and all allopatric mildews. All Po. plantaginis and Ampelomyces strains used in this experiment were collected in September 2015, and so are temporally contemporary. Each mildew–hyperparasite isolate combination was replicated between five and 24 times once any contaminated or failed mildew‐inoculations were trimmed from the data (see Table S2, Supporting information).
Assessing metapopulation‐wide and among‐population Ampelomyces prevalence
To assess patterns of hyperparasite prevalence at variable spatial scales, we conducted two sampling regimes in the Åland archipelago in the summer and autumn of 2014.
In September 2014, 70 Po. plantaginis populations were surveyed and screened for Ampelomyces as part of an ongoing large‐scale metapopulation survey (Ojanen et al. 2013; Jousimo et al. 2014) (Fig. 1). First, survey assistants scored each pathogen population for host plant abundance and mildew coverage. Following positive identification of mildew, populations were revisited a few days later and up to five leaves from five infected plants at ≥1 m apart were collected, dried and processed for DNA extraction. Prior to drying, up to ten leaves from each population were examined under a dissecting microscope for the presence of Ampelomyces pycnidia. When found, these were isolated onto cell‐free agar culture for experimentation (see Fungal and Plant material section above). All leaf samples were screened for Ampelomyces infection using the qPCR method described below, and any mildew population exhibiting at least one Ampelomyces‐positive sample was considered to be an infected population.
Figure 1.
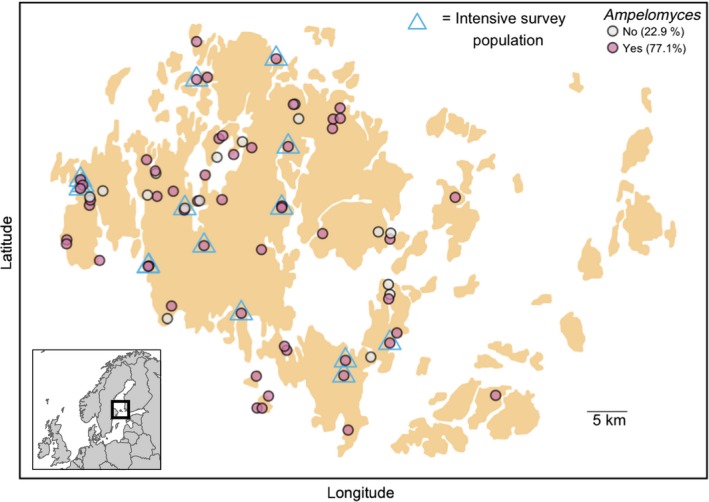
Ampelomyces is common at the metapopulation level. Map of the 70 Po. plantaginis populations in Åland that were screened during 2014. Of these, 54 were found to be infected by the Ampelomyces hyperparasite (pink circles). The 15 sites indicated with blue triangles were intensively surveyed across the whole summer of 2014.
Spatial and host genetic factors as determinates of within‐population hyperparasite spread
To assess within‐population hyperparasite prevalence patterns, we intensively sampled fifteen discrete populations of Po. plantagins infecting Pl. lanceolata up to eight times from early July to late August 2014. These populations were selected based upon their previous mildew infection status; that is, they have been infected in the previous year, and their location within the Åland archipelago; they covered a broad range of geographical regions (Fig. 1, blue triangles).
At each survey time, a maximum of thirty mildew‐infected Plantago plants, that were at least 1.5 m apart, were identified and scored for the presence/absence of mildew. We also scored the coverage of infected and uninfected Plantago in a 3 m diameter around the focal plant. Plant positions were recorded using GPS triangulation that was then corrected by eye on detailed topographical maps in the field‐site. Up to five infected leaves were sampled from each plant throughout the survey period. We scaled the number and timing of samples depending upon the mildew infection severity of the plant, to avoid removing the mildew from the field completely. Leaf samples were dried prior to DNA extraction and subsequent SNP genotyping of the mildew and qPCR screening for Ampelomyces (see below for molecular methods).
From these survey data, we calculated indices of within‐population connectivity (S i) to mildew‐infected sites and Ampelomyces‐infected sites for each focal plant at each survey time point. First, we calculate connectivity to all mildew‐infected plants, regardless of hyperparasite infection status, as a measure of pathogen aggregation and dispersal potential within the population. This is given by
where d ij is the Euclidian distance between focal plants i and j. We estimate dispersal distance (1/α) as 2 m, based on model simulations (Ovaskainen & Laine 2006) and empirical demonstrations (Tack et al. 2014a) that show the majority of Po. plantaginis spores land within 1 m of a progenitor infection and that dispersal over 2 m is extremely rare. p j is the abundance of powdery mildew infection at location j (percentage coverage of Pl. lanceolata within a 1.5 m radius of the focal plant × the percentage of those Pl. lanceolata infected with powdery mildew). Our second index describes the connectivity of focal plants to Ampelomyces‐infected sites within the population by including the binary response of Ampelomyces (1 = infected, 0 = uninfected) as a multiplier of p j. We assume that Ampelomyces has at least a similar dispersal kernel to its host, and that the extent of mildew coverage also represents the maximum possible Ampelomyces coverage in a given focal circle, and thus, we keep α and p j constant in both calculations. The square root of both indices is used in analyses.
To reduce the dimensionality of our data, we chose to estimate Ampelomyces prevalence at the ‘peak epidemic’ survey time for each focal plant, and thus, we took the maximum values found for all the variables mentioned above for each focal plant in every population. In this way, we account for the epidemics in each of the 15 populations occurring at different rates. Furthermore, given that Ampelomyces is dependent upon its host mildew for spread and survival, peak epidemic phase more realistically represents the risk of Ampelomyces invasion than, for example, the mean values for these metrics or by choosing an arbitrary time point at which to analyse all the data.
Genotyping of powdery mildew hosts
DNA from leaf samples was extracted using E.Z.N.A plant DNA extraction kit at the institute of biotechnology, University of Helsinki. Samples were subsequently genotyped at 19 SNP loci (see Table S3, Supporting information) with the Sequenome iPlex platform at the Finnish Institute of Molecular Medicine Finland (FIMM) (see Tollenaere et al. 2012 for more details). SNP calling was performed with typer4 software (Table S3, Supporting information). Because we sampled the haploid phase of Po. plantaginis, samples which were called as heterozygous for any locus are determined to be coinfected with at least two mildew multilocus genotypes. Any sample that failed to call all 19 SNP loci was removed from genotype analysis.
To analyse Ampelomyces presence by MLG diversity, we used the ‘poppr’ package in r (cran core team) to correct for the clonal nature of the mildew and to calculate indices of MLG diversity and richness at the population level. Several indices of MLG diversity were calculated for each of these 15 populations, all of which showed strong colinearity. For this reason in our analyses, we chose to use only Shannon's H index of diversity and estimated MLG richness based on rarefaction curves (N = 10) as computed by the ‘poppr’ package.
Screening for Ampelomyces infection
Because only heavily established Ampelomyces infections can be observed by eye, and microscope detection is extremely laborious, we used an Ampelomyces ‐specific qPCR screening technique developed by (Tollenaere et al. 2014) to detect hyperparasite infections in the field. Briefly, each sample was run for 30 cycles (30 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C) with the primers AmpITS_F (GCTGCCAATTGCTTTGAGAT) and AmpITS_R (GATGAAGAACGCAGCGAAAT) which target an Ampelomyces‐specific sequence within the rDNA ITS gene were used. This was followed by a melting curve analysis from 45 °C to 95 °C by 0.5 °C increment every 5 s. Samples with a mean C T value ≤24 (over three replicate reactions with SD < 0.2) were determined to possess an established Ampelomyces infection (see Supporting information of Tollenaere et al. 2014 for establishment of this threshold).
Statistical analyses
Statistical analyses were conducted in r (R core team 2016), and used additional packages ‘lme4’, ‘ordinal’, ‘poppr’, ‘mgcv’ and ‘car’. We broadly analysed our data within the generalized linear, generalized additive and cumulative link model frameworks. Minimum adequate models were derived from saturated models through stepwise simplification and selection based on likelihood ratio or χ2 tests of nested models (Crawley 2012). Nonsignificant factors are reported as the output of these model comparisons. The effect of significant independent variables is derived from analysis of the minimum adequate model with the ‘car’ package where possible or through model simplification when not. Where necessary, overdispersion was tested and accounted for by fitting a cloglog link function (fixed‐effect models) or an observation‐level random effect (mixed‐effect models). Before analysing our laboratory experiment, the data were trimmed to remove any leaf on which powdery mildew had not sporulated by 12 DPM, as these were regarded as failed pathogen inoculations which were unable to host Ampelomyces infection.
Ampelomcyes infectivity (pycnidia present at 16 DPM) in both inoculation experiments was modelled with mixed‐effect GLMs with a binomial distribution of errors and logit link function using package ‘lme4’. Mildew identity, Ampelomyces identity, their interaction and mildew growth at the time of hyperparasite inoculation (8 DPM) were coded as fixed effects. Experimental block was included as a random intercept. To test for local adaptation in our second experiment, we included a ‘sympatry/allopatry’ fixed effect to our model and furthermore included ‘sympatry/allopatry’ nested within ‘Ampelomyces strain identity’ as an additional random effect to account for overall variation in infectivity among Ampelomyces strains. Models of our second experiment failed to converge due to rank deficiencies if we included the interaction between mildew and Ampelomyces strains. Therefore, we included a separate factorial variable with a level for each pathogen–hyperparasite combination as a fixed effect to account for variation among strain combinations.
Ampelomyces infection severity was analysed for all lesions on which Ampelomyces infection was observed. These data were analysed with a cumulative link mixed‐effect model (clmm) to account for the ordinal nature of the Ampelomyces infection severity scale. Similar model structures as before were used but with Ampelomyces severity level (1–3) as the dependent variable and replacing mildew growth at 8DPM with mildew growth at the time of scoring (16DPM).
To analyse the variation in among‐population Ampelomyces prevalence in the field, we use a generalized linear model with a quasibinomial distribution of errors to account for overdispersion of the response variable. The proportion of leaf samples hosting Ampelomyces in each population was set as the dependent variable. Fixed independent variables were: estimated mildew MLG richness, Shannon's H index of MLG diversity, the proportion of leaf samples presenting signals of coinfection (heterozygous SNP calls at ≥1 loci), mean mildew coverage across focal plants in the population and the population's connectivity index relative to the entire pathogen metapopulation (as calculated in Penczykowski et al. 2014).
To assess patterns of within‐population prevalence of Ampelomyces, we analysed presence/absence of the hyperparasite in each leaf sample within a generalized additive model framework. We include two measures of hyperparasite spatial clustering in our analyses. First, within‐population connectivity of Ampelomyces‐infected plants is included as a fixed effect to test whether the hyperparasite is aggregated within its already aggregated mildew host. That is to say, does the Ampelomyces infection prevalence across neighbouring plants influence the probability that it will infect a given focal plant? Second, we include the spline of each focal plant's longitude and latitude as an additive fixed effect to account for spatial variation in infection risk across each population. That is to say, are Ampelomyces infections aggregated in space irrespective of how many hyperparasite infected and uninfected plants are in that location? We also include the maximum‐recorded amount of mildew surrounding each focal plant to determine whether relative pathogen abundance determines Ampelomyces infection success. We include focal plant identity nested within‐population identity as a random intercept to account for repeated sampling of the same host plants and the aggregation of host plants within discrete populations in the archipelago. Because our field data contain a large number of rare and singleton MLG types (75 MLGs, see Fig. 5D), we were unable to incorporate this information as a multi‐level factor in the analysis detailed above. Instead, we performed a second analysis on a truncated data set from which any sample with a rare MLG (<5 instances found in the full dataset) or evidence of coinfection was removed. This resulted in a reduced data set representing samples from 20 MLG types. Otherwise, the initial model structure for this analysis was the same as described previously. Numerical covariates in both of these analyses were centred and scaled before they were modelled.
Results
Hyperparasite and pathogen genotype combination determines infection outcome
Our laboratory cross‐inoculation experiment of allopatric pathogen–hyperparasite isolates revealed that Ampelomyces infectivity was significantly dependent upon the mildew genotype (χ2 = 38.13, d.f. = 5, P < 0.001), hyperparasite genotype (χ2 = 51.57, d.f. = 3, P < 0.001) and their interaction (χ2 = 19.21, d.f. = 8, P = 0.0138) (Fig. 2). Hyperparasite infectivity was also significantly, positively correlated with the developmental stage of the mildew lesion at the time of hyperparasite inoculation (χ2 = 9.113, d.f. = 1, P = 0.003). The severity of Ampelomyces infection was analysed at 16 DPM, at which point there was a significant effect of both mildew and hyperparasite strain identity (mildew genotype: LRT = 94.289, d.f. = 4, P < 0.001, hyperparasite genotype: LRT= 257.2, d.f. = 2, P < 0.001), but no significant interaction between these factors (LRT=1.77, d.f. = 2, P = 0.2). Ampelomyces infection severity was also positively correlated with the mildew infection severity at the time of scoring (LRT = 27.714, d.f. = 1, P < 0.001).
Figure 2.
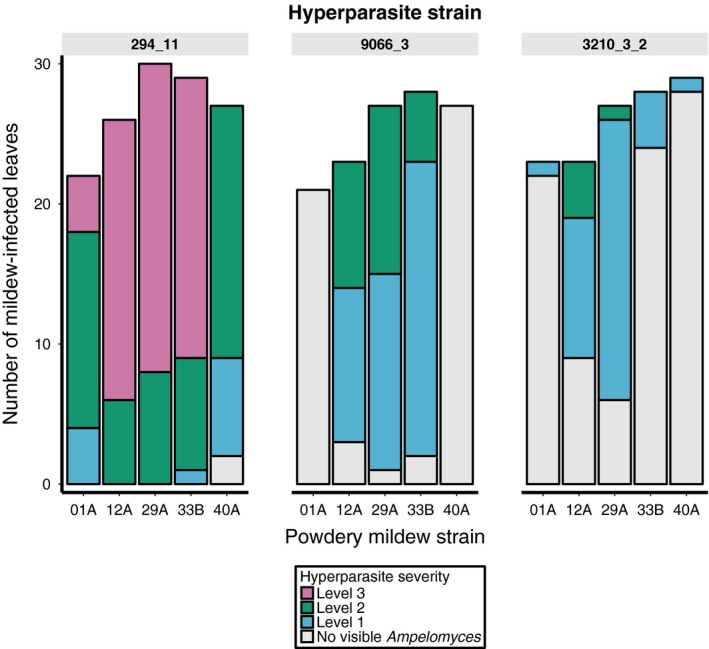
Hyperparasite infectivity and infection severity varied among pathogen–hyperparasite genotype combinations. Hyperparasite strain 294_11 performed better than both other strains in terms of both the proportion of mildew lesions it infected, and the extent of infection after 16 days of growth. Mildew strains also vary in their susceptibility to hyperparasite attack, which is dependent upon the identity of the hyperparasite. For instance, mildew 33B is highly susceptible to 294_11 and 9066_3, but is able to resist 3210_3_2.
Hyperparasite vary in their degree of local adaptation to their mildew host
Our test for local adaptation found that hyperparasites were more successful at infecting sympatric pathogen strains than allopatric ones (χ2 = 4.69, d.f. = 1, P = 0.03) (Fig. 3). However, there was variation in this trend, with a significant impact of mildew genotype (χ2 = 55.609, d.f. = 6 P < 0.001) and Ampelomyces strain (χ2 = 23.74, d.f. = 5 P < 0.001) on hyperparasite infectivity. There was no significant effect of mildew and Ampelomyces strain combination on hyperparasite infectivity (χ2 = 3.3, d.f. = 5, P = 0.654). As above, the lesion size of the mildew at the time of Ampelomyces inoculation had a significant, positive impact on hyperparasite infection success (χ2 = 33.06, d.f. = 1, P < 0.001). Hyperparasites significantly affected sympatric mildew isolates more severely than allopatric ones (LRT = 6.547, d.f. = 1, P = 0.011), and infection severity was also significantly dependent upon the identity of both the mildew (LRT = 25.23, d.f. = 5, P < 0.001) and the Ampelomyces (LRT = 19.67, d.f. = 6, P = 0.003).
Figure 3.
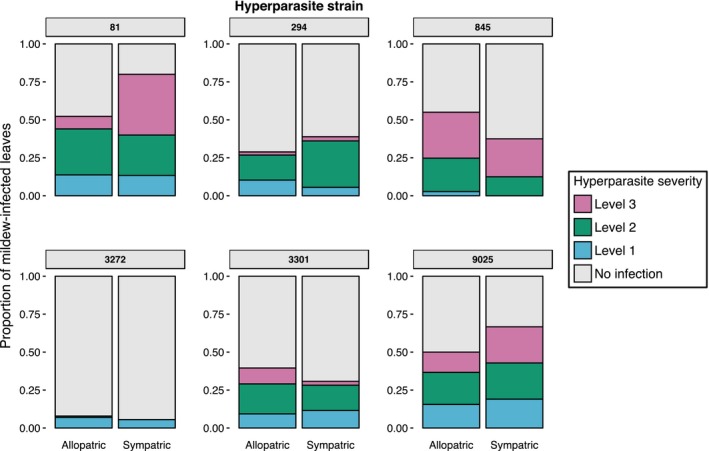
Ampelomyces can be locally adapted to its mildew host. Hyperparasite infectivity and infection severity strongly varied among hyperparasite strains (individual plots) and between sympatric/allopatric mildew hosts. Statistical analysis suggests that Ampelomyces was more infective against sympatric mildew than the mean of all allopatric combinations; however, the presence and intensity of this effect varied greatly among hyperparasite genotypes. This could indicate that selection for local adaptation varies in strength across the metapopulation, or that the strength of selection is highly dynamic over evolutionary time.
Hyperparasite prevalence varies at different spatial scales
Metapopulation‐scale sampling of the Åland archipelago found that 54 of 70 (77.1%) Po plantaginis populations supported Ampelomyces infection, indicating that the hyperparasite is widespread across the archipelago. However, in the 15 populations that were intensively sampled throughout the epidemic season, we found that hyperparasite prevalence ranged between 0 and 45%. Ampelomyces prevalence in these populations was not significantly associated with mildew genetic diversity (χ2 = −0.006, d.f. = 1, P = 0.9), MLG richness: (χ2 = −0.052, d.f. = 1, P = 0.91) or mildew population size (χ2 = −0.436, d.f. = 1, P = 0.729). However, the proportion of leaf samples supporting coinfecting mildew MLGs and the connectivity of the populations within the mildew metapopulation have a marginally positive effect on hyperparasite prevalence (mildew coinfection: χ2 = −11.26, d.f. = 1, P = 0.067, population connectivity: χ2 = −12.232, d.f. = 1, P = 0.056).
Spatial structure determines within‐population prevalence
Analysis of our intensive field surveys (Fig. 4 and Table 1) data revealed that a focal plant's connectivity to Ampelomyces‐infected plants within a population positively correlated with the probability that it also hosts the hyperparasite (z = 5.310731 P < 0.001, Fig. 5A). In addition, leaves that supported multiple co‐infecting mildew MLGs were significantly more likely to also host the hyperparasite (z = 2.104731,1, P = 0.032, Fig. 5B). Furthermore, the spatial location of plants within populations significantly affected their probability of hosting Ampelomyces (approx. significance of smooth terms: χ2 = 8.101, e d.f. = 2, P = 0.02, Fig. 4). However, patterns of within‐population Ampelomyces occurrence were not significantly associated with the amount of mildew surrounding focal plants (χ2 = 1.1027, d.f. = 1, P = 0.293, Fig. 5C). In addition, within‐population connectivity of mildew‐infected plants also has no significant bearing on whether they were infected with the hyperparasite (χ2 = 3.176, d.f. = 1, P = 0.075).
Figure 4.
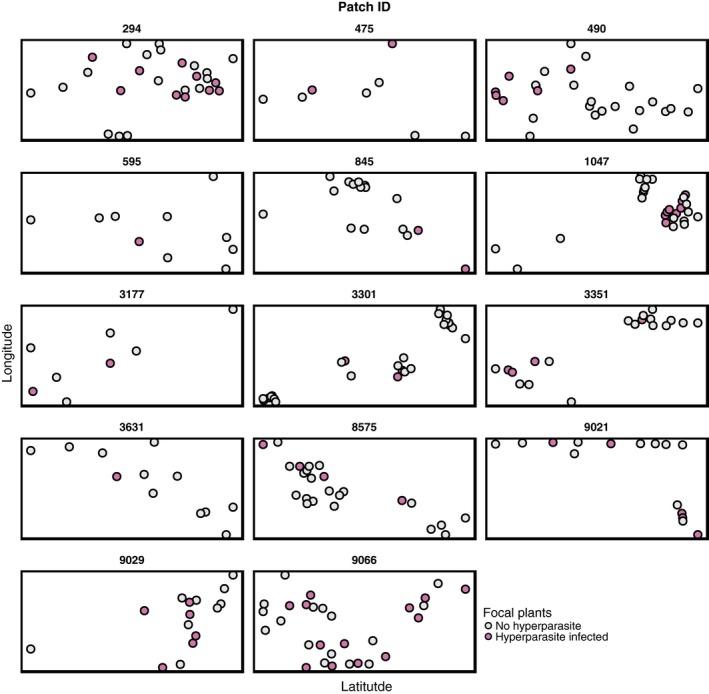
Ampelomyces infections are spatially clustered among mildew‐infected plants within populations. Facets represent geometric area of the 14 intensively surveyed Po. plantaginis populations in which at least one mildew‐infected leaf supported a hyperparasite infection. Circles denote the locations of mildew‐infected Plantago lanceolata. Pink circles denote host plants from which at least one mildew sample was found to support the hyperparasite.
Table 1.
Factors contributing to Ampelomyces infection in natural field populations analyzed with generalised linear additive models
| Estimate ± SE | Test statistic | P‐value | |
|---|---|---|---|
| Variables dropped through model selection | |||
| Focal circle mildew coverage1,707 | — | χ2 = 1.107 | 0.293 |
| Mildew connectivity1,707 | — | χ2 = 1.722 | 0.189 |
| Variables retained after model selection | |||
| Intercept1,733 | −2.329 ± 0.231 | z = −10.075 | <0.001 |
| Coinfection1,733 | 0.649 ± 0.303 | z = 2.141 | 0.032 |
| Ampelomyces connectivity1,733 | 1.273 ± 0.215 | z = 5.931 | <0.001 |
| Spline(long, lat) | χ2 = 8.101 | 0.017 | |
| Random effects | Variance | ||
| Focal plant (within population)259,707 | 4.6 | — | — |
| Population14,707 | 0.025 | — | — |
Significant P‐values (<0.05) are in bold.
Figure 5.
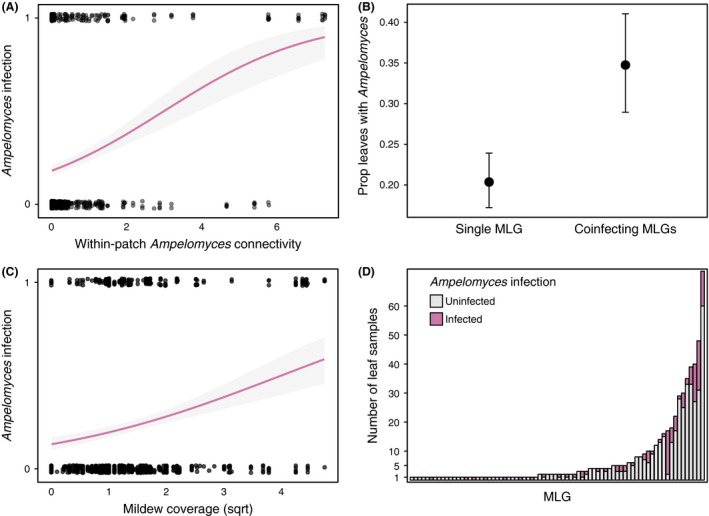
Factors influencing within‐population Ampelomyces prevalence. Ampelomyces infections are significantly positively correlated with the connectivity of Ampelomyces‐infected plants (A) and also significantly more common on leaves supporting >1 mildew MLG (B). There is a positive, but not significant correlation between the amount of mildew surrounding a focal plant and its probability of supporting Ampelomyces (C). Mildew MLG identity does not significantly predict Ampelomyces presence, although hyperparasite infectivity does vary across MLGs (D).
Separate analysis of Ampelomyces infection in the most common MLG types in our data set revealed no significant association between mildew identity and hyperparasite infection (χ2 = 9.46, d.f. = 1, P = 0.221). However, we still see a significant effect of within‐population Ampelomyces connectivity (z = 5.222352, P < 0.002) even in this truncated data set.
Discussion
Understanding how pathogens and parasites interact with their own natural enemies will be essential if we are to effectively predict and control outbreaks of infectious disease. Here, we assess the contribution of both genetic and spatial components to a hyperparasite–pathogen interaction. We show that under laboratory conditions, the establishment and severity of a hyperparasite infection varies depending upon the identity of both antagonists, suggesting that main genotype and G×G dynamics govern the outcome of hyperparasitism. Furthermore, we find evidence for local adaptation of the hyperparasite to its sympatric pathogen host strain across a spatially fragmented landscape, although the strength of this varies across populations. However, when exploring hyperparasite–pathogen interactions in the field, we do not find a strict association of Ampelomyces with specific host strains. This result reinforces previous population genetic work which found that Ampelomyces strains are not strictly associated with their host lineage (Pintye et al. 2012). Rather, we find that hyperparasite infection is more likely when host genetic diversity is high, that is multiple mildew genotypes coinfect the same plant. Furthermore, we show that the hyperparasite can be spatially aggregated and relatively rare (<40% prevalence) within local populations of its pathogen host, but is very common at the metapopulation scale (77.1% of populations).
Our data suggest that Ampelomyces may be dispersal‐limited within pathogen populations, or sensitive to local environmental conditions. In contrast, Ampelomyces is relatively common at the metapopulation level. This may be partially explained by the timing of the respective surveys; July–August for within‐population prevalence, early September for the metapopulation‐wide survey. Although, an alternative explanation for this dichotomy is that Ampelomyces may be present upon multiple mildew hosts in Åland, which allows it to cover a large geographical area and to infect many Po. plantaginis populations despite having limited spread at the local level. Several studies have shown that many Ampelomyces isolates can readily host‐shift (Liang et al. 2007; Kiss et al. 2011; Angeli et al. 2012b), so the hyperparasite infections that we observe may be the result of a spillover from Ampelomyces reservoirs in other mildew species (Power & Mitchell 2004). However, this would not explain why we see limitations to Ampelomyces spread once it has spilled‐over to a Po. plantaginis population, nor would it explain why we see the same Ampelomyces isolate performing differently against different mildew hosts in the laboratory.
Interestingly, our laboratory and field observations present contrasting results regarding the importance of genetic factors in governing hyperparasite infection. Our laboratory experiments strongly suggest that both main genotype and G×G interactions dictate hyperparasite infectivity and infection severity. However, in the field we find no significant effect of mildew host genotype on Ampelomyces prevalence patterns. Rather, we find that the hyperparasite is spatially clustered among mildew‐infected plants, which alludes towards an environmentally derived barrier to infection spread. This discrepancy may indicate that Ampelomyces epidemiology in nature is strongly influenced by ecological factors before any genetic filtering can occur. Indeed, the degree of aggregation appears to vary among populations (Fig. 4), and so is likely to be a function of the multiple environmental factors that affect dispersal, such as prevailing wind strength and direction, rainfall, humidity and the presence of physical barriers. It is also possible that the severity and specificity of any G×G interactions and local adaptation effects are sensitive to such environmental factors. Similar issues have been encountered in other parasitic interactions, where the environment can influence the outcome of genetic interactions between organisms (Laine 2008; Bryner & Rigling 2011; Tack et al. 2015). Furthermore, the discrepancy between our laboratory and field observations may also be partially due to the sheer genetic diversity present in our pathogen populations. For instance, it is possible that several distinct mildew genotypes within a given population may be equally susceptible or resistant to the resident Ampelomyces. This could mask any signal of host association, particularly if many mildew genotypes are rare and thus underrepresented in our data.
Although we have found no evidence that the hyperparasite is associated with particular pathogen genotypes in nature, our field observations do demonstrate that Ampelomyces is most likely to be found on plants that are coinfected by multiple mildew genotypes. In addition, we found that the amount of mildew coinfection within a population positively correlated with the prevalence of the hyperparasite. Interestingly, this effect appears to be independent of the absolute amount of mildew available for the hyperparasite to infect, as there was no significant correlation between hyperparasite infection and pathogen coverage of either the focal plant or within a 3 metre radius. These results confirm a similar pattern found by (Tollenaere et al. 2014) and strongly suggest that some property of coinfecting pathogens makes them more suitable as hosts for the hyperparasite. If Ampelomyces infection is indeed determined in a G×G manner, as our experimental results suggest, then coinfections may simply double the probability that one of the two mildews present is a suitable host. Alternatively, mildew physiology may alter under coinfection in a way that increases their susceptibility to hyperparasite infection. For example, mildews may invest in sexual reproduction or competition with coinfecting conspecifics rather than defence against the hyperparasite.
Despite the lack of genetic filtering in our field observations, our laboratory results are, to our knowledge, the first to demonstrate any degree of genotype‐by‐genotype interaction and local adaptation in a horizontally transmitted hyperparasite system. G×G interactions and local adaptation are key processes driving disease dynamics and governing infection risks in natural pathogen populations (Lambrechts et al. 2006; Tack et al. 2012), but these processes require a degree of fidelity between host–pathogen strains to be maintained over evolutionary time. Many hyperparasites are likely to be under selection to utilize multiple host species and/or to disperse over long distances in order to maintain an effective population size (Nair et al. 2016). Thus, we do not necessarily expect G×G interactions or local adaptation to arise in hyperparasite systems. Ampelomyces has been shown to readily host‐shift among mildew species under laboratory conditions (Liang et al. 2007; Kiss et al. 2011; Angeli et al. 2012b), and molecular work indicates that strains are only very loosely associated with distinct mildew species (Liang et al. 2007; Park et al. 2010; Kiss et al. 2011). Therefore, our finding that Ampelomyces exhibits a G×G relationship within genetically distinct isolates of one species of powdery mildew is unexpected. Indeed, the only other evidence for a pathogen–hyperparasite G×G interaction has been found between a pathogenic fungus and a virus that can vertically transmit within lineages of its host, thus sidestepping the issue of host scarcity (Bryner & Rigling 2011). This may be testament to how rapidly co‐evolutionary forces can shape host–pathogen interactions and generate diversity within populations, even at higher trophic levels.
Our laboratory experiments also revealed a signal of local adaptation of the hyperparasite to its sympatric mildew host when compared to the average of allopatric combinations. This effect was seen both in measures of infectivity and infection severity, but was highly variable among populations. Similarly to our laboratory evidence for G×G dynamics, the extent to which Ampelomyces local adaptation governs hyperparasite epidemiology in nature is unclear and may be minimal. However, this does allude to a co‐evolutionary relationship between pathogen and hyperparasite. Co‐evolutionary relationships such as these can be highly dynamic over short periods of time and across metapopulations. Thus, the variation in hyperparasite adaptation that we see among populations may be a direct result of temporal fluctuations in the strength and synchrony in co‐evolutionary selection (Thompson 2005; Blanquart et al. 2013; Koskella 2014). Combining both a sympatry/allopatry and time‐shift approach to future studies in this system may reveal the evolutionary dynamics more fully.
Several studies have documented that the infection severity of Ampelomyces isolates can vary depending on the species of mildew host (Liang et al. 2007; Kiss et al. 2011; Angeli et al. 2012b), but our study is the first to robustly demonstrate phenotypic variation of the hyperparasite within a single host species at an intermediate spatial scale (5–50 km). Previous work on this system has found extremely low levels of diversity in the Ampelomyces strains infecting Po. plantaginis using conservative genetic markers (Tollenaere et al. 2014). In the light of this, our findings suggest that relatively rapid co‐evolutionary processes are able to maintain phenotypic diversity within highly related groups of hyperparasite strains. Therefore, to fully explore within‐Ampelomyces variation, and thus accurately track mildew–hyperparasite associations across the natural metapopulation, future studies should utilize population genetic techniques that can detect variation in rapidly evolving loci. A suite of Ampelomyces genes recently identified in host recognition and infection present a promising resource for such work (Siozios et al. 2015).
Our results also demonstrate that powdery mildew strains of a single species deriving from a single metapopulation vary in their susceptibility to the hyperparasite. Whether this variation represents adaptive resistance to Ampelomyces attack, or is a by‐product of the variation present in the Po. plantaginis metapopulation is unknown. Both empirical and theoretical work have shown that variation in susceptibility to natural enemies across spatially structured populations can fundamentally alter epidemiological and evolutionary patterns of infectious disease (Salvaudon et al. 2008; Laine 2011; Jousimo et al. 2014). However, this has not been explicitly considered in the case of pathogens that must both defend against hyperparasite attack and also infect and spread among their own hosts. The consequences of evolving resistance to hyperparasites for pathogen epidemiology, and even the existence of nonadaptive variation in susceptibility among pathogen populations, are of significant interest when considering hyperparasites as effective biocontrol measures (Parratt & Laine 2016).
The risk of disease incidence and severity of infectious outbreaks can vary greatly over spatially structured host populations (Ostfeld et al. 2005). Determining the factors, both genetic and ecological, that drive this variation is imperative if we are to successfully predict and tackle the emergence of dangerous human, agricultural and wildlife pathogens. Given that hyperparasites can fundamentally alter disease virulence (Abo‐Foul et al. 1996; Nuss 2005), epidemiology (Verhaar et al. 1996; Andersen et al. 2012; Springer et al. 2013; Tollenaere et al. 2014) and evolution (Taylor et al. 1998), they may represent a key top‐down moderator of disease dynamics. Here, we have shown that hyperparasite infectivity and virulence can vary greatly across intermediate spatial scales due to genetic variation in both the hyperparasite itself and its host. We also show that pathogen spatial configuration is still a key determinant of local‐level hyperparasite spread in nature, even if hyperparasites are ubiquitous in the environment. These findings have implications for selecting hyperparasite strains when considering the use of these organisms as biocontrol measures for human and agricultural epidemics (Swinton & Gilligan 1999; Nobrega et al. 2015).
The study was conceived and designed by S.R.P. and A.‐L.L. Intensive field surveys were designed and conducted by S.R.P., R.M.P. and B.B. Pathogen SNP genotyping and analysis was conducted by S.R.P. and B.B. Laboratory experiments and data analyses were conducted by S.R.P. and R.M.P. The main manuscript was written by S.R.P. and A.‐L.L., and all authors read and contributed to the final draft.
Data accessibility
All data presented in this manuscript are available on the Dryad digital repository: doi:10.5061/dryad.5fh23.
Supporting information
Table S1. Fungal strains used in laboratory experiments.
Table S2. Replication within each treatment of the two inoculation experiments.
Table S3. SNP markers and their typer4 calling parameters used to genotype Podosphaera plantaginis.
Table S4. MLG genotypes of Podosphaera plantaginis strains used in laboratory inoculation experiments.
Acknowledgments
We would like to thank Pauliina Hyttinen, Sara Neggazi and Hanna Parri for conducting the field surveys, collecting fungal samples and processing genetic samples. We also thank Aku Korhonen and Angela Sims for assistance with qPCR, Krista Raveala for her help in maintaining and conducting laboratory experiments and Torsti Schulz and Elina Numminen for their statistical advice. We acknowledge funding from the Academy of Finland (Center‐of‐Excellence in Metapopulation Biology 2015‐2017; 284601) and the European Research Council (ERC; Independent Starting Grant PATHEVOL; 281517) to A.‐L.L.
References
- Abo‐Foul S, Raskin VI, Sztejnberg A, Marder JB (1996) Disruption of chlorophyll organization and function in powdery mildew‐diseased cucumber leaves and its control by the hyperparasite Ampelomyces quisqualis . Phytopathology, 86, 195–199. [Google Scholar]
- Andersen SB, Ferrari M, Evans HC et al (2012) Disease dynamics in a specialized parasite of ant societies. PLoS One, 7, e36352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, May RM (1991) Infectious Diseases of Humans: Dynamics and Control. Oxford University Press, Oxford. [Google Scholar]
- Angeli D, Maurhofer M, Gessler C, Pertot I (2012a) Existence of different physiological forms within genetically diverse strains of Ampelomyces quisqualis . Phytoparasitica, 40, 37–51. [Google Scholar]
- Angeli D, Puopolo G, Maurhofer M, Gessler C, Pertot I (2012b) Is the mycoparasitic activity of Ampelomyces quisqualis biocontrol strains related to phylogeny and hydrolytic enzyme production? Biological Control, 63, 348–358. [Google Scholar]
- Bangham J, Kim K‐W, Webster CL, Jiggins FM (2008) Genetic variation affecting host‐parasite interactions: different genes affect different aspects of sigma virus replication and transmission in Drosophila melanogaster . Genetics, 178, 2191–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan JR, Crute IR, Clarke DD (1993) Diversity and variation in expression of resistance to Erysiphe‐fischeri in Senecio‐vulgaris . Plant Pathology, 42, 647–653. [Google Scholar]
- Blanquart F, Kaltz O, Nuismer SL, Gandon S (2013) A practical guide to measuring local adaptation. Ecology Letters, 16, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Braun U (1995) The Powdery Mildews (Erysiphales) of Europe. VEB Gustav Fischer Verlag, Jena. [Google Scholar]
- Bryner SF, Rigling D (2011) Temperature‐dependent genotype‐by‐genotype interaction between a pathogenic fungus and its hyperparasitic virus. The American Naturalist, 177, 65–74. [DOI] [PubMed] [Google Scholar]
- Caffi T, Legler SE, Bugiana R, Rossi V (2013) Combining sanitation and disease modelling for control of grapevine powdery mildew. European Journal of Plant Pathology, 135, 817–829. [Google Scholar]
- Crawley MJ (2012) The R Book. John Wiley & Son, West Sussex. [Google Scholar]
- Falk SP, Gadoury DM, Pearson RC, Seem RC (1995) Partial central of grape powdery mildew by the mycoparasite Ampelomyces‐quisqualis . Plant Disease, 79, 483–490. [Google Scholar]
- Gandon S, Michalakis Y (2002) Local adaptation, evolutionary potential and host‐parasite coevolution: interactions between migration, mutation, population size and generation time. Journal of Evolutionary Biology, 15, 451–462. [Google Scholar]
- Gilligan CA (2002) An epidemiological framework for disease management. Advances in Botanical Research, 38, 1–64. [Google Scholar]
- Gómez JM, Abdelaziz M, Camacho JPM, Munoz‐Pajares AJ, Perfectti F (2009) Local adaptation and maladaptation to pollinators in a generalist geographic mosaic. Ecology Letters, 12, 672–682. [DOI] [PubMed] [Google Scholar]
- Greischar MA, Koskella B (2007) A synthesis of experimental work on parasite local adaptation. Ecology Letters, 10, 418–434. [DOI] [PubMed] [Google Scholar]
- Hoeksema JD, Forde SE (2008) A meta‐analysis of factors affecting local adaptation between interacting species. American Naturalist, 171, 275–290. [DOI] [PubMed] [Google Scholar]
- Holt RD, Hochberg ME (1998) The coexistence of competing parasites. Part II—hyperparasitism and food chain dynamics. Journal of Theoretical Biology, 193, 485–495. [DOI] [PubMed] [Google Scholar]
- Jackson JA, Tinsley RC (2005) Geographic and within‐population structure in variable resistance to parasite species and strains in a vertebrate host. International Journal for Parasitology, 35, 29–37. [DOI] [PubMed] [Google Scholar]
- Jousimo J, Tack AJM, Ovaskainen O et al (2014) Ecological and evolutionary effects of fragmentation on infectious disease dynamics. Science, 344, 1289–1293. [DOI] [PubMed] [Google Scholar]
- Kawecki TJ, Ebert D (2004) Conceptual issues in local adaptation. Ecology Letters, 7, 1225–1241. [Google Scholar]
- Kiss L (1997) Genetic diversity in Ampelomyces isolates, hyperparasites of powdery mildew fungi, inferred from RFLP analysis of the rDNA ITS region. Mycological Research, 101, 1073–1080. [Google Scholar]
- Kiss L, Nakasone KK (1998) Ribosomal DNA internal transcribed spacer sequences do not support the species status of Ampelomyces quisqualis, a hyperparasite of powdery mildew fungi. Current Genetics, 33, 362–367. [DOI] [PubMed] [Google Scholar]
- Kiss L, Russell JC, Szentiványi O, Xu X, Jeffries P (2004) Biology and biocontrol potential of Ampelomyces mycoparasites, natural antagonists of powdery mildew fungi. Biocontrol Science and Technology, 14, 635–651. [Google Scholar]
- Kiss L, Pintye A, Kovács GM et al (2011) Temporal isolation explains host‐related genetic differentiation in a group of widespread mycoparasitic fungi. Molecular Ecology, 20, 1492–1507. [DOI] [PubMed] [Google Scholar]
- Koskella B (2014) Bacteria‐phage interactions across time and space: merging local adaptation and time‐shift experiments to understand phage evolution. The American Naturalist, 184, S9–21. [DOI] [PubMed] [Google Scholar]
- Kyle CJ, Rico Y, Castillo S et al (2014) Spatial patterns of neutral and functional genetic variations reveal patterns of local adaptation in raccoon (Procyon lotor) populations exposed to raccoon rabies. Molecular Ecology, 23, 2287–2298. [DOI] [PubMed] [Google Scholar]
- Laine A‐L (2004) Resistance variation within and among host populations in a plant: pathogen metapopulation: implications for regional pathogen dynamics. Journal of Ecology, 92, 990–1000. [Google Scholar]
- Laine A‐L (2007) Detecting local adaptation in a natural plant‐pathogen metapopulation: a laboratory vs. field transplant approach. Journal of Evolutionary Biology, 20, 1665–1673. [DOI] [PubMed] [Google Scholar]
- Laine A‐L (2008) Temperature‐mediated patterns of local adaptation in a natural plant‐pathogen metapopulation. Ecology Letters, 11, 327–337. [DOI] [PubMed] [Google Scholar]
- Laine A‐L (2011) Context‐dependent effects of induced resistance under co‐infection in a plant‐pathogen interaction. Evolutionary Applications, 4, 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine A‐L, Hanski I (2006) Large‐scale spatial dynamics of a specialist plant pathogen in a fragmented landscape. Journal of Ecology, 94, 217–226. [Google Scholar]
- Lambrechts L, Fellous S, Koella JC (2006) Coevolutionary interactions between host and parasite genotypes. Trends in Parasitology, 22, 12–16. [DOI] [PubMed] [Google Scholar]
- Legler SE, Caffi T, Kiss L, Pintye A, Rossi V (2011) Methods for screening new Ampelomyces strains to be used as biocontrol agents against grapevine powdery mildew. IOBC/WPRS Bulletin, 67, 149–154. [Google Scholar]
- Legler SE, Pintye A, Caffi T et al (2016) Sporulation rate in culture and mycoparasitic activity, but not mycohost specificity, are the key factors for selecting Ampelomyces strains for biocontrol of grapevine powdery mildew (Erysiphe necator). European Journal of Plant Pathology, 144, 723–736. [Google Scholar]
- Liang C, Yang J, Kovács GM et al (2007) Genetic diversity of Ampelomyces mycoparasites isolated from different powdery mildew species in China inferred from analyses of rDNA ITS sequences. Fungal Diversity, 24, 225–240. [Google Scholar]
- Lively CM (1999) Migration, virulence, and the geographic mosaic of adaptation by parasites. American Naturalist, 153, S34–S47. [DOI] [PubMed] [Google Scholar]
- Nair A, Fountain T, Ikonen S, Ojanen SP, van Nouhuys S (2016) Spatial and temporal genetic structure at the fourth trophic level in a fragmented landscape. Proceedings of the Royal Society B: Biological Sciences, 283, 20160668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega FL, Costa AR, Kluskens LD, Azeredo J (2015) Revisiting phage therapy: new applications for old resources. Trends in Microbiology, 23, 185–191. [DOI] [PubMed] [Google Scholar]
- Nuss DL (2005) Hypovirulence: Mycoviruses at the fungal|[ndash]|plant interface. Nature Reviews. Microbiology, 3, 632–642. [DOI] [PubMed] [Google Scholar]
- Ojanen SP, Nieminen M, Meyke E, Pöyry J, Hanski I (2013) Long‐term metapopulation study of the Glanville fritillary butterfly (Melitaea cinxia): survey methods, data management, and long‐term population trends. Ecology and Evolution, 3, 3713–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostfeld R, Glass G, Keesing F (2005) Spatial epidemiology: an emerging (or re‐emerging) discipline. Trends in Ecology & Evolution, 20, 328–336. [DOI] [PubMed] [Google Scholar]
- Ovaskainen O, Laine A‐L (2006) Inferring evolutionary signals from ecological data in a plant‐pathogen metapopulation. Ecology, 87, 880–891. [DOI] [PubMed] [Google Scholar]
- Park M‐J, Choi Y‐J, Hong S‐B, Shin H‐D (2010) Genetic variability and mycohost association of Ampelomyces quisqualis isolates inferred from phylogenetic analyses of ITS rDNA and actin gene sequences. Fungal Biology, 114, 235–247. [DOI] [PubMed] [Google Scholar]
- Parratt SR, Laine A‐L (2016) The role of hyperparasitism in microbial pathogen ecology and evolution. The ISME Journal, 10, 1815–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penczykowski RM, Walker E, Soubeyrand S, Laine A‐L (2014) Linking winter conditions to regional disease dynamics in a wild plant‐pathogen metapopulation. The New Phytologist, 205, 1142–1152. [DOI] [PubMed] [Google Scholar]
- Pintye A, Bereczky Z, Kovács GM et al (2012) No indication of strict host associations in a widespread mycoparasite: grapevine powdery mildew (Erysiphe necator) is attacked by phylogenetically distant Ampelomyces strains in the field. Phytopathology, 102, 707–716. [DOI] [PubMed] [Google Scholar]
- Pintye A, Ropars J, Harvey N et al (2015) Host phenology and geography as drivers of differentiation in generalist fungal mycoparasites. PLoS One, 10, e0120703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power AG, Mitchell CE (2004) Pathogen spillover in disease epidemics. The American Naturalist, 164, 79–89. [DOI] [PubMed] [Google Scholar]
- R Core Team (2016) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria: URL https://www.R-project.org/. [Google Scholar]
- Romero D, Rivera ME, Cazorla FM, De Vicente A, Perez‐Garcia A (2003) Effect of mycoparasitic fungi on the development of Sphaerotheca fusca in melon leaves. Mycological Research, 107, 64–71. [DOI] [PubMed] [Google Scholar]
- Salvaudon L, Giraud T, Shykoff JA (2008) Genetic diversity in natural populations: a fundamental component of plant‐microbe interactions. Current Opinion in Plant Biology, 11, 135–143. [DOI] [PubMed] [Google Scholar]
- Schulte RD, Makus C, Hasert B, Michiels NK, Schulenburg H (2011) Host‐parasite local adaptation after experimental coevolution of Caenorhabditis elegans and its microparasite Bacillus thuringiensis . Proceedings of the Royal Society B: Biological Sciences, 278, 2832–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MW (2002) Epidemic modelling and disease forecasting In: Plant Pathologists Pocketbook (eds Waller JM, Lenné JM, Waller SJ.), pp. 252–265. Plant Pathologist's Pocketbook, Wallingford. [Google Scholar]
- Shishkoff N, McGrath MT (2002) AQ10 biofungicide combined with chemical fungicides or AddQ spray adjuvant for control of cucurbit powdery mildew in detached leaf culture. Plant Disease, 86, 915–918. [DOI] [PubMed] [Google Scholar]
- Siozios S, Tosi L, Ferrarini A et al (2015) Transcriptional reprogramming of the mycoparasitic fungus Ampelomyces quisqualis during the powdery mildew host‐induced germination. Phytopathology, 105, 199–209. [DOI] [PubMed] [Google Scholar]
- Slatkin M (1985) Gene flow in natural‐populations. Annual Review of Ecology and Systematics, 16, 393–430. [Google Scholar]
- Springer YP (2007) Clinal resistance structure and pathogen local adaptation in a serpentine flax‐flax rust interaction. Evolution; International Journal of Organic Evolution, 61, 1812–1822. [DOI] [PubMed] [Google Scholar]
- Springer JC, Baines ALD, Fulbright DW, Chansler MT, Jarosz AM (2013) Hyperparasites influence population structure of the chestnut blight pathogen, Cryphonectria parasitica . Phytopathology, 103, 1280–1286. [DOI] [PubMed] [Google Scholar]
- Storfer A (1999) Gene flow and local adaptation in a sunfish‐salamander system. Behavioral Ecology and Sociobiology, 46, 273–279. [Google Scholar]
- Sullivan RF, White JF (2000) Phoma glomerata as a mycoparasite of powdery mildew. Applied and Environmental Microbiology, 66, 425–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susi H, Laine A‐L (2013) Pathogen life‐history trade‐offs revealed in allopatry. Evolution, 67, 3362–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinton J, Gilligan CA (1999) Selecting hyperparasites for biocontrol of dutch elm disease. Proceedings: Biological Sciences, 266, 437–445. [Google Scholar]
- Szentivanyi O, Kiss L, Russell JC et al (2005) Ampelomyces mycoparasites from apple powdery mildew identified as a distinct group based on single‐stranded conformation polymorphism analysis of the rDNA ITS region. Mycological Research, 109, 429–438. [DOI] [PubMed] [Google Scholar]
- Tack AJM, Thrall PH, Barrett LG, Burdon JJ, Laine A‐L (2012) Variation in infectivity and aggressiveness in space and time in wild host‐pathogen systems: causes and consequences. Journal of Evolutionary Biology, 25, 1918–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack AJM, Hakala J, Petaejae T, Kulmala M, Laine A‐L (2014a) Genotype and spatial structure shape pathogen dispersal and disease dynamics at small spatial scales. Ecology, 95, 703–714. [DOI] [PubMed] [Google Scholar]
- Tack AJM, Horns F, Laine A‐L (2014b) The impact of spatial scale and habitat configuration on patterns of trait variation and local adaptation in a wild plant parasite. Evolution, 68, 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack AJM, Laine A‐L, Burdon JJ, Bissett A, Thrall PH (2015) Below‐ground abiotic and biotic heterogeneity shapes above‐ground infection outcomes and spatial divergence in a host‐parasite interaction. New Phytologist, 207, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Jarosz AM, Fulbright DW, Lenski RE (1998) The acquisition of hypovirulence in host‐pathogen systems with three trophic levels. The American Naturalist, 151, 343–355. [DOI] [PubMed] [Google Scholar]
- Thompson JN (2005) The Geographic Mosaic of Coevolution. University of Chicago Press, Chicago, IL. [Google Scholar]
- Thrall PH, Burdon JJ, Bever JD (2002) Local adaptation in the Linum marginale‐Melampsora lini host‐pathogen interaction. Evolution; International Journal of Organic Evolution, 56, 1340–1351. [DOI] [PubMed] [Google Scholar]
- Tollenaere C, Laine A‐L (2013) Investigating the production of sexual resting structures in a plant pathogen reveals unexpected self‐fertility and genotype‐by‐environment effects. Journal of Evolutionary Biology, 26, 1716–1726. [DOI] [PubMed] [Google Scholar]
- Tollenaere C, Susi H, Nokso‐Koivisto J et al (2012) SNP design from 454 sequencing of Podosphaera plantaginis transcriptome reveals a genetically diverse pathogen metapopulation with high levels of mixed‐genotype infection. PLoS One, 7, e52492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollenaere C, Pernechele B, Mäkinen HS et al (2014) A hyperparasite affects the population dynamics of a wild plant pathogen. Molecular Ecology, 23, 5877–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaar MA, Hijwegen T, Zadoks JC (1996) Glasshouse experiments on biocontrol of cucumber powdery mildew (Sphaerotheca fuliginea) by the Mycoparasites verticillium lecanii and Sporothrix rugulosa . Biological Control, 6, 353–360. [Google Scholar]
- Wolinska J, King KC (2009) Environment can alter selection in host‐parasite interactions. Trends in Parasitology, 25, 236–244. [DOI] [PubMed] [Google Scholar]
- Zhan J, Thrall PH, Burdon JJ (2014) Achieving sustainable plant disease management through evolutionary principles. Trends in Plant Science, 19, 570–575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Fungal strains used in laboratory experiments.
Table S2. Replication within each treatment of the two inoculation experiments.
Table S3. SNP markers and their typer4 calling parameters used to genotype Podosphaera plantaginis.
Table S4. MLG genotypes of Podosphaera plantaginis strains used in laboratory inoculation experiments.
Data Availability Statement
All data presented in this manuscript are available on the Dryad digital repository: doi:10.5061/dryad.5fh23.