

Abstract
Background
The study hypothesis is that administration of inhaled NO plus oxygen to subjects with submassive pulmonary embolism (PE) will improve RV systolic function; reduce RV strain and necrosis while improving patient dyspnea more than treatment with oxygen alone.
Methods
This paper describes the rationale and protocol for a registered (NCT01939301), a nearly completed Phase II, three-center, randomized, double blind, controlled trial. Eligible patients have pulmonary imaging-proven acute PE. Subjects must be normotensive, have RV dysfunction on echocardiography or elevated troponin or brain natriuretic peptide and no fibrinolytics. Subjects receive NO plus oxygen or placebo for 24 hours (±3 h) with blood sampling before and after treatment, and mandatory echocardiography and high sensitivity troponin post treatment to assess the composite primary endpoint. The sample size of N=78 was predicated on 30% more NO-treated patients having a normal high sensitivity troponin (<14 pg/mL) and a normal RV on echocardiography at 24 hours with α=0.05 and β=0.20. Safety was ensured by continuous spectrophotometric monitoring of %methemoglobinemia, and a predefined protocol to respond to emergent changes in condition. Blinding was ensured by identical tanks, software and physical shielding of the device display and query of the clinical care team to assess blinding efficacy.
Results
We have enrolled 78 patients over a 31 month period. No patient has been withdrawn stopped as a result of a safety concern, and no patient has had a serious adverse event related to NO.
Conclusions
We present methods and a protocol for the first double-blinded, randomized trial of inhaled NO to treat PE.
Keywords: nitric oxide, randomized trial, pulmonary embolism, troponin, echocardiography
Background and Scientific Rationale
Current standard treatment for acute pulmonary embolism (PE) focuses on reducing clot burden with anticoagulant and fibrinolytic agents1. However, experimental models of PE have shown that the majority of the increased pulmonary vascular resistance (PVR) during PE comes not from the obstructing clot, but rather from vasoconstriction2. Acute PE can abruptly increase the systolic pressure in the right ventricle (RV), producing turbulent flow across the tricuspid and pulmonic valves immediately proximal to the pulmonary vasculature, a compartment that depends on steady state production of nitric oxide (NO) to maintain its low resistance (Figure 1). The turbulent flow across the tricuspid and pulmonic valves results in hemolysis with subsequent release of free hemoglobin and heme (both of which scavenge NO), as well as release of arginase-1 (which reduces plasma L-arginine, the substrate of endothelial NO synthase). 3-6 Taken together, these influences may reduce NO availability to pulmonary vascular smooth muscle cells, thus further increasing the PVR and leading to a vicious cycle of increased PVR, increased RV pressures and more hemolysis. In addition to these pulmonary vascular effects, acutely elevated RV pressure causes shear injury to the RV muscle, resulting in severe inflammatory injury within 18 hours.7,8 This cycle can cause RV failure, shock and death, occurring most commonly within 24 hours of acute PE.9 Lastly, even if the acute PE does not lead to mortality, serious long-term complications may arise. In fact, survivors of PE that produces acute RV dysfunction may go on to experience chronic shortness of breath and exercise intolerance.10 These observations provide a rationale for limiting increases in PVR in acute PE. Given the NO deficiency state occurring in acute PE, administration of NO makes biological sense. Indeed, a phase I pilot study of inhaled NO in eight patients with PE found no evidence of clinical worsening with inhaled NO.11 These observations prompted the inhaled NO for PE (iNOPE) trial.
Figure 1.
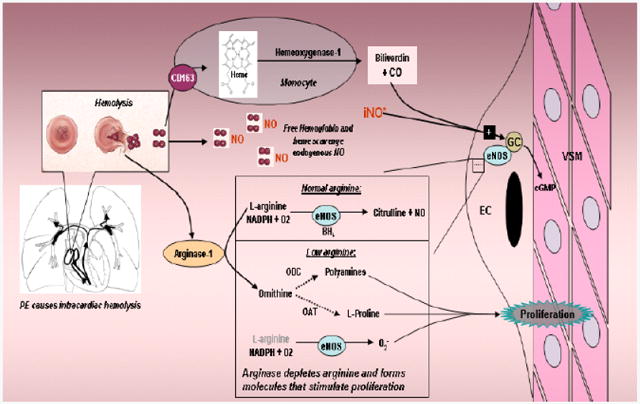
Diagram of study hypothesis and integration of the clinical study and the basic science study. PE causes intracardiac hemolysis which disrupts NO signaling by scavenging NO and releasing arginase which depletes L-arginine, producing a negative feedback cycle of increased pulmonary VSM constriction, increased PVR, higher RV pressures and more hemolysis. Inhaled NO (iNO*) will break this cycle.
Trial conduct and registrations
The study protocol and execution was guided by a multidisciplinary group, representing disciplines of emergency care (JAK, AEJ, MAP), critical care and pulmonology (TL), cardiology (RM) and clinical trials specialist (CLH). The study was overseen in part by representatives from the NIH, and Mallinckrodt Pharmaceuticals (formerly IKARIA, Inc.). The safety and efficacy of the trial was overseen by a four-person independent data safety monitoring board in accordance with an NIH-approved independent charter (see appendix). The data coordinating center was overseen by an independent statistician (Anthony Perkins, MS). The trial was being conducted under an investigational new drug exemption from FDA in accordance with a letter from the Food and Drug Administration (FDA), responding to our January 24, 2013 1571 submission. This trial was registered on Clinicaltrials.gov on August 28, 2013 (NCT01939301). Trial oversight was in accordance with the Code of Federal Regulations (21CFR312), Good Clinical Practice Guidelines and International Conference on Harmonization Guidelines. The iNOPE protocol and informed consent forms were approved by the Institutional Review Boards of Indiana University School of Medicine and the University of Mississippi Medical Center in 2013.
Study design
iNOPE is a multicenter randomized, double-blind, controlled trial of inhaled NO + oxygen versus nitrogen + oxygen in subjects with acute submassive PE and RV dysfunction. Study drug or placebo was administered with a commercially available, FDA-cleared device (INOvent, ® Mallinckrodt Pharmaceuticals, Clinton, NJ). A prior randomized trial of inhaled NO for acute chest syndrome in sickle cell anemia patients had demonstrated the feasibility of blinding.12
Trial funding
The trial was sponsored by the National Institutes of Health (UM1HL113203-01 to JAK) and by an investigator initiated grant from Ikaria Pharmaceuticals, who manufactured the INOvent® device in 2013; Ikaria was acquired by Mallinckrodt Pharmaceuticals in 2015. Additional funding and the COBAS device for high sensitivity troponin T measurements was obtained through an investigator initiated contract with Roche. The sponsors had no role in the protocol design have had and will have no role in data analysis presentation or manuscript writing.
Inclusion and exclusion criteria
Patients were enrolled under the scientific premise that the presence of RV dysfunction worsens outcome from PE.13,14 Given that the hypothesized biological basis of action for NO is pulmonary vasodilation with reduction in RV pressure, the inclusion criteria required evidence of RV dysfunction. RV dysfunction was indicated by either abnormal biomarker values (including an elevated BNP > 90 pg/mL or an FDA cleared, laboratory troponin I concentration elevated above the 99th percentile at a coefficient of variability <10%, generally >0.1 ng/mL), evidence of RV dilation and/or hypokinesis on echocardiography, or an RV:LV ratio >1.0 as observed on CT scan. 13-15 To avoid confounding effects of fibrinolysis, which appears to resolve RV dysfunction more rapidly than anticoagulation alone, we excluded patients who had recently received fibrinolysis by any route. To avoid possible drug synergy that might cause hypotension, we excluded patients taking organic nitrates, phosphodiesterase 5 inhibitors, or soluble guanylate cyclase stimulators. The explicit inclusion and exclusions are in Box 1.
Box 1. Inclusion and exclusion criteria for iNOPE.
Inclusion
Age ≥18
Pulmonary imaging-proven PE, as interpreted by local radiologists.
- At least one predictor of RV dysfunction
- echocardiography with RV dilation or hypokinesis,
- estimated RVSP > 40 mm Hg,
- RV>LV on CTPA,
- elevated troponin I (>0.1 ng/mL) or natriuretic peptide (BNP> 90 pg/mL)
- screening bedside cardiac ultrasound with color flow capability that shows RV dysfunction (must be included with another RV dysfunction predictor),
- RV strain on ECG (Defined by a Daniel score >8)
Plan to admit to a bed with telemetry capability
Exclusion
Vasopressor support at time of enrollment
Pregnancy
Plan by clinical care team to use lytics or surgical embolectomy
Plan by clinical care team to use platelet inhibiting drugs
Contraindication to anticoagulation
Altered mental status such that the patient is unable to provide informed consent
Inability to use a nasal cannula
Comfort care measures instituted
Supplemental oxygen requirements greater than can be administered via nasal cannula in order to maintain SpO2>80%
Pneumothorax with decompression
Recent use of drugs known to increase cGMP
Use of nitroprusside or nitroglycerin within the last 4 hours
Use of any other long-acting nitrates within the past 24 hours
Use of a fibrinolytic medicine within the past 3 days
At the discretion of the Principal Investigator
Patients were enrolled at three hospitals (Methodist Hospital and The Sidney and Louis Eskenazi Hospital in Indianapolis, IN, and the University of Mississippi Medical Center Hospital in Jackson, MS). Daily screening consisted of a research specialist review of the formal dictation report of either contrast enhanced computerized tomographic angiography of the chest or ventilation-perfusion scintillation lung scans, identified by standing query of electronic orders. The research specialists, with input and assistance as needed from a site PI, then further reviewed each apparently positive study for evidence of RV strain as required in the inclusion criteria and for other eligibility criteria, followed by physical approach for possible informed consent. Patients were approached in the emergency department, intensive and intermediate care units, and rarely in ward setting.
Primary efficacy endpoint
As a phase II study, the endpoint was primarily physiological, in the form of RV function. All patients had a formal transthoracic echocardiogram and high sensitivity troponin measurement that was paid for by the study, both performed after 24 hours of treatment with NO or placebo. The study authors reasoned that an effective treatment should be more likely to completely normalize RV function after 24 hours of treatment compared with placebo. All patients were treated with an accepted regimen of systemic anticoagulation such as low molecular weight heparin, unfractionated heparin, or a direct acting anti-Xa antagonist. The formal definition of normal RV function and viability requires normal RV size (<42 mm in diastole) and TAPSE >16 mm and normal right ventricular index of myocardial performance (RIMP) (<0.40 using spectral Doppler or < 0.55 using tissue Doppler) and normal fractional area of contraction (FAC) (>33%) and a serum high sensitivity troponin T hsTnT <14 pg/mL (Roche COBAS, Indianapolis, IN) as a marker of RV myocyte necrosis.16,17 Missing values will be considered normal.
Study Hypothesis
We hypothesize that the administration of inhaled NO + oxygen to subjects with submassive PE will improve RV systolic function, reduce RV strain and necrosis (as assessed by hsTnT), and improve dyspnea more than nitrogen + oxygen treatment. Clinical efficacy will be defined as the percentage of subjects in each group who have a normal RV size, TAPSE and RIMP and FAC on the echocardiogram and a hsTnT concentration <14 pg/ml after 24 hours of treatment with inhaled NO + oxygen (intervention) or nitrogen + oxygen (placebo).
The clinical trial also has three secondary endpoints and secondary clinical hypotheses to be tested:
NO + oxygen will reduce RV strain as assessed by composite of serum BNP<90 pg/mL RV hypokinesis and a Borg score ≤2 (very slight dyspnea) assessed at the end of 24 hours of treatment.
Test if NO + oxygen reduces blood viscosity and attenuates platelet-associated clotting kinetics observed on thromboelastography (TEG).
Test if NO + oxygen improves perceived dyspnea and perception of wellness with a standardized survey at 3 months (SF36 and PEmb QoL).18
The primary safety endpoint was the clinical care team's decision to unblind because of perception of clinical deterioration during treatment. Secondary safety endpoints include need for rescue therapy, including need for mechanical or noninvasive ventilatory support, systemic or catheter based fibrinolysis, or catheter or surgical embolectomy. In-hospital serious adverse events were followed for 5 days after treatment ends. At 28 days, patients were assessed by for potential study drug related adverse events. Stopping criteria were planned according to the method of Lan and DeMets if patients had met an adverse event or clinical deterioration criteria, defined as: respiratory distress prompting the clinical care team to plan for mechanical or noninvasive ventilatory support, hypoxemia (SpO2 < 90% for 15 min), or circulatory shock (SBP< 90 mm Hg for 15 min in consecutive measurements), new neurologic signs or any ventricular arrhythmias or sustained supraventricular arrhythmia with hypotension or refractory to intravenous medication for more than one hour, and methemoglobin (MtHb) levels >10% for more than one hour. Pulse oximetry (SpO2%), transcutaneous mtHb and carboxyhemoglobin were measured with the Masimo Rainbow® Cooximeter device (Masimo Corporation, Irving, CA).
Ancillary basic science study
Under the UM funding mechanism from NHLBI, the study has several preplanned basic science studies being performed on blood samples from patients. These measurements are summarized as follows:
Platelet mitochondrial oxygen consumption (Oxygraph-2k Oroboros Instruments, Innsbruck, Austria) and clotting propensity, assessed with whole blood thromboelastography with ADP and arachidonic acid agonists (Haemoscope 5000, Haemonetics, Braintree, MA) together with measurements of cellular apoptosis. The hypothesis is platelets will be hyperactivated and apoptotic in PE patients compared with healthy controls, and that platelet hyperactivation will be attenuated with NO treatment.
Measurements of hemolysis markers LDH, free hemoglobin, haptoglobin, hemopexin and carboxyhemoglobin. The hypothesis is that there will be a more complete reduction in these biomarkers with NO treatment.
Plasma nitrite, nitrate and asymmetric dimethyl arginine (ADMA, an inhibitor of endothelial nitric oxide synthase, eNOS) concentrations. The hypothesis is PE patients have decreased nitrite+nitrate and elevated ADMA concentrations at baseline compared with healthy controls.5
Sequencing of the eNOS gene to test for common sequence variations affecting treatment responses. Buffy coat will be recovered and stored at -80°C for genotypes that predict response to NO therapy. Two common polymorphisms(- 786T>C and 894G>T) will be assessed with a 5′-allele discrimination PCR assay using primer sequences previously described by our group and others.19,20 Both polymorphisms impair eNOS synthesis of endogenous NO, impair endothelial-induced vascular smooth muscle relaxation, increase red cell fragility and increase the risk of PE in Caucasians and Asians.20-22 The hypothesis is that patients with these variations will have improved response to inhaled NO.
Measurement of cell free circulating plasma nucleic acids (total plasma cell free DNA and RNA content), which we hypothesized to increase with PE, possibly as a result of neutrophil apoptosis and RV necrosis.23 We hypothesize that RV myolysis will also liberate CF DNA/RNA which contributes to thrombin generation and increased blood viscosity in the right heart.24 Of fundamental importance to this hypothesis, the RV venous drainage returns to right atrium via the coronary sinus where it can potentially synergize with platelet-derived thrombin generation to increase blood viscosity and RV workload. In addition to measuring total CF DNA/RNA, with quantitative rtPCR, we will provide evidence of association with RV damage by quantifying three RNAs: 1. Troponin T (gene ID X74819), a gene specifically overexpressed in the heart.25 2. Natriuretic peptide precursor B (gene ID 4879), a gene that we have previously shown by microarray analysis to be specifically upregulated in the RV outflow tract with PE26 and 3. microRNA 134, found to be a good diagnostic biomarker for PE that is specifically increased in the plasma of humans27. The hypothesis is that there will be a more complete and rapid reduction with NO treatment.
Summary of study protocol
The study measurements are shown in Table 1, and the overall flow of the study is diagrammed in Figure 2. At baseline, a blood sample was obtained before initiation of study drug to allow for a high sensitivity troponin I measurement and for ancillary studies. Blood was drawn, stored, and processed according to study standard operating procedures. Because of funding limitations, echocardiography has to be performed as part of usual care and therefore cannot be performed as a study procedure before the study drug wass delivered. Study drug was delivered by a commercial device (INOvent®) via nasal cannula induced at 2 ppm/min, increased to 50 ppm and maintained for 24h (+/- 3 hrs.) with oxygen delivered at 3-5 LPM as the carrying gas for study drug delivery.
Table 1. Study Events Schedule.
| Procedure | Time point | ||||||
|---|---|---|---|---|---|---|---|
| Screening/enrollment | Pre-drug | During 24 Hours of drug | Post Study Drug Completion (±3 h) | Sooner of day 5 or Discharge | 28-Day Phone Follow-up | 3 Months | |
| Informed Consent | X | ||||||
| Inclusion/Exclusion Review | X | ||||||
| Physical Exam | Physician to complete after ICS is obtained (pre physical exam) AND within 12 hours after study dug is complete (post physical exam). | ||||||
| Doppler Echo | X (optional) | X | |||||
| Blood draw | X | X | |||||
| Blood Pressure | X | X | |||||
| Borg Score | X | X | X | ||||
| SaO2% | X | Continuously | |||||
| MtHb% | X | Continuously | |||||
| Adverse Events Assessment | X | X | X | X | |||
| Outcome Assessment | X | ||||||
| Dyspnea & Quality of Life Questionnaire | X | ||||||
Figure 2.
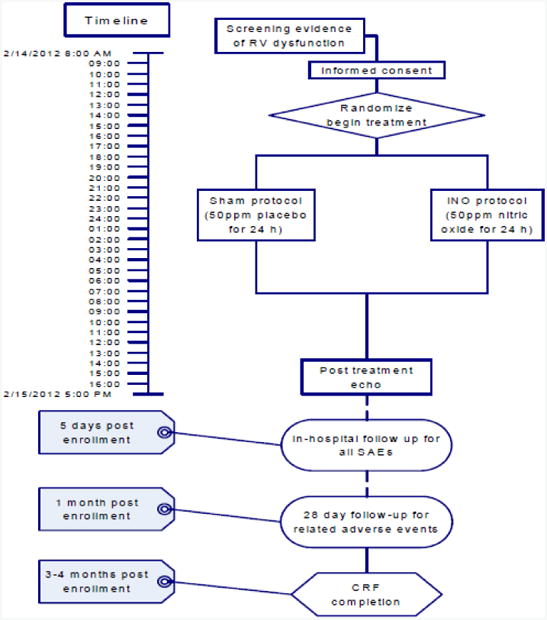
Timeline and key measurements of the clinical study. Echo #1was optional. O2 delivery by nasal cannula can vary depending upon patient comfort.
A study-funded transthoracic Doppler-echocardiogram was obtained within 3 hours of weaning of the study drug. Echocardiography was performed by registered diagnostic cardiac ultrasonographers operating from an ICAEL-certified echocardiography laboratory. Echocardiography images were interpreted for the primary outcomes (RV size, RIMP, TAPSE and FAC) using explicit criteria and recorded on a standard form by a central cardiologist-echocardiography expert (RM). A second blood sample was obtained after 24 (+/-3h) of NO, before study drug wean for the high sensitivity tropoinin and BNP measurements required for the aims. Subjects were followed for serious adverse events through their inpatient stay (through hospital day 5 or discharge, whichever was first) and until 28 days for study drug-related adverse events. Subjects will also be contacted at 3 months for completion of a questionnaire on dyspnea symptoms and quality of life (SF36 and PEmb QOL surveys).
Titration dosing and weaning protocol
Titration and weaning procedures were performed by trained research personnel with on-site assistance from the manufacturers of the INOvent® device. NO was titrated at 2 ppm/min over 25 min to a dose of 50 ppm via nasal cannula as described in Figure 3. Persons administering iNO included RRTs or study-specific research coordinators trained by personnel from Ikaria. In the phase I study, the PI used 25 ppm NO delivered by a specially designed face mask for four hours. However, several patients had considerable discomfort and requested to remove the mask.11 Anticipating patient noncompliance with a mask, especially overnight, the authors therefore elected to use a nasal cannula for NO delivery. The 50 ppm dose was chosen since the nadir in PVR found in dose-response studies of secondary pulmonary hypertension was observed at that dose, and since up to 80 ppm was well tolerated in a recent clinical trial of NO for acute chest syndrome in subjects with sickle cell disease.12,28 Subjects in that trial were treated for up to 72 hours. We chose 24 hours in our protocol because the majority of deaths directly attributed to PE occur within 24 hours, and because placebo groups in RCTs of fibrinolytic agents that measured PA pressures at 2-72 hours post treatment found 24 hours sufficient for PVR to decrease.29-33 From the outset, we justified the three hour variance based on expectations of imprecise time of arrival of echocardiography, and to prevent the weaning procedure from interrupting of standard care procedures. Blood samples were drawn immediately prior to weaning to measure plasma nitrite and nitrate concentrations to assess NO delivery. After patients received 24 hours (+/- 3h) of inhaled NO at 50 ppm and were then weaned at 2ppm per minute to 0 ppm NO over 25 min.
Figure 3.
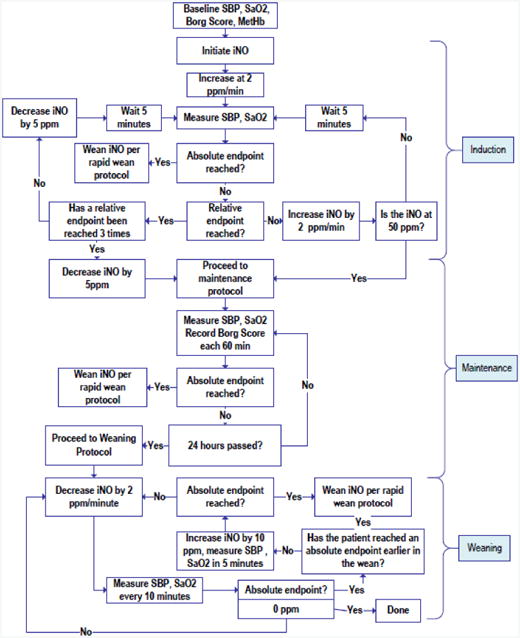
Protocol for administering and weaning study drug. Relative endpoints included persistent symptoms or signs, that in the judgment of the investigator, appeared to be worsened by increasing the inhaled NO ppm and improved by decreasing the inhaled NO ppm. Absolute endpoints included: respiratory distress prompting the clinical care team to plan for mechanical or noninvasive ventilatory support, hypoxemia (SaO2 < 90% for 15 min), circulatory shock (SBP< 90 mm Hg for 15 min in consecutive measurements), new neurologic signs or any ventricular arrhythmias or sustained supraventricular arrhythmia with hypotension or refractory to intravenous medication for more than one hour, ECG changes, MtHb >10% for more than one hour, or persistent vomiting).
The rapid wean protocol was an immediate halving of the inspired concentration and assessment of clinical status; if the patient were improved, the NO was reduced to 0 ppm.
Patient safety concerns
Treatment with 50 ppm NO for 24 hours (+/- 3 h) may cause mtHb values to exceed 5%. This was expected to occur in 1-2 subjects; mtHb levels were therefore continuously monitored. NO could theoretically cause hypotension, and for this reason, we exclude use of drugs known to increase cGMP. Blood pressure values were monitored closely. NO treatment could also theoretically increase risk of lung injury by producing peroxynitrite in the alveoli. We continue to monitor for evidence of acute lung injury for 5 days and report any to the DSMB. We anticipated that 5 to 10 participants would experience a dry nose from the oxygen delivered by nasal cannula for 24 hours (+/- 3 h).
The primary safeguard to prevent immediate harm during treatment was the algorithm presented in Figure 3. The algorithm has multiple feedback loops that raise or lower the iNO concentration in response to the subject's status. To improve safety, one member of the study team (PI or their designee) was not be blinded to treatment, and was available throughout the treatment. The DSMB meets regularly to review unblinded data and uses explicit stopping criteria for evidence of harm or futility (see DSMB charter in supplement). The SpO2%, mtHb% and COHb% was continuously monitored during NO delivery, and Borg score and blood pressure was monitored hourly for the first four hours and then per hospital policy. The protocol allows for one tank per patient, and each tank has up to 100 hours of drug delivery, at no charge to the patient. In the event of severe symptoms, the recommended procedure was to unblind the patient, and if the patientreceiving NO, to decrease the inspired NO to 25 ppm and observe for either clinical worsening or improvement, and allow that response to guide decisions about continuing or discontinuing NO. If the patient were receiving nitrogen + oxygen, then standard care would have proceeded. For the care of suspected worsening related directly to PE, we provided a suggested “rescue algorithm” that follows ACCP guidelines. It is possible for NO + oxygen to combine to form the irritating gas, NO2. The machine that delivers the treatment will continuously monitor for this and will sound an alarm if >2 ppm of NO2 were formed.
Sample size, power and statistical analysis
Sample size was predicated on the effect size defined as a 30% difference in the independent proportions of patients in each group who reach the primary efficacy endpoint (denoted p1 and p2 below), which was justified as the minimum effect size to warrant a phase III trial. Prior work that has performed repeated echocardiograms in patients receiving heparin only allow the assertion of a >90% probability that fewer than 15% of patients will normalize their RV size and function within 24 hours of standard treatment.34-36 The nature of the binomial distribution used in the test for independent proportions results in a power to detect a difference that varies on sample and effect size but also the values of p1 and p2 as diagrammed in Figure 4.37 Accordingly, in a conservative case estimate of p1=0.15 and p2,=0.45, n=35 per group or N=70 total was required to yield a power of 0.80. To account for possible withdrawals or unexpectedly high p1, we have estimated need for n=39 per group or N=78.
Figure 4.
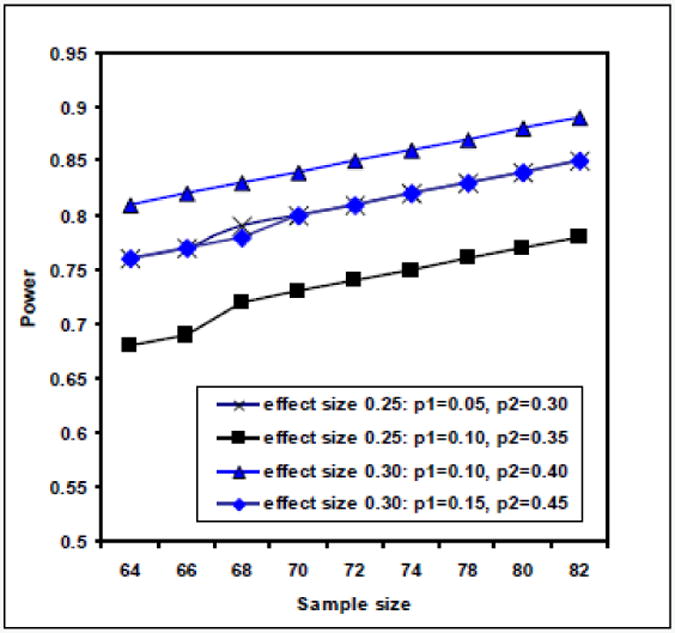
Power estimates assuming two effect sizes of 0.25 and 0.30 each with two different assumptions for p1 and p2
Sample size/power calculation:
α = 0.05 (probability of type I error); Zα/2 = 1.96; β = 0.20 (probability of type II error); Zβ = 0.842
p1 = proportion of patients treated with placebo who normalize RV size and function at 24 hours
p2= proportion of patients treated with NO who normalize RV size and function at 24 hours; p = (p1+p2)/2; δeffect = p1-p2
N = [Zα/2(2p(1-p))1/2 + Zβ(p1(1-p1) +p2(1-p2)) ½]2/δeffect2 =78
Intent to treat was defined as follows: presence of the necessary data to assess the 24 hour primary efficacy endpoint (death, or survival with either echocardiography positive, hsTnT positive, or both echocardiography an hsTnT normal as described above); otherwise the patient will be censored from analysis. Patients who discontinue their original assigned group will be analyzed in the original group, and the denominator used to calculate the proportion of patients reaching the primary efficacy endpoint in that group.
Randomization and Blinding
The randomization and blinding procedure was also described by a specific guidance document and is also explained with photographs in the supplement. This was a randomized, double-blinded trial. The identity of the study drug being delivered to the subjects (i.e., either NO + oxygen or nitrogen + oxygen) was concealed according to the randomization and blinding guidance document. Briefly, this required one non-research team person (referred to as the blinder) to receive and conceal study drug canisters from the industry source (Mallinckrodt Pharmaceuticals) and label them externally with the patient ID that covered the label from the industry source which indicated the container contents. One canister was used per patient and except for differing identification numbers, (e.g., “ESKZ-10”), all canisters appear identical. The patient ID was linked to treatment allocation in a list maintained by the study statistician, the safety monitor, and the blinder, who all can be aware of the assignment. Subjects, caretakers, and the research team have and will not be aware of the identity of the canisters unless they open the unblinding envelope. During titration, treatment and weaning, the INOvent® delivery system conceals the identity of the inspired study drug using Mallinckrodt Pharmaceutical's proprietary blinding software, and an opaque metal hood with a narrow vertical window was flipped down during drug delivery. While the patient was on protocol, an observer looking through this window could only see that the patient was receiving “50 ppm NO”, regardless of treatment assignment by the randomization sequence. The blinding shield was intended to prevent any curious person from scrolling through menus and possibly seeing the NO2 feedback monitoring system, which would potentially unblind the treatment (although this would require a password that even study personnel did not know). Sealed envelopes contain subject ID-specific enrollment cards and indicate what the subject did receive. Enrollment packets contain a checklist, data collection templates, informed consent document, and tubes for blood draws. Each unblinding envelope was a pre-sealed, opaque envelope labeled with the subject ID and the notation “Open only to unblind” with a letter explaining the treatment assignment. The letter asks the name of the unblinder and reason for unblinding. This envelope was readily available to all staff.
Discontinuation of study drug: Absolute endpoints include respiratory distress prompting the clinical care team to plan for mechanical or noninvasive ventilatory support, hypoxemia (SaO2 < 90% for 15 min) requiring intervention, circulatory shock (SBP< 90 mm Hg for 15 min in consecutive measurements) requiring fluids or vasopressors, any ventricular arrhythmias or sustained supraventricular arrhythmia with hypotension or refractory to intravenous medication for more than one hour, ECG changes, MtHb >10% for more than one hour, or persistent vomiting. Relative endpoints in Figure 3 include persistent symptoms or signs, that in the judgment of the investigator, were worsened by reduction in inhaled NO ppm and improved by increasing the inhaled NO ppm.
Results
The first patient was enrolled in March 2014, and to date, the study has enrolled 82 patients with 8 screen failures and two voluntary withdrawals leaving 78 patients treated per protocol with the last patient enrolled in late October, 2016. To reach this number, approximately 1500 patients with PE have been screened, and 155 were approached for informed consent, yielding a consent rate of approximately 50% per patient approached. No patient has been discontinued from iNO for a safety reason. No patient experienced hypotension or any other adverse event during NO titration. Two patients have been unblinded, in one case in a patient who died during an attempted clot extractionprocedure 48 hours after treatment. This patient had metastatic adenocarcinoma of the colon and underwent attempted extraction of a large right atrial thrombus using the Angiovac device and suffered cardiac arrest during the procedure. and in a second patient who had worsened symptoms during treatment. Four patients treated per protocol (4/78) have died within 28 days. No serious adverse event has been found to be probably or definitely related to study drug. The DSMB has met as scheduled, including the final meeting that allowed the study to proceed to its final sample size. We anticipate study completion by the end of calendar year 2016.
Discussion
This paper presents the rationale and protocol for the first phase II study of NO + oxygen to treat acute PE. To our knowledge, this is the first detailed description of the methodology to conduct a randomized, placebo controlled, double-blind trial using inhaled nitric oxide at 50 ppm via nasal cannula. To maintain blinding, we use tanks supplied by the manufacturers of the commercial device (INOvent®) that are identical for placebo and NO. Because INOvent machines have a display that measures both NO and NO2, this portion of the display was concealed with a flip-down hood during drug delivery. To ensure integrity of blinding, we surveyed the clinical care team. For safety, we created a predefined titration and weaning algorithm, shown in Figure 3, and a procedure for handling emergent change in condition, consisting of unblinding of an envelope attached to the INOvent® machine, and a rapid wean protocol. Although two patients voluntarily withdrew because of the perception that they could be discharged sooner, no patient was stopped on protocol because of a safety concern. Use of the Masimo Rainbow device has allowed continuous transcutaneous spectrophotometric monitoring of the blood percentage of methemoglobinemia. No patient has had methemoglobin percentage value that exceeded 10% for longer than 10 minutes. No patient has had an adverse event thought to be related to NO. With 78/78 patients enrolled, it appears this research protocol is both feasible and safe for patients. This work serves as a reference for future investigators to use NO in similarly ill patients (e.g., acute exacerbation of pulmonary hypertension from other causes) or the use of other inhaled agents (e.g. epoprostenol) for patients with acute PE.
Supplementary Material
Acknowledgments
Funding provided by an investigator initiated research by Mallinckrodt and National Institutes of Health (UM1HL113203) to JAK.
Abbreviations
- ADMA
asymmetric dimethyl arginine
- CTEPH
chronic thromboembolic pulmonary hypertension
- CRF
case report form
- ED
emergency department
- eNOS
endothelial nitric oxide synthase
- FDA
Food and Drug Administration
- GC
guanylate cyclase
- ICAEL
International committee for accreditation of echocardiography laboratories
- IRB
institutional review board
- mtHb
met hemoglobin
- NO
nitric oxide
- OAT
ornithine aminotransferase
- ODC
ornithine decarboxylase
- PCR
polymerase chain reaction
- PE
pulmonary embolism
- PVR
pulmonary vascular resistance
- RCT
randomized controlled trial
- rtPCR
Reverse transcriptase polymerase chain reaction
- RVSP
right ventricular systolic pressure
- RRT
registered respiratory therapist
- SBP
systolic blood pressure
- SNP
single nucleotide polymorphism
- VSM
vascular smooth muscle
Footnotes
Author contributions: JAK: study design, protocol, funding, data collection, analysis, manuscript preparation: CLH, AEJ, RM, TL: protocol, data collection, analysis, manuscript preparation
Conflicts of interest: No other authors have a conflict to report
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jeffrey A. Kline, Indiana University School of Medicine, Dept. of Emergency Medicine, 720 Eskenazi Avenue, Fifth Third Faculty Office Building, 3rd Floor Emergency Medicine Office, Indianapolis, IN 46202, 317-880-3869.
Cassandra L. Hall, Indiana University School of Medicine, Dept. of Emergency Medicine, 1701 N. Senate Blvd., AG001, Indianapolis, IN 46202, 317-962-1190.
Alan E. Jones, Department of Emergency Medicine, University of Mississippi Medical Center, 2500 N. State St., Jackson, MS 39216, 601-984-5543.
Michael A. Puskarich, Department of Emergency Medicine, University of Mississippi Medical Center, 2500 N. State St., Jackson, MS 39216, 601-984-5543
Ronald Mastouri, Indiana University School of Medicine, Department of Medicine, Division of, Cardiology, 317-962-0539.
Tim Lahm, Division of Pulmonary, Allergy, Critical, Care, Occupational and Sleep Medicine, Indiana University School of Medicine, Richard L. Roudebush VA Medical, Center, Walther Hall, room C400, 980 W. Walnut Street, Indianapolis, IN 46202.
References
- 1.Kearon C, Akl EA, Ornelas J, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149:315–352. doi: 10.1016/j.chest.2015.11.026. [DOI] [PubMed] [Google Scholar]
- 2.Watts JA, Gellar MA, Fulkerson MB, Kline JA. Pulmonary vascular reserve during experimental pulmonary embolism: Effects of a soluble guanylate cyclase stimulator, BAY 41-8543. Crit Care Med. 2011;39:2700–2704. doi: 10.1097/CCM.0b013e318226678e. [DOI] [PubMed] [Google Scholar]
- 3.Insenser M, Montes-Nieto R, Martinez-Garcia MA, et al. Identification of reduced circulating haptoglobin concentration as a biomarker of the severity of pulmonary embolism: a nontargeted proteomic study. PLoS One. 2014;9:e100902. doi: 10.1371/journal.pone.0100902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kline JA, Steuerwald M, Watts JA, Courtney DM, Bonkovsky HL. Leukocyte Expression of Heme Oxygenase-1 [hmox1] Varies Inversely with Severity of Tricuspid Regurgitation in Acute Pulmonary Embolism. Thromb Res. 2015;136:769–774. doi: 10.1016/j.thromres.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kline JA, Watts J, Courtney D, Lee Y, Hwang S. Severe pulmonary embolism decreases plasma L-arginine. Eur Respir J. 2013 doi: 10.1183/09031936.00171913. [DOI] [PubMed] [Google Scholar]
- 6.Watts JA, Gellar MA, Fulkerson MB, Das SK, Kline JA. Arginase depletes plasma l-arginine and decreases pulmonary vascular reserve during experimental pulmonary embolism. Pulm Pharmacol Ther. 2011 doi: 10.1016/j.pupt.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Watts JA, Gellar MA, Obraztsova M, Kline JA, Zagorski J. Role of inflammation in right ventricular damage and repair following experimental pulmonary embolism in rats. Int J Exp Path. 2008;89:389–399. doi: 10.1111/j.1365-2613.2008.00610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watts JA, Zagorski J, Gellar MA, Stevinson BG, Kline JA. Cardiac inflammation contributes to right ventricular dysfunction following experimental pulmonary embolism in rats [abstract] J Mol Cell Cardiol. 2006;41:296–307. doi: 10.1016/j.yjmcc.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Stein PD, Matta F, Alrifai A, Rahman A. Trends in case fatality rate in pulmonary embolism according to stability and treatment. Thromb Res. 2012;130:841–846. doi: 10.1016/j.thromres.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Sista AK, Miller L, Kahn SR, Kline JA. Persistent Right Ventricular Dysfunction, Functional Capacity Limitation, Exercise Intolerance, and Quality of Life Impairment Following Pulmonary Embolism: Systematic Review With Meta-analysis. Vascular Medicine. doi: 10.1177/1358863X16670250. In press. [DOI] [PubMed] [Google Scholar]
- 11.Kline JA, Hernandez-Nino J, Garrett JS, Jones AE. Pilot study of a protocol to administer Inhaled nitric oxide to treat severe acute submassive pulmonary embolism. Emerg Med J. 2014;31:459–463. doi: 10.1136/emermed-2013-202426. [DOI] [PubMed] [Google Scholar]
- 12.Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305:893–902. doi: 10.1001/jama.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konstantinides SV, Torbicki A, Agnelli G, et al. ESC Guidelines on the diagnosis and management of acute pulmonary embolism: The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC) Endorsed by the European Respiratory Society (ERS) Eur Heart J. 2014;2014 [Google Scholar]
- 14.Jaff MR, McMurtry MS, Archer SL, et al. Management of Massive and Submassive Pulmonary Embolism, Iliofemoral Deep Vein Thrombosis, and Chronic Thromboembolic Pulmonary Hypertension: A Scientific Statement From the American Heart Association. Circulation. 2011;1:1788–1830. doi: 10.1161/CIR.0b013e318214914f. [DOI] [PubMed] [Google Scholar]
- 15.Becattini C, Agnelli G, Germini F, Vedovati MC. Computed tomography to assess risk of death in acute pulmonary embolism: a meta-analysis. Eur Respir J. 2014;43:1678–1690. doi: 10.1183/09031936.00147813. [DOI] [PubMed] [Google Scholar]
- 16.Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23:685–713. doi: 10.1016/j.echo.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Lankeit M, Jimenez D, Kostrubiec M, et al. Predictive value of the high-sensitivity troponin T assay and the simplified pulmonary embolism severity index in hemodynamically stable patients with acute pulmonary embolism: a prospective validation study. Circulation. 2011;124:2716–2724. doi: 10.1161/CIRCULATIONAHA.111.051177. [DOI] [PubMed] [Google Scholar]
- 18.van EJ, den Exter PL, Kaptein AA, et al. Quality of life after pulmonary embolism as assessed with SF-36 and PEmb-QoL. Thromb Res. 2013;132:500–505. doi: 10.1016/j.thromres.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 19.Kruse L, Mitchell AM, Camargo CA, Jr, Hernandez J, Kline JA. Frequency of thrombophilia-related genetic variations in patients with idiopathic pulmonary embolism in an urban emergency department. Clinical Chemistry. 2006;52:1026–1032. doi: 10.1373/clinchem.2005.061861. [DOI] [PubMed] [Google Scholar]
- 20.Ragia G, Nikolaidis E, Tavridou A, et al. Endothelial nitric oxide synthase gene polymorphisms -786T > C and 894G > T in coronary artery bypass graft surgery patients. Hum Genomics. 2010;4:375–383. doi: 10.1186/1479-7364-4-6-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fatini C, Mannini L, Sticchi E, et al. eNOS gene affects red cell deformability: role of T-786C, G894T, and 4a/4b polymorphisms. Clin Appl Thromb Hemost. 2005;11:481–488. doi: 10.1177/107602960501100417. [DOI] [PubMed] [Google Scholar]
- 22.Li YY, Zhai ZG, Yang YH, et al. Association of the 894G>T polymorphism in the endothelial nitric oxide synthase gene with risk of venous thromboembolism in Chinese population. Thromb Res. 2011;127:324–327. doi: 10.1016/j.thromres.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 23.Arnalich F, Maldifassi MC, Ciria E, et al. Plasma levels of mitochondrial and nuclear DNA in patients with massive pulmonary embolism in the emergency department: a prospective cohort study. Crit Care. 2013;17:R90. doi: 10.1186/cc12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kannemeier C, Shibamiya A, Nakazawa F, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Megy K, Audic S, Claverie JM. Heart-specific genes revealed by expressed sequence tag (EST) sampling. Genome Biol. 2002;3:RESEARCH0074. doi: 10.1186/gb-2002-3-12-research0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zagorski J, Obraztsova M, Gellar MA, Kline JA, Watts JA. Transcriptional changes in right ventricular tissues are enriched in the outflow tract compared with the apex during chronic pulmonary embolism in rats. Physiol Genomics. 2009;39:61–71. doi: 10.1152/physiolgenomics.00076.2009. [DOI] [PubMed] [Google Scholar]
- 27.Xiao J, Jing ZC, Ellinor PT, et al. MicroRNA-134 as a potential plasma biomarker for the diagnosis of acute pulmonary embolism. J Transl Med. 2011;9:159. doi: 10.1186/1479-5876-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solina AR, Ginsberg SH, Papp D, et al. Dose response to nitric oxide in adult cardiac surgery patients. J Clin Anesth. 2001;13:281–286. doi: 10.1016/s0952-8180(01)00270-7. [DOI] [PubMed] [Google Scholar]
- 29.Tibbutt DA, Davies JA, Anderson JA, Fletcher EWL, Hamill J, et al. Comparison by controlled clinical trial of streptokinase and heparin in treatment of life-threatening pulmonary embolism. Br Am J. 1974;1:343–347. doi: 10.1136/bmj.1.5904.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.PIOPED Investigators. Tissue plasminogen activator for the treatment of acute pulmonary embolism. Chest. 1990;97:528–533. doi: 10.1378/chest.97.3.528. [DOI] [PubMed] [Google Scholar]
- 31.Konstantinides S, Tiede N, Geibel A, Olschewski M, Just H, Kasper W. Comparison of alteplase versus heparin for resolution of major pulmonary emoblism. Amer Jrnl of Cardiology. 1998;82:966–970. doi: 10.1016/s0002-9149(98)00513-x. [DOI] [PubMed] [Google Scholar]
- 32.Sasahara AA, Hyers TM, Cole CM. The urokinase pulmonary embolism trial: A national cooperative study. Circulation. 1973;47:II-66–II-89. [PubMed] [Google Scholar]
- 33.Dalla-Volta S, Palla A, Santolicandro A, et al. PAIMS 2: Alteplase combined with heparin versus heparin in the treatment of acute pulmonary embolism. Plasminogen activator Italian multicenter study 2. Journal of the American College of Cardiology. 1992;20:520–526. doi: 10.1016/0735-1097(92)90002-5. [DOI] [PubMed] [Google Scholar]
- 34.Becattini C, Agnelli G, Salvi A, et al. Bolus tenecteplase for right ventricle dysfunction in hemodynamically stable patients with pulmonary embolism. Thromb Res. 2010;125:e82–e86. doi: 10.1016/j.thromres.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 35.Kooter AJ, Ijzerman RG, Kamp O, Boonstra AB, Smulders YM. No effect of epoprostenol on right ventricular diameter in patients with acute pulmonary embolism: a randomized controlled trial. BMC Pulm Med. 2010;10:18–2. doi: 10.1186/1471-2466-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rydman R, Soderberg M, Larsen F, Caidahl K, Alam M. Echocardiographic evaluation of right ventricular function in patients with acute pulmonary embolism: a study using tricuspid annular motion. Echocardiography. 2010;27:286–293. doi: 10.1111/j.1540-8175.2009.01015.x. [DOI] [PubMed] [Google Scholar]
- 37.Bristol DR. Sample sizes for constructing confidence intervals and testing hypotheses. Statistics in Medicine. 1989;8:803–811. doi: 10.1002/sim.4780080705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.