

Abstract
DNA amplification is a powerful mutational mechanism that is a hallmark of cancer and drug resistance. It is therefore important to understand the fundamental pathways that cells employ to avoid over‐replicating sections of their genomes. Recent studies demonstrate that, in the absence of RecG, DNA amplification is observed at sites of DNA double‐strand break repair (DSBR) and of DNA replication arrest that are processed to generate double‐strand ends. RecG also plays a role in stabilising joint molecules formed during DSBR. We propose that RecG prevents a previously unrecognised mechanism of DNA amplification that we call reverse‐restart, which generates DNA double‐strand ends from incorrect loading of the replicative helicase at D‐loops formed by recombination, and at arrested replication forks.
Keywords: DNA amplification, double‐strand break repair, RecG
Abbreviations
cSDR, constitutive stable DNA replication
DSBR, double‐strand break repair
DSBs, DNA double‐strand breaks
iSDR, inducible stable DNA replication
SIOD, Schimke immunoosseous dysplasia
Over the years since its discovery, different hypotheses have been put forward to explain the function of RecG in bacteria. These have ranged from branch migration and resolution of Holliday junctions 1, 2, 3, 4, 5, 6 via the promotion and inhibition of RecA‐mediated strand exchange 7, 8 to replication fork reversal 9, 10, 11, 12, 13, 14, 15. However, evidence has recently emerged that RecG is implicated in stabilising joint molecules 16 and in controlling DNA amplification by a mechanism that involves over‐replication associated with DNA double‐strand break repair (DSBR) 17, 18, 19, 20, 21, 22, 23, 24. These observations place RecG at the interface of DNA replication and DNA repair. But what is the function of RecG? Four hypotheses have been proposed to account for the role of RecG in preventing over‐replication. In two of these, RecG prevents the formation of DNA double‐strand ends that are associated with the generation of new origin‐independent replication forks by two different mechanisms 17, 21. In the third hypothesis, RecG catalyses the formation of double‐strand ends that are associated with the elimination of new origin‐independent replication forks 23. And in the fourth hypothesis, RecG prevents a form of origin‐independent DNA replication known as constitutive stable DNA replication (cSDR), which is initiated at R‐loops 25.
For many years, no eukaryotic homologue or orthologue of the bacterial RecG protein had been identified. However, recently several candidates have been proposed. These include the mitochondrial helicase Irc3 of Saccharomyces cerevisiae 26, the plastid and mitochondrial helicase RECG of Physcomitrella patens 27, the mitochondrial helicase RECG1 of Arabidopsis thaliana 28 and the human nuclear helicase SMARCAL1 29. All of these genes are implicated in the maintenance of DNA stability and all the plastid and mitochondrial genes show partial cross‐complementation with recG. Irc3 and SMARCAL1 catalyse similar reactions to purified RecG on replication fork and Holliday junction substrates in vitro. SMARCAL1 is a particularly attractive orthologue of RecG as it is a nuclear DNA damage response protein that is a substrate for phosphorylation by ATR 30, 31 and travels with the replication fork 32. Cells lacking SMARCAL1 are prone to accumulate DSBs 32 and patients with a biallelic deficiency in SMARCAL1 have the Schimke immunoosseous dysplasia (SIOD) disease that includes cancer predisposition 33, 34. It is interesting to note that SMARCAL1 is required to accurately and effectively replicate telomeric DNA 35, 36, 37. This is the DNA of eukaryotic chromosomes that is predicted to be most sensitive to replication restart because a stalled replication fork at this location cannot be rescued by a convergent fork from another replication origin.
In this review, we firstly discuss the importance of DNA double‐strand breaks (DSBs) in DNA amplification. We then describe the evidence that RecG and RuvABC catalyse alternative steps in DNA repair by homologous recombination. This is followed by an overview of the biochemical activity of RecG and a discussion of whether the replication fork reversal reaction, which has been well documented to be catalysed by RecG in vitro, is implicated in DNA repair in vivo. We then discuss the recent evidence that RecG and RuvABC collaborate to stabilise joint molecules. Finally, we discuss the evidence that RecG prevents DNA amplification at DSBs and arrested DNA replication forks and assess the strengths of the four models that have been proposed to account for the function of RecG. Readers are encouraged to consult two recent reviews that take different perspectives. In the first of these, Piero Bianco concentrates on the biochemical activities of the protein with a particular emphasis on recent single‐molecule approaches to studying replication fork reversal catalysed by RecG 38. In the second, Christian Rudolph and colleagues discuss chromosome replication in the absence of RecG concentrating on the hypothesis that replication fork collisions are responsible for ‘pathological’ patterns of DNA replication and on the role of replication fork traps (where the Tus protein binds ter sites) in this context 39.
In eukaryotic cells, DSBs associated with DNA replication stimulate DNA amplification highlighting the importance of understanding the sources of replication‐dependent DSBs and their association with over‐replication.
DNA amplification, the formation of an abnormally high copy number of one or more genomic regions, is a characteristic of cancer and of the evolution of tumours that resist treatment with anticancer drugs 40, 41, 42, 43. It is also a mechanism that bacteria use to evolve resistance to antibiotics 44. There is evidence that in eukaryotes DNA amplification is stimulated by impaired S‐phase checkpoint activities and by chromosomal sites and treatments that elevate the frequency of DNA double‐strand ends associated with DNA replication 45, 46, 47, 48, 49, 50. These amplification events are frequently associated with altered deoxynucleoside triphosphate pools and DNA replication stress leading to the early stages of cancer development 50, 51, 52, 53, 54. For these reasons, it is critical to understand the pathways by which DNA double‐strand ends are formed as a consequence of DNA replication and how these events may be associated with DNA amplification. Many of these pathways of DNA double‐strand end formation have been initially investigated in prokaryotic systems but are not exclusive to prokaryotes. As depicted in Fig. 1, the pathways of replication‐dependent DSB formation include: (A) replication fork reversal 55, 56, 57, (B) replication fork collapse 58, (C) replication fork rear‐ending 59, (D) secondary structure cleavage 60, 61, (E) replication fork restart at a 3′ flap 21, (F) template‐switching with replication fork reversal 23, and (G) reverse‐restart of an arrested replication fork 17. Depending of the pathway, RecG has been proposed to promote the formation of double‐strand ends (in pathways A and F) or to prevent the formation of double‐strand ends (in pathways E and G). Pathways E and G postulate over‐replication associated with the formation of DSBs invoking a direct link between DSBs and DNA amplification in Escherichia coli. We will evaluate below the arguments for and against the proposed in vivo roles of RecG.
Figure 1.
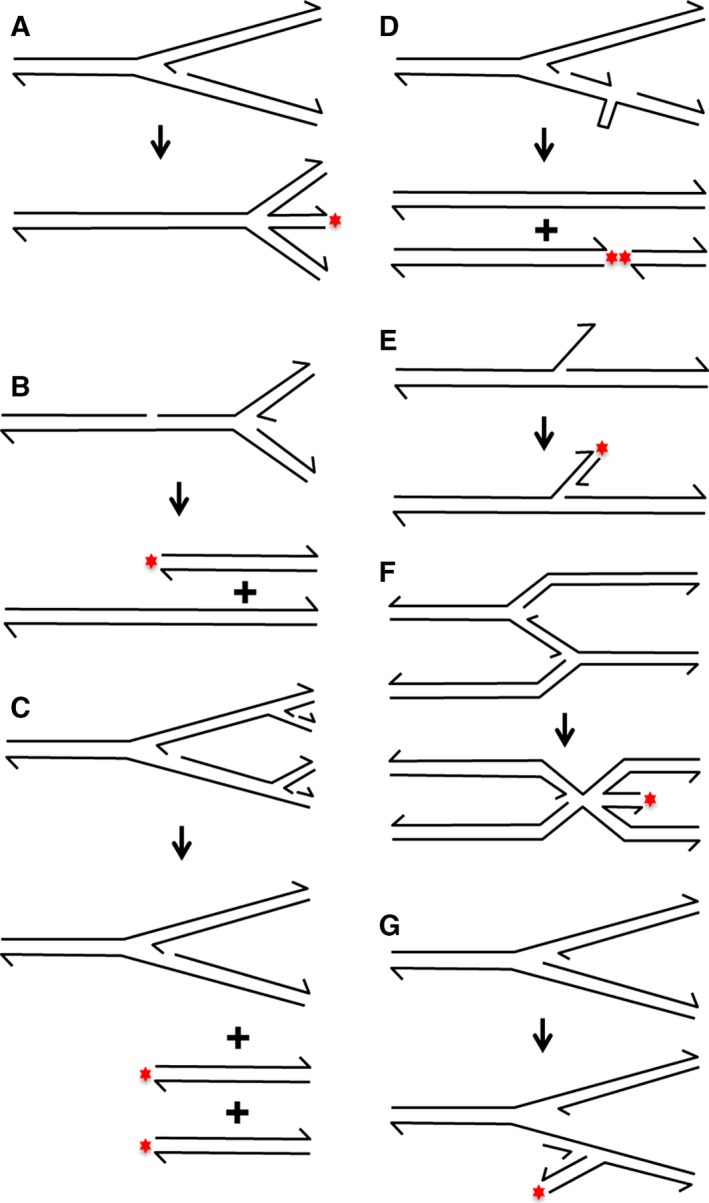
Sources of DNA double‐strand breaks formed during DNA replication. Red stars indicate the positions of DNA double‐strand ends. (A) Replication fork reversal. A four‐way ‘chicken‐foot’ structure can be generated when parental DNA strands re‐pair and newly replicated strands anneal. This forms a DNA double‐strand end and a Holliday junction, which may be cleaved to generate a broken chromosome 55, 56, 57. (B) Replication fork collapse. A one‐ended DSB can be generated when a DNA replication fork encounters a nick on one of the template strands 58. (C) Replication fork rear‐ending. Two one‐ended DSBs can be formed when a DNA replication fork is arrested and the subsequent DNA replication forks replicate this arrested fork 59. (D) Secondary structure cleavage. A DNA secondary structure, such as a hairpin, may form during DNA replication. A two‐ended DSB can be generated when a structure‐specific nuclease, such as SbcCD (Rad50/Mre11), cleaves this sequence 60. (E) Replication fork restart at a 3′ flap. A one‐ended DSB may be formed if a 3′ flap is generated during the termination of DNA replication and acts as a template for initiation of DNA synthesis and the assembly of a new replication fork 21. (F) Template‐switching with replication fork reversal. Template‐switching may occur when two replication forks collide. The two newly replicated strands would then act as reciprocal templates, which would result in DNA over‐replication. To eliminate this over‐replication, one of the replication forks might reverse, forming a DNA double‐strand end that can be degraded 23. (G) Reverse‐restart of an arrested replication fork. Following replication fork arrest, incorrect loading of the replicative helicase to a newly replicated DNA strand would result in the establishment of a new fork proceeding in the reverse direction. This reaction would generate a DNA double‐strand end 17.
RecG and RuvABC catalyse alternative steps in DNA repair and recombination
The recG gene was first identified by Storm and collaborators as a recombination‐deficient mutant of E. coli K12 62. Cells with the recG162 or recG258 mutation were more sensitive to UV, ionising radiation and mitomycin C, and displayed reduced conjugational and P1 transductional efficiency 1, 62, 63. More recent in vivo studies have confirmed the involvement of RecG in DSBR. Cells lacking RecG are sensitive to breaks induced by the I‐SceI homing endonuclease 5, the EcoKI endonuclease 6 and cleavage of a 246 bp palindrome by the SbcCD DNA hairpin endonuclease 60. The observation that (like RecA) RecG plays a role in several different homologous recombination pathways in E. coli suggests that it plays a fundamental role in DNA repair 63. But, what does RecG do? Further understanding of the role of RecG came from genetic studies combining the recG mutation with other mutations in genes encoding proteins involved in DNA repair and recombination 1, 63. recG mutants showed a modest additional sensitivity to UV when combined with either the recB (RecB subunit of the RecBCD enzyme, exonuclease IV, implicated in DNA double‐strand end unwinding, resection and RecA loading during DSBR) or recJ (RecJ 5′–3′ exonuclease, implicated in gap extension during single‐strand gap repair) but not the recF mutation (RecF component of RecFOR, implicated in RecA loading during single‐strand gap repair). However, more striking observations were obtained when recG was combined with ruv mutations (RuvABC implicated in the branch migration and cleavage of Holliday junctions). Double ruvA recG, ruvB recG and ruvC recG mutants exhibited a more dramatic increase in sensitivity to UV and ionising radiation, and a greater defect in recombination after conjugation or transduction when compared to either of the single mutants. These results suggest that RecG and RuvABC catalyse two alternative steps in the repair of DSBs by homologous recombination, potentially during the resolution of Holliday junctions 1. This idea was supported by the study of rusA mutants that suppress the recombination deficiency phenotype of ruvA mutants. These suppressor strains have activated the expression of a Holliday junction resolvase gene encoded within a cryptic prophage 4. The suppression observed in these ruvA rusA double mutants requires the presence of RecG, further suggesting that the alternative pathways catalysed by RuvABC or RecG might be for the resolution of Holliday junctions 4. However, we describe below an alternative hypothesis to explain the redundancy of RecG and RuvABC.
RecG protein unwinds and remodels branched DNA molecules in vitro
Purified RecG protein has 3′–5′ helicase and nucleic acid translocase activities. In vitro, it can bind and unwind synthetic model Holliday junctions and various other types of branched DNA substrates including replication forks, D‐loops and R‐loops 2, 3, 8, 9, 10, 64, 65, 66, 67, 68. Unlike most other helicases, this enzyme unwinds DNA by translocating on dsDNA rather than on ssDNA. In vitro, RecG works as a monomer 69, 70 and efficiently catalyses the re‐pairing of template strands in substrates mimicking replication forks. Interestingly, RecG promoted unwinding reactions occur preferentially on substrates mimicking replication forks with a nascent strand annealed to the lagging‐strand template 9, 15.
RecG catalyses replication fork reversal (also known as replication fork regression) in vitro on a substrate containing both nascent strands (Fig. 1A) 9, 10, 11, 12, 13, 14. This RecG‐catalysed replication fork reversal reaction has been observed using an oligonucleotide substrate with nascent strands annealed to both the leading‐ and lagging‐strand templates 14, a replication fork in supercoiled plasmid DNA 71 and a replication fork blocked at a DNA lesion in an in vitro replication system where the DNA polymerase and the replicative helicase remain associated with the DNA 11. These studies have led to the opinion that replication fork reversal is an important biochemical activity of RecG 9, 11, 12, 14, 15, 38, 66, 67, 70, 71, 72, 73, 74, 75, 76, 77. RecG can catalyse this reaction thanks to its unusual structure 70. This 76‐kDa enzyme possesses a unique translocation by RecG motif, which is located between the wedge and the helicase domains of the protein and contributes to the unwinding of branched molecules by forming a helical hairpin motif 78. For a more detailed discussion of the structure of the RecG protein, readers are referred to the recent review 38.
RecG does not catalyse replication fork reversal in vivo
In 1976, two papers proposed a mechanism for nonmutagenic replication bypass of a DNA lesion that involved reannealing of replicated template DNA strands and extrusion and pairing of newly synthesised DNA strands 55, 57. Over two decades later, a study of E. coli rep mutants provided evidence for the occurrence of this replication fork reversal reaction in cells with undamaged DNA but with compromised DNA replication 56. It was proposed that the RecG‐catalysed replication fork reversal reaction observed in vitro might also happen following UV irradiation in vivo 14. The absence of this pathway in recG mutants would permit re‐pairing of template strands to help repair DNA lesions 14. However, none of the studies of replication fork reversal to date, using different ways of compromising DNA replication, has revealed any situation where RecG is required for the reaction in vivo 56, 79, 80, 81, 82, 83, 84. Furthermore, a subsequent investigation showed little evidence that RecG promotes replication fork reversal following UV irradiation 85. This generated a conundrum. Why would RecG be so good at catalysing replication fork reversal in vitro but unable to catalyse the reaction in vivo? A clue to this might be the observation that when PriA is present, RecG initiates the re‐pairing of parental strands but only proceeds as far as bringing the 3′ end of the nascent leading‐strand to the fork junction point, whereupon the DNA is bound by PriA in a fork‐stabilising configuration (Fig. 2) 86. We shall return to this observation later.
Figure 2.
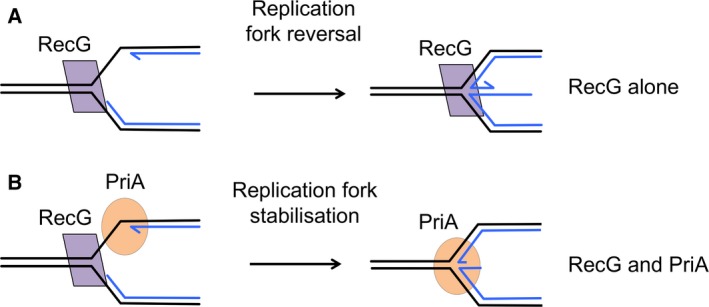
In vitro RecG alone catalyses replication fork reversal but RecG and PriA together stabilise the fork. (A) Replication fork reversal in vitro. RecG has a preference for replication fork substrates with a 5′ nascent strand at the fork. It binds the double‐stranded template strands and unwinds the new strands by moving the fork backwards. As the template strands re‐pair, the new strands anneal and extrude from the fork, forming a DNA double‐strand end in a replication fork reversal reaction 9, 10, 11, 12, 13, 14, 15. (B) Replication fork stabilisation in vitro. When RecG and PriA are both present, RecG begins to re‐pair the template strands while displacing the 5′ ending nascent strand at the fork. PriA is bound to the 3′ ending nascent strand ready to start the reaction to assemble DnaB and initiate DNA replication. The RecG reaction stops when it encounters PriA and the 3′ ending nascent strand. 86.
RecG and RuvABC collaborate to stabilise joint molecules during DSBR
As described above, there is good evidence that RecG and RuvABC catalyse alternative steps in the pathway of recombination, which would explain the high DNA damage sensitivity and recombination deficiency of a ruv recG double mutant. Since RuvABC is known to act as a branch migration and Holliday junction resolution complex 87, it was attractive to hypothesise that this redundancy arose from two alternative pathways of resolution of Holliday junctions. One possibility was that RecG with the help of a topoisomerase might catalyse the dissolution of structures containing two Holliday junctions as had originally been proposed for bacteriophage lambda recombination 88 and has been shown in eukaryotic chromosomes by a combination of BLM, TopoIIIα and Rmi1 (see 89). However, a substantial proportion of chromosome dimers is generated among recombinants formed in the absence of RuvABC, indicating that crossing over has taken place in conditions where the hypothetical RecG‐mediated resolution pathway would be operating 5, 6. This observation is not compatible with a dissolution pathway catalysed by RecG as topoisomerases do not catalyse crossing over and has prompted two alternative hypotheses. First, an unknown nuclease could participate in the RecG pathway of resolution 5 and second, resolution could be mediated by the next round of chromosomal DNA replication passing through the Holliday junction 6.
On the assumption that RuvABC and RecG catalyse alternative pathways of Holliday junction resolution, it was logical to look for evidence of accumulation of Holliday junction intermediates in a ruvAB recG double mutant. However, very surprisingly this double mutant failed to accumulate Holliday junction intermediates while a ruvAB mutant readily did (Fig. 3A) 16. This result clearly showed that RuvABC is responsible for the resolution of Holliday junctions in cells containing RecG. However, few joint molecules of any kind were detected in a strain lacking both RuvAB and RecG. Clearly, the presence of either RuvAB or RecG is required to generate stable joint molecules (including molecules with Holliday junctions) in the first place 16. This led Mawer and Leach to suggest that the branch migration activities of RuvAB and/or RecG might provide alternative ways of stabilising an initially formed and otherwise unstable form of joint molecule, thus explaining the genetic redundancy observed previously. Since joint molecules could not be stably recovered in the absence of RuvAB and RecG, it was hypothesised that initially formed intermediates generated in the absence of these proteins might consist of D‐loops that could be destabilised by a helicase. Further work revealed that this helicase is PriA 17.
Figure 3.
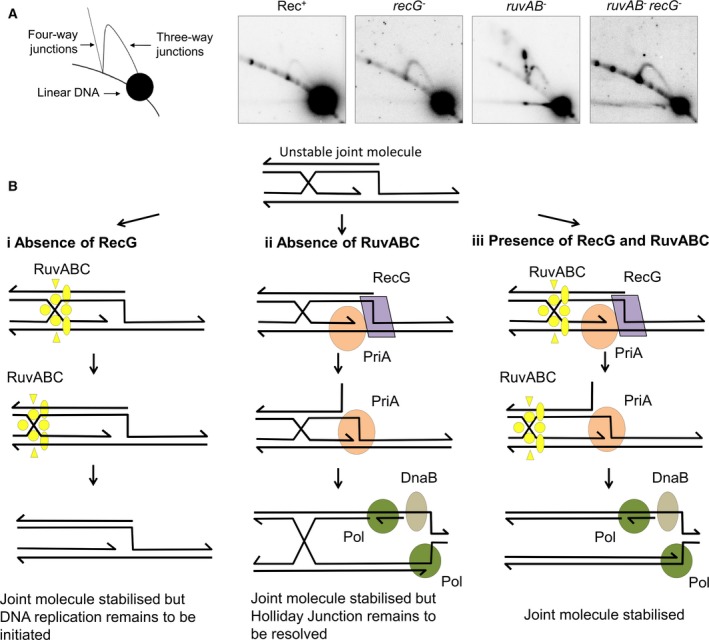
Stabilisation of joint molecules by RuvABC and RecG. (A) DSBR intermediates visualised by 2D gel electrophoresis. RuvAB and RecG do not simply provide alternative pathways for the resolution of Holliday junctions, as previously suggested. Four‐way Holliday junction intermediates accumulate in the absence of RuvAB but not in the absence of RecG. The accumulation of Holliday junctions in the absence of RuvAB requires the presence of RecG 16. Data reproduced with permission from PLoS Genetics. (B) Role of RuvABC and RecG in the stability of joint molecules (i) Joint molecule stabilisation by RuvABC. In the absence of RecG, RuvAB migrates the Holliday junction away from the site of initiation of DSBR and leads to its cleavage by RuvC. Both branch migration and cleavage stabilise the joint molecule. (ii) Joint molecule stabilisation by RecG. In the absence of RuvABC, RecG manipulates the replication fork end of the D‐loop to allow PriA to bind in its 3′ end‐binding fork‐stabilising mode. This allows the initiation of DNA replication that stabilises the joint molecule. (iii) Joint molecule stabilisation by RuvABC and RecG. In the presence of both RuvABC and RecG, both the Holliday junction and replication fork ends of the D‐loop are stabilised.
The stabilisation of initially formed joint molecules, consisting of D‐loops generated by the RecA protein, through the branch migration activities of RuvAB and RecG is readily understandable. Given that RuvAB branch migrates Holliday junctions prior to their resolution by RuvC, it is highly probable that the stabilising activity of RuvABC operates at the Holliday junction end of a D‐loop by extending the region of base pairing between the recombining duplexes, leading to their covalent exchange following cleavage and ligation (Fig. 3Bi). However, the site of action of RecG is less clearly defined by the biochemistry of the enzyme, since this protein can catalyse both the migration of Holliday junctions and the remodelling of replication forks. During DSBR both of these structures are present, one at each end of a D‐loop. A clue as to the nature of the RecG substrate in vivo comes from the observation that a class of suppressors of the recG recombination‐deficient phenotype carries mutations in PriA, either reducing or eliminating the helicase activity of the protein 90. PriA plays a critical role in the reloading of DnaB, the replicative helicase, onto various DNA structures 91, 92, 93, 94. It does so by binding to a replication fork substrate with a 3′ end at the fork junction in a configuration whereupon the fork is stabilised and the helicase activity of PriA is switched off 95. The helicase‐defective mutants of priA that suppress the recombination‐deficient phenotype of recG mutants are indeed competent for catalysing replication restart 96. This suppression, coupled with the observation that RecG delivers PriA to a replication fork substrate in its 3′ end‐binding mode 86, argue strongly for a joint molecule stabilising role of RecG associated with the replication fork end of a D‐loop (Fig. 3Bii). Accordingly, we propose that D‐loops are stabilised in the presence of RuvABC and RecG by activities at both DNA junctions (Fig. 3Biii). Furthermore, we conclude that this overlap in function could be responsible for the genetic redundancy of recG and ruvABC mutants.
RecG controls DNA amplification during DSBR and at arrested replication forks
It has long been known that there is a link between RecG and DNA replication. cSDR is induced in the absence of RecG 97. cSDR is a form of DNA synthesis 98, 99 that requires RecA 100, 101, transcription 102, 103 and is stimulated in rnhA mutants 102. It is therefore proposed to originate from persistent R‐loops that may be generated through the action of RecA. recG rnhA double mutants are not viable and it has been proposed that RecG either unwinds persistent R‐loops or prevents their formation through opposing the action of RecA 97. Inducible stable DNA replication (iSDR) is also elevated in the absence of RecG 104, 105. iSDR requires the induction of the SOS response 106, the action of RecBCD 105, 107 and is insensitive to inhibition of transcription 108, consistent with resulting from DSBs. The reader is directed to the review 108 for a more detailed description of cSDR and iSDR.
During DSBR in E. coli, the RecBCD enzyme resects broken ends for distances of up to several kilobases 109. It is therefore essential that the degraded DNA is restored. This is normally carried out by establishing DNA replication initiated through the action of PriA 60, arguing for the loading of the replicative helicase DnaB and the replicative DNA polymerase PolIII. However, in the absence of RecG, DNA over‐replication is observed following DNA damage 19, 20, 21. At a site‐specific DNA break, this over‐replication flanks the site of DSBR 17. Furthermore, even in the absence of DNA damage, recG mutants over‐replicate the terminus region of their chromosome between termination sites terA and terB 17, 18, 20, 22, 23. This over‐replication is mediated by PriA and PriB and is suppressed by combining the recG mutation with PriA‐helicase mutations 22. These results suggest that the replicative helicase DnaB loads onto DNA substrates generated in this region.
Four alternative hypotheses have been proposed, none of which is free from limitations, to explain the observation that DNA amplification is prevented by RecG.
First (Fig. 4A), DNA amplification is a consequence of DNA flaps that are hypothesised to arise when replication forks collide 18, 19, 21, 22. It is hypothesised that replication fork collisions frequently give rise to 3′ flaps that can be converted into 5′ flaps by RecG, and then these 5′ flaps are degraded by 5′–3′ exonucleases 18, 21. In the absence of RecG, the 3′ flaps persist and are converted into new replication forks through the action of PriA 18, 21. The existence of 3′ flaps is supported by the preference of RecG for processing 3′ flaps over 5′ flaps 15, 86 and the observation of DNA over‐replication in the terminus region of the chromosome of a triple 3′–5′ exonuclease mutant, xseA xonA sbcDC 22. The products of the xseA, xonA and sbcDC genes (exonuclease VII, exonuclease I and SbcCD exo/endonuclease respectively) are the major 3′–5′ exonucleases in E. coli. They participate in several DNA repair and genome stability pathways and the reader is directed to review 110 for a more detailed discussion of their functions. The over‐replication in the xseA xonA sbcDC mutant is very interesting and does indeed suggest the existence of a pathway of DNA amplification involving 3′ overhangs. However, contrary to the prediction of the model that RecG can remove 3′ flaps by converting them to 5′ flaps, RecG is unable to prevent this pathway as the amplification observed in the xseA xonA sbcDC mutant occurs in the presence of RecG. Conversely in the absence of RecG, the three 3′–5′ exonucleases are not able to prevent over‐replication. Therefore, either a single 3′ flap processing pathway is delicately balanced between the activities of RecG on one hand and the three 3′–5′ exonucleases on the other, or there are two separate pathways operating on different substrates. The synthetic lethality of a recG xseA xonA sbcDC quadruple mutant provides some indirect evidence for the existence of a single substrate but it is not conclusive since the phenotype of DNA over‐replication in the terminus region is not lethal and the cause of lethality of the quadruple mutant is unknown 18. Furthermore, although a priA300 helicase defective mutation suppresses the DNA damage sensitivity of a recG mutant, it does not suppress the DNA damage sensitivity of an xseA xonA sbcDC mutant 18, presenting a counter‐argument in favour of the existence of two distinct substrates. In this first model, PriA is hypothesised to recruit DnaB without acting in PriA's 3′ end‐binding and fork‐stabilising mode, which does not fit easily with the biochemical observation that RecG remodels a replication fork substrate to favour PriA binding in its 3′ end‐binding mode 86. The DNA ends generated during this process should be at multiple positions where collisions happen between replication forks and should be pointing in both directions but in fact they are primarily generated at ter sites where they are unidirectional 17. Finally, a complete inversion of chromosome replication is observed in a dnaA recG tus rpo* mutant 22 where replication forks cannot form at the origin of DNA replication so there is no prediction of fork collisions in the chromosome terminus region, from where replication is nevertheless observed to originate.
Figure 4.
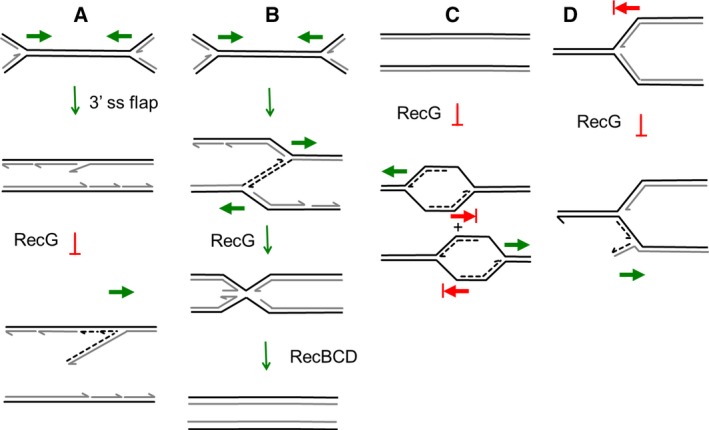
Four different models proposed to explain how RecG controls DNA amplification. (A) Fork collision and restart at a 3′ flap. When two replication forks (moving in the directions of the green arrows) collide, it is hypothesised that in the absence of RecG a 3′ flap is generated that leads to the assembly of a replication fork. In the presence of RecG, the 3′ flap is converted into a 5′ flap that can be degraded by 5′–3′ exonucleases 18, 19, 21, 22. (B) Fork collision and template‐switching followed by replication fork reversal. When two replication forks (moving in the directions of the green arrows) collide, it is hypothesised that template switching occurs leading to over‐replication. This is corrected by RecG‐dependent replication fork reversal and DNA degradation at one (or both) of the replication forks 23. (C) cSDR and termination at Tus/ter blocks. It is proposed that, in the absence of RecG, cSDR initiates at sites of transcription around the genome leading to replication forks that are blocked by Tus/ter. This results principally in over‐replication of the region between termination sites (at the positions of blocked red arrows) as cSDR forks are removed by colliding with origin‐initiated replication forks 25. (D) Reverse‐restart of an arrested replication fork. At an arrested replication fork (at the position of the blocked red arrow) RecG prevents the assembly of the replicative helicase on the newly synthesised lagging‐strand. In the absence of RecG, this loading is permitted and backwards‐directed DNA replication occurs 17.
Second (Fig. 4B), DNA amplification is caused by replication forks sliding past each other in the terminus region of the chromosome 23. This reaction is corrected by RecG that catalyses replication fork reversal on one (or both) of the replication forks, generating one or more DNA double‐strand ends that can be degraded by RecBCD. This hypothesis differs from the first hypothesis in two principal respects. First, RecG is predicted to generate DNA double‐strand ends rather than to remove a precursor of DNA double‐strand ends and second the sliding of replication forks past each other requires a rather complex double DNA template switch. We now know that there is an increase in the frequency of DNA double‐strand ends that bind RecA protein in the terminus region of the chromosome of a recG mutant 17, which is not predicted by this model. As with the first hypothesis, this model does not explain the inversion of chromosome replication observed in a dnaA recG tus rpo* mutant, since this model also predicts that over‐replication of the terminus region requires the meeting of replication forks coming from the origin, which are absent in this mutant 22.
Third (Fig. 4C), DNA amplification in the terminus region is simply a consequence of cSDR that is allowed to occur in a recG mutant and proceeds through the terminus region until it reaches a Tus/ter block 25. cSDR may indeed contribute in some ways to the pattern of DNA replication observed in a recG mutant. However, this hypothesis does not explain the origin of the DNA double‐strand ends that bind RecA at ter sites in a recG mutant 17. Furthermore, the unusual replication observed in a recG mutant is different from that observed in an rnhA mutant as only the former can be suppressed by a priA300 helicase‐defective mutant 111. These observations argue against the involvement of cSDR in the terminus over‐replication formed in the absence of recG. In contrast, the stimulation of iSDR in a recG mutant could be related to the over‐replication observed in the absence of RecG as proposed by the first and fourth hypotheses. iSDR occurs as a consequence of DSBR by homologous recombination and the recombination deficiency of recG mutants is known to be suppressed by priA300 112.
Fourth (Fig. 4D), DNA amplification is caused by the incorrect loading of PriA at a site of replication fork arrest or at a newly formed replication fork 17, leading to the formation of a backwards‐directed replication fork. This reverse‐restart hypothesis is based on two observations. (a) RecG loads PriA onto a model replication fork in the 3′ end‐binding and fork‐stabilising mode 86, predicted to facilitate the loading of DnaB to restart the fork correctly. (b) DNA double‐strand ends bound to RecA protein are detected at the sites of initiation of DNA amplification at an induced DSB and in the terminus region of the chromosome between terA and terB 17. As attractive as this model is, it does not explain all the previous observations either. For example, it does not explain the observation of DNA amplification in the terminus region of a RecG+ cell in the absence of the 3′–5′ exonucleases. It also does not directly explain the inversion of chromosome replication observed in a dnaA recG tus rpo* mutant 22. However, DSBs have been observed surrounding the dif site 17, 113. These breaks could provide the DNA replication initiation sites that would allow this inversion of chromosome replication to occur according to this model.
Only the first and fourth hypotheses propose that over‐replication occurs as a consequence of DNA double‐strand ends that are generated in the absence of RecG. The detection of RecA bound to DNA double‐strand ends in the terminus region, which is specifically enhanced in a recG mutant, provides support for these two models. This stimulation of DSBR is also consistent with iSDR being induced in a recG mutant.
Conclusions and perspectives
It is clear that RecG prevents DNA amplification at a site of induced DSBR in the lacZ gene 17. This is also the case in the terminus region of the E. coli chromosome where DNA amplification in the absence of RecG is similarly associated with DSBR 17. These observations are only in accordance with hypotheses one and four (Fig. 4A,D). We favour the simple explanation, prevention of reverse‐restart, that is described in Fig. 4D. RecG directs the correct loading of PriA, at replication forks that have lost (or not yet acquired) the DNA replication machinery. Appropriate binding of PriA allows DNA replication to proceed correctly via loading of the replicative helicase DnaB. In the presence of RecG, the formation of normal replication forks is predicted to occur at sites of DSBR where they are required to replace the DNA lost during resection. In the absence of RecG, PriA and DnaB can be loaded incorrectly to replications forks that have been created by DSBR or replication forks that have arrested and lost their replisomes. Incorrect loading of DnaB leads to DNA amplification (Fig. 4D) 17.
However, if this explanation is not correct and DNA double‐strand ends arise as a consequence of replication fork collisions in the absence of RecG (Fig. 4A), then these collisions must occur primarily at terA and terB sites in a recG mutant as this is where RecA binding to DNA double‐strand ends is detected by ChIP 17. The ChIP data reveal that RecA binding is at one‐ended DNA breaks all pointing in one of the two possible directions at each of the ter sites 17. This implies that any fork collision occurring at a ter site would have to lead to a specific orientation of break. This may be possible if the direction of replication fork movement upon collision with a ter site can determine the strand on which the hypothetical 3′ single‐strand is generated.
Why a xseA xonA sbcDC triple 3′–5′ exonuclease mutant stimulates DNA amplification in the terminus region of the chromosome remains to be determined. Does this amplification arise from the same pathway as the over‐replication in a recG mutant, or is it mediated by a separate pathway controlled by 3′ overhangs? How DNA replication is initiated in the terminus region of a dnaA recG tus rpo* mutant also remains to be determined. Is this replication initiated by the DSBs detected on the two sides of the dif site 17, 113? Further investigations are required to answer these questions.
DNA replication restart is stringently restricted in eukaryotic cells. However, one might predict that such a pathway could exist to ensure completion of replication between the most telomere proximal origin of replication and the end of the chromosome. One might also predict that, even in the absence of a pathway for restart, incorrect loading of a replicative helicase at the site of a stalled replication fork, to allow reverse‐restart, should be prevented to avoid DNA amplification. Perhaps this is where SMARCAL1 plays a role in maintaining genome stability.
Author contributions
Both authors have contributed to writing the manuscript.
Acknowledgements
We thank Elise Darmon and Ielyzaveta Iurchenko for critical reading of the manuscript and the MRC (UK) for funding.
Edited by Wilhelm Just
References
- 1. Lloyd RG (1991) Conjugational recombination in resolvase‐deficient ruvC mutants of Escherichia coli K‐12 depends on recG . J Bacteriol 173, 5414–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lloyd RG and Sharples GJ (1993) Processing of recombination intermediates by the RecG and RuvAB proteins of Escherichia coli . Nucleic Acids Res 21, 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lloyd RG and Sharples GJ (1993) Dissociation of synthetic Holliday junctions by E. coli RecG protein. EMBO J 12, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mandal TN, Mahdi AA, Sharples GJ and Lloyd RG (1993) Resolution of Holliday intermediates in recombination and DNA repair: indirect suppression of ruvA, ruvB, and ruvC mutations. J Bacteriol 175, 4325–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meddows TR, Savory AP and Lloyd RG (2004) RecG helicase promotes DNA double‐strand break repair. Mol Microbiol 52, 119–132. [DOI] [PubMed] [Google Scholar]
- 6. Wardrope L, Okely E and Leach D (2009) Resolution of joint molecules by RuvABC and RecG following cleavage of the Escherichia coli chromosome by EcoKI. PLoS One 4, e6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitby MC and Lloyd RG (1995) Branch migration of three‐strand recombination intermediates by RecG, a possible pathway for securing exchanges initiated by 3′‐tailed duplex DNA. EMBO J 14, 3302–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Whitby MC, Ryder L and Lloyd RG (1993) Reverse branch migration of Holliday junctions by RecG protein: a new mechanism for resolution of intermediates in recombination and DNA repair. Cell 75, 341–350. [DOI] [PubMed] [Google Scholar]
- 9. Abd Wahab S, Choi M and Bianco PR (2013) Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J Biol Chem 288, 26397–26409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buss JA, Kimura Y and Bianco PR (2008) RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res 36, 7029–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gupta S, Yeeles JT and Marians KJ (2014) Regression of replication forks stalled by leading‐strand template damage: I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the holliday junctions formed by RecG preferentially. J Biol Chem 289, 28376–28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manosas M, Perumal SK, Bianco P, Ritort F, Benkovic SJ and Croquette V (2013) RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun 4, 2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGlynn P and Lloyd RG (1999) RecG helicase activity at three‐ and four‐strand DNA structures. Nucleic Acids Res 27, 3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGlynn P and Lloyd RG (2000) Modulation of RNA polymerase by (p)ppGpp reveals a RecG‐dependent mechanism for replication fork progression. Cell 101, 35–45. [DOI] [PubMed] [Google Scholar]
- 15. McGlynn P and Lloyd RG (2001) Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci USA 98, 8227–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mawer JS and Leach DR (2014) Branch migration prevents DNA loss during double‐strand break repair. PLoS Genet 10, e1004485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Azeroglu B, Mawer JS, Cockram CA, White MA, Hasan AM, Filatenkova M and Leach DR (2016) RecG directs DNA Synthesis during double‐strand break repair. PLoS Genet 12, e1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rudolph CJ, Mahdi AA, Upton AL and Lloyd RG (2010) RecG protein and single‐strand DNA exonucleases avoid cell lethality associated with PriA helicase activity in Escherichia coli . Genetics 186, 473–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rudolph CJ, Upton AL, Briggs GS and Lloyd RG (2010) Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 9, 210–223. [DOI] [PubMed] [Google Scholar]
- 20. Rudolph CJ, Upton AL, Harris L and Lloyd RG (2009) Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73, 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rudolph CJ, Upton AL and Lloyd RG (2009) Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol 74, 940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rudolph CJ, Upton AL, Stockum A, Nieduszynski CA and Lloyd RG (2013) Avoiding chromosome pathology when replication forks collide. Nature 500, 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wendel BM, Courcelle CT and Courcelle J (2014) Completion of DNA replication in Escherichia coli . Proc Natl Acad Sci USA 111, 16454–16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lloyd RG and Rudolph CJ (2016) 25 years on and no end in sight: a perspective on the role of RecG protein. Curr Genet 62, 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gowrishankar J (2015) End of the beginning: elongation and termination features of alternative modes of chromosomal replication initiation in bacteria. PLoS Genet 11, e1004909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaidutsik I, Sedman T, Sillamaa S and Sedman J (2016) Irc3 is a mitochondrial DNA branch migration enzyme. Sci Rep 6, 26414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Odahara M, Masuda Y, Sato M, Wakazaki M, Harada C, Toyooka K and Sekine Y (2015) RECG maintains plastid and mitochondrial genome stability by suppressing extensive recombination between short dispersed repeats. PLoS Genet 11, e1005080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wallet C, Le Ret M, Bergdoll M, Bichara M, Dietrich A and Gualberto JM (2015) The RECG1 DNA translocase is a key factor in recombination surveillance, repair, and segregation of the mitochondrial DNA in Arabidopsis. Plant Cell 27, 2907–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Betous R, Couch FB, Mason AC, Eichman BF, Manosas M and Cortez D (2013) Substrate‐selective repair and restart of replication forks by DNA translocases. Cell Rep 3, 1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bansbach CE, Betous R, Lovejoy CA, Glick GG and Cortez D (2009) The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23, 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Postow L, Woo EM, Chait BT and Funabiki H (2009) Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem 284, 35951–35961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Betous R, Mason AC, Rambo RP, Bansbach CE, Badu‐Nkansah A, Sirbu BM, Eichman BF and Cortez D (2012) SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baradaran‐Heravi A, Raams A, Lubieniecka J, Cho KS, DeHaai KA, Basiratnia M, Mari PO, Xue Y, Rauth M, Olney AH et al (2012) SMARCAL1 deficiency predisposes to non‐Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo . Am J Med Genet A 158A, 2204–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carroll C, Badu‐Nkansah A, Hunley T, Baradaran‐Heravi A, Cortez D and Frangoul H (2013) Schimke immunoosseous dysplasia associated with undifferentiated carcinoma and a novel SMARCAL1 mutation in a child. Pediatr Blood Cancer 60, E88–E90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox KE, Marechal A and Flynn RL (2016) SMARCAL1 resolves replication stress at ALT telomeres. Cell Rep 14, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Poole LA and Cortez D (2016) SMARCAL1 and telomeres: replicating the troublesome ends. Nucleus 7, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Poole LA, Zhao R, Glick GG, Lovejoy CA, Eischen CM and Cortez D (2015) SMARCAL1 maintains telomere integrity during DNA replication. Proc Natl Acad Sci USA 112, 14864–14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bianco PR (2015) I came to a fork in the DNA and there was RecG. Prog Biophys Mol Biol 117, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dimude JU, Midgley‐Smith SL, Stein M and Rudolph CJ (2016) Replication termination: containing fork fusion‐mediated pathologies in Escherichia coli . Genes (Basel) 7, 7080040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- 41. Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M and Trent J (1983) Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 305, 245–248. [DOI] [PubMed] [Google Scholar]
- 42. Sequist LV, Waltman BA, Dias‐Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3, 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A and McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- 44. Sandegren L and Andersson DI (2009) Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat Rev Microbiol 7, 578–588. [DOI] [PubMed] [Google Scholar]
- 45. Brown PC, Tlsty TD and Schimke RT (1983) Enhancement of methotrexate resistance and dihydrofolate reductase gene amplification by treatment of mouse 3T6 cells with hydroxyurea. Mol Cell Biol 3, 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coquelle A, Pipiras E, Toledo F, Buttin G and Debatisse M (1997) Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell 89, 215–225. [DOI] [PubMed] [Google Scholar]
- 47. Tanaka H, Cao Y, Bergstrom DA, Kooperberg C, Tapscott SJ and Yao MC (2007) Intrastrand annealing leads to the formation of a large DNA palindrome and determines the boundaries of genomic amplification in human cancer. Mol Cell Biol 27, 1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanaka H, Tapscott SJ, Trask BJ and Yao MC (2002) Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc Natl Acad Sci USA 99, 8772–8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tlsty TD, Brown PC and Schimke RT (1984) UV radiation facilitates methotrexate resistance and amplification of the dihydrofolate reductase gene in cultured 3T6 mouse cells. Mol Cell Biol 4, 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kondratova A, Watanabe T, Marotta M, Cannon M, Segall AM, Serre D and Tanaka H (2015) Replication fork integrity and intra‐S phase checkpoint suppress gene amplification. Nucleic Acids Res 43, 2678–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS and Kerem B (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Halazonetis TD, Gorgoulis VG and Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- 53. Poli J, Tsaponina O, Crabbe L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A and Pasero P (2012) dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zeman MK and Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Higgins NP, Kato K and Strauss B (1976) A model for replication repair in mammalian cells. J Mol Biol 101, 417–425. [DOI] [PubMed] [Google Scholar]
- 56. Seigneur M, Bidnenko V, Ehrlich SD and Michel B (1998) RuvAB acts at arrested replication forks. Cell 95, 419–430. [DOI] [PubMed] [Google Scholar]
- 57. Fujiwara Y and Tatsumi M (1976) Replicative bypass repair of ultraviolet damage to DNA of mammalian cells: caffeine sensitive and caffeine resistant mechanisms. Mutat Res 37, 91–110. [DOI] [PubMed] [Google Scholar]
- 58. Kuzminov A (2001) Single‐strand interruptions in replicating chromosomes cause double‐strand breaks. Proc Natl Acad Sci USA 98, 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bidnenko V, Ehrlich SD and Michel B (2002) Replication fork collapse at replication terminator sequences. EMBO J 21, 3898–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eykelenboom JK, Blackwood JK, Okely E and Leach DR (2008) SbcCD causes a double‐strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell 29, 644–651. [DOI] [PubMed] [Google Scholar]
- 61. Leach DR (1994) Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. BioEssays 16, 893–900. [DOI] [PubMed] [Google Scholar]
- 62. Storm PK, Hoekstra WP, de Haan PG and Verhoef C (1971) Genetic recombination in Escherichia coli. IV. Isolation and characterization of recombination‐deficiency mutants of Escherichia coli K12. Mutat Res 13, 9–17. [DOI] [PubMed] [Google Scholar]
- 63. Lloyd RG and Buckman C (1991) Genetic analysis of the recG locus of Escherichia coli K‐12 and of its role in recombination and DNA repair. J Bacteriol 173, 1004–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fukuoh A, Iwasaki H, Ishioka K and Shinagawa H (1997) ATP‐dependent resolution of R‐loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction‐specific helicase. EMBO J 16, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zegeye ED, Balasingham SV, Laerdahl JK, Homberset H and Tonjum T (2012) Mycobacterium tuberculosis RecG binds and unwinds model DNA substrates with a preference for Holliday junctions. Microbiology 158, 1982–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thakur RS, Basavaraju S, Khanduja JS, Muniyappa K and Nagaraju G (2015) Mycobacterium tuberculosis RecG protein but not RuvAB or RecA protein is efficient at remodeling the stalled replication forks: implications for multiple mechanisms of replication restart in mycobacteria . J Biol Chem 290, 24119–24139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thakur RS, Basavaraju S, Somyajit K, Jain A, Subramanya S, Muniyappa K and Nagaraju G (2013) Evidence for the role of Mycobacterium tuberculosis RecG helicase in DNA repair and recombination. FEBS J 280, 1841–1860. [DOI] [PubMed] [Google Scholar]
- 68. Beyene GT, Balasingham SV, Frye SA, Namouchi A, Homberset H, Kalayou S, Riaz T and Tonjum T (2016) Characterization of the Neisseria meningitidis Helicase RecG. PLoS One 11, e0164588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McGlynn P, Mahdi AA and Lloyd RG (2000) Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res 28, 2324–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Singleton MR, Scaife S and Wigley DB (2001) Structural analysis of DNA replication fork reversal by RecG. Cell 107, 79–89. [DOI] [PubMed] [Google Scholar]
- 71. McGlynn P, Lloyd RG and Marians KJ (2001) Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc Natl Acad Sci USA 98, 8235–8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bolt EL and Lloyd RG (2002) Substrate specificity of RusA resolvase reveals the DNA structures targeted by RuvAB and RecG in vivo . Mol Cell 10, 187–198. [DOI] [PubMed] [Google Scholar]
- 73. McGlynn P and Lloyd RG (2002) Genome stability and the processing of damaged replication forks by RecG. Trends Genet 18, 413–419. [DOI] [PubMed] [Google Scholar]
- 74. Gregg AV, McGlynn P, Jaktaji RP and Lloyd RG (2002) Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol Cell 9, 241–251. [DOI] [PubMed] [Google Scholar]
- 75. Robu ME, Inman RB and Cox MM (2004) Situational repair of replication forks: roles of RecG and RecA proteins. J Biol Chem 279, 10973–10981. [DOI] [PubMed] [Google Scholar]
- 76. Briggs GS, Mahdi AA, Weller GR, Wen Q and Lloyd RG (2004) Interplay between DNA replication, recombination and repair based on the structure of RecG helicase. Philos Trans R Soc Lond B Biol Sci 359, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bhattacharjee SM (2010) Interfacial instability and DNA fork reversal by repair proteins. J Phys Condens Matter 22, 155102. [DOI] [PubMed] [Google Scholar]
- 78. Mahdi AA, Briggs GS, Sharples GJ, Wen Q and Lloyd RG (2003) A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J 22, 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Baharoglu Z, Petranovic M, Flores MJ and Michel B (2006) RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J 25, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. De Septenville AL, Duigou S, Boubakri H and Michel B (2012) Replication fork reversal after replication‐transcription collision. PLoS Genet 8, e1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Flores MJ, Bierne H, Ehrlich SD and Michel B (2001) Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. EMBO J 20, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grompone G, Ehrlich D and Michel B (2004) Cells defective for replication restart undergo replication fork reversal. EMBO Rep 5, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Grompone G, Seigneur M, Ehrlich SD and Michel B (2002) Replication fork reversal in DNA polymerase III mutants of Escherichia coli: a role for the beta clamp. Mol Microbiol 44, 1331–1339. [DOI] [PubMed] [Google Scholar]
- 84. Michel B, Boubakri H, Baharoglu Z, LeMasson M and Lestini R (2007) Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 6, 967–980. [DOI] [PubMed] [Google Scholar]
- 85. Khan SR and Kuzminov A (2012) Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein‐catalyzed disintegration in Escherichia coli . J Biol Chem 287, 6250–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tanaka T and Masai H (2006) Stabilization of a stalled replication fork by concerted actions of two helicases. J Biol Chem 281, 3484–3493. [DOI] [PubMed] [Google Scholar]
- 87. West SC (1997) Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet 31, 213–244. [DOI] [PubMed] [Google Scholar]
- 88. Thaler DS, Stahl MM and Stahl FW (1987) Tests of the double‐strand‐break repair model for red‐mediated recombination of phage lambda and plasmid lambda dv. Genetics 116, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bizard AH and Hickson ID (2014) The dissolution of double Holliday junctions. Cold Spring Harb Perspect Biol 6, a016477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Al‐Deib AA, Mahdi AA and Lloyd RG (1996) Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K‐12. J Bacteriol 178, 6782–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Masai H, Tanaka T and Kohda D (2010) Stalled replication forks: making ends meet for recognition and stabilization. BioEssays 32, 687–697. [DOI] [PubMed] [Google Scholar]
- 92. Liu J and Marians KJ (1999) PriA‐directed assembly of a primosome on D loop DNA. J Biol Chem 274, 25033–25041. [DOI] [PubMed] [Google Scholar]
- 93. Marians KJ (1999) PriA: at the crossroads of DNA replication and recombination. Prog Nucleic Acid Res Mol Biol 63, 39–67. [DOI] [PubMed] [Google Scholar]
- 94. Huang YH and Huang CY (2014) Structural insight into the DNA‐binding mode of the primosomal proteins PriA, PriB, and DnaT. Biomed Res Int 2014, 195162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tanaka T, Mizukoshi T, Sasaki K, Kohda D and Masai H (2007) Escherichia coli PriA protein, two modes of DNA binding and activation of ATP hydrolysis. J Biol Chem 282, 19917–19927. [DOI] [PubMed] [Google Scholar]
- 96. Sandler SJ, Samra HS and Clark AJ (1996) Differential suppression of priA2:kan phenotypes in Escherichia coli K‐12 by mutations in priA, lexA, and dnaC. Genetics 143, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hong X, Cadwell GW and Kogoma T (1995) Escherichia coli RecG and RecA proteins in R‐loop formation. EMBO J 14, 2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kogoma T and Lark KG (1970) DNA replication in Escherichia coli: replication in absence of protein synthesis after replication inhibition. J Mol Biol 52, 143–164. [DOI] [PubMed] [Google Scholar]
- 99. Kogoma T and Lark KG (1975) Characterization of the replication of Escherichia coli DNA in the absence of protein synthesis: stable DNA replication. J Mol Biol 94, 243–256. [DOI] [PubMed] [Google Scholar]
- 100. Kogoma T, Skarstad K, Boye E, von Meyenburg K and Steen HB (1985) RecA protein acts at the initiation of stable DNA replication in rnh mutants of Escherichia coli K‐12. J Bacteriol 163, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Torrey TA and Kogoma T (1982) Suppressor mutations (rin) that specifically suppress the recA+ dependence of stable DNA replication in Escherichia coli K‐12. Mol Gen Genet 187, 225–230. [DOI] [PubMed] [Google Scholar]
- 102. Kogoma T (1978) A novel Escherichia coli mutant capable of DNA replication in the absence of protein synthesis. J Mol Biol 121, 55–69. [DOI] [PubMed] [Google Scholar]
- 103. von Meyenburg K, Boye E, Skarstad K, Koppes L and Kogoma T (1987) Mode of initiation of constitutive stable DNA replication in RNase H‐defective mutants of Escherichia coli K‐12. J Bacteriol 169, 2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Asai T and Kogoma T (1994) Roles of ruvA, ruvC and recG gene functions in normal and DNA damage‐inducible replication of the Escherichia coli chromosome. Genetics 137, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Asai T, Sommer S, Bailone A and Kogoma T (1993) Homologous recombination‐dependent initiation of DNA replication from DNA damage‐inducible origins in Escherichia coli . EMBO J 12, 3287–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kogoma T, Torrey TA and Connaughton MJ (1979) Induction of UV‐resistant DNA replication in Escherichia coli: induced stable DNA replication as an SOS function. Mol Gen Genet 176, 1–9. [DOI] [PubMed] [Google Scholar]
- 107. Magee TR and Kogoma T (1990) Requirement of RecBC enzyme and an elevated level of activated RecA for induced stable DNA replication in Escherichia coli . J Bacteriol 172, 1834–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kogoma T (1997) Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev 61, 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dillingham MS and Kowalczykowski SC (2008) RecBCD enzyme and the repair of double‐stranded DNA breaks. Microbiol Mol Biol Rev 72, 642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lovett ST (2011) The DNA exonucleases of Escherichia coli . EcoSal Plus 4.4.7, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dimude JU, Stockum A, Midgley‐Smith SL, Upton AL, Foster HA, Khan A, Saunders NJ, Retkute R and Rudolph CJ (2015) The consequences of replicating in the wrong orientation: bacterial chromosome duplication without an active replication origin. MBio 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Jaktaji RP and Lloyd RG (2003) PriA supports two distinct pathways for replication restart in UV‐irradiated Escherichia coli cells. Mol Microbiol 47, 1091–1100. [DOI] [PubMed] [Google Scholar]
- 113. Cockram CA, Filatenkova M, Danos V, El Karoui M and Leach DR (2015) Quantitative genomic analysis of RecA protein binding during DNA double‐strand break repair reveals RecBCD action in vivo . Proc Natl Acad Sci USA 112, E4735–E4742. [DOI] [PMC free article] [PubMed] [Google Scholar]