

Abstract
The axon initial segment (AIS) is a specialized domain essential for neuronal function, the formation of which begins with localization of an ankyrin‐G (AnkG) scaffold. However, the mechanism directing and maintaining AnkG localization is largely unknown. In this study, we demonstrate that in vivo knockdown of microtubule cross‐linking factor 1 (MTCL1) in cerebellar Purkinje cells causes loss of axonal polarity coupled with AnkG mislocalization. MTCL1 lacking MT‐stabilizing activity failed to restore these defects, and stable MT bundles spanning the AIS were disorganized in knockdown cells. Interestingly, during early postnatal development, colocalization of MTCL1 with these stable MT bundles was observed prominently in the axon hillock and proximal axon. These results indicate that MTCL1‐mediated formation of stable MT bundles is crucial for maintenance of AnkG localization. We also demonstrate that Mtcl1 gene disruption results in abnormal motor coordination with Purkinje cell degeneration, and provide evidence suggesting possible involvement of MTCL1 dysfunction in the pathogenesis of spinocerebellar ataxia.
Keywords: axon initial segment, microtubule cross‐linking factor 1, microtubules, Purkinje cells
Subject Categories: Neuroscience
Introduction
Neurons are highly polarized cells that develop and maintain distinct processes, specifically axons and dendrites. Axonal identity in terms of structure and function is maintained by a specialized domain at the proximal end of the axon: the axon initial segment (AIS; Rasband, 2010). Presence of the AIS is critical for action potential generation (Kole & Stuart, 2012) and maintenance of axonal polarity by preventing mixing of axonal and somatodendritic components (Leterrier & Dargent, 2014). These AIS functions are dependent on dense clustering of a variety of specific protein components in the vicinity of the proximal axon membrane. Among them, voltage‐gated Na channels (Nav) allow action potential generation (Kole et al, 2008), while the cytoskeletal scaffold, ankyrin‐G (AnkG), and the actin cytoskeleton play essential roles in maintenance of axonal polarity (Sobotzik et al, 2009; Song et al, 2009; Watanabe et al, 2012). Although structural architecture of the AIS complex has been extensively studied, the underlying mechanisms regulating assembly and maintenance of the complex are not yet fully understood.
In developing neurons, formation of the AIS is thought to be triggered by AnkG localization to the proximal axon (Grubb & Burrone, 2010). This initial AnkG localization is essential for subsequent recruitment of many other AIS components, including beta‐IV spectrin, Nav, and neuronal cell adhesion molecule (NrCAM; Zhou et al, 1998; Komada & Soriano, 2002; Hedstrom et al, 2007; Yang et al, 2007). AnkG depletion from developing neurons in vitro results in loss of localization of these AIS components (Zhou et al, 1998; Jenkins & Bennett, 2001; Ango et al, 2004; Hedstrom et al, 2007). In mice, genetic disruption of AnkG in cerebellar Purkinje cells also results in loss of AIS, which is accompanied by axonal polarity defects (Zhou et al, 1998; Jenkins & Bennett, 2001; Sobotzik et al, 2009) and induction of ataxia coupled with Purkinje cell degeneration (Zhou et al, 1998). Conversely, initial AnkG localization is not affected by depletion of other AIS components such as beta‐IV spectrin and neurofascin‐186 (NF186) (Yang et al, 2007; Zonta et al, 2011). AnkG localization is also important for AIS maintenance in mature neurons, and depletion of AnkG from mature neurons causes dispersion of AIS components (Hedstrom et al, 2008). Galiano et al (2012) demonstrated that AnkG localization at the proximal axon is restricted by the distal axonal cytoskeleton comprising AnkB, αII‐ and βII‐spectrin. Nonetheless, the factors and mechanisms responsible for directing and maintaining AnkG localization are largely unknown.
In electron microscopic studies, one of the criteria to identify AIS is the presence of distinctive MT fascicles (Palay et al, 1968). In AIS, MTs are aligned in parallel with their plus‐end pointing toward the distal axon and are linked together by a thin material, the identity of which has not yet been determined (Palay et al, 1968; Peters et al, 1968; Baas et al, 1988; Kevenaar & Hoogenraad, 2015). In addition, MTs in AIS are highly stabilized and enriched in posttranslational modifications including acetylation (Hammond et al, 2010; Song et al, 2013). Importantly, a recent study demonstrated that, before initial AnkG localization, MTs are already formed into bundles in the proximal axon of developing neurons, and progressively coated with actin and many AIS components including AnkG (Jones et al, 2014). Thus, formation of uniformly oriented and stabilized MT bundles might be a critical event for AIS formation and establishment of neuronal function. However, evidence supporting this idea has been lacking until recently, when several studies demonstrated the importance of various MT‐regulating proteins in AnkG localization or axonal polarity (Leterrier et al, 2011; Yau et al, 2014; van Beuningen et al, 2015; Freal et al, 2016; Kuijpers et al, 2016). MT involvement was also suggested by a study demonstrating that disruption of MT stability and acetylation in developing neurons impairs initial AnkG localization (Tapia et al, 2010; Sanchez‐Ponce et al, 2011). These results support the importance of AIS MT bundles for AIS development, although the underlying mechanism of how such characteristic MT bundles are organized within this specialized neuronal region is not known.
Microtubule cross‐linking factor 1 (MTCL1) is a MT‐regulating protein identified as a binding protein of the cell polarity‐regulating kinase, PAR‐1b/MARK2 (Sato et al, 2013). MTCL1 has coiled‐coil motifs in the central part of the protein and two MT‐binding domains (MTBDs) at the N‐ and C‐terminal regions. The N‐terminal MTBD cross‐links MTs with the aid of the downstream coiled‐coil region (Sato et al, 2013), whereas the C‐terminal MTBD is rich in basic residues (also known as the KR‐region) and shows MT‐stabilizing activity (Sato et al, 2014). Knockdown experiments in cultured cells show an indispensable role for MTCL1 in organization of non‐centrosomal MTs and cell polarity. In epithelial cells, MTCL1 is crucial for development of lateral MT bundles with their specific columnar cell shape (Sato et al, 2013). In less polarized cells, such as HeLa cells, it plays essential roles in cross‐linking and stabilizing Golgi‐derived MTs, and thereby critically contributes to Golgi‐ribbon formation and polarized cell migration (Sato et al, 2014). In this study, we analyzed Mtcl1 gene‐trap and knockout mice to explore the physiological role of MTCL1 in vivo. Consistent with preferential expression of MTCL1 in cerebellar Purkinje cells, mice lacking normal MTCL1 display abnormal motor coordination. Subsequent analysis revealed that MTCL1 is essential for postnatal development of AIS in Purkinje cells. By performing in utero electroporation knockdown and rescue experiments, we also show that MTCL1 plays an indispensable role in maintenance of AnkG localization by mediating AIS MT bundle formation. We also present data indicating possible involvement of a MTCL1 mutation in pathogenesis of spinocerebellar ataxia.
Results
Disruption of the Mtcl1 gene results in abnormal motor coordination in mice
To investigate MTCL1 function in vivo, we generated Mtcl1 gene‐trap mice, in which a gene‐trap vector has been inserted within intron 4 of the Mtcl1 gene (Fig 1A and B, and Appendix Fig S1). Although homozygous gene‐trap mice (hereafter, “GT mice”) were born at the predicted Mendelian frequency of 25% (153/610 mice) and with a normal life span, their body weight was ~20% lower than wild‐type (WT) littermates (WT 6.2 ± 1.47 g SD, GT: 5.0 ± 0.71 g SD) at 4 weeks of age (Fig 1C). In addition, GT female mice, but not male, were infertile, suggesting an impairment of sexual development. Most significantly, GT mice exhibited abnormal motor coordination at 3–4 weeks of age in a sex‐independent manner (Movie EV1). Because this last phenotype is consistent with preferential expression of MTCL1 in the cerebellum (in addition to the lung and testis; Fig 1D), we chose to further examine MTCL1 function within the cerebellum. In cerebellar extracts from GT mice, anti‐MTCL1 antibody (raised against the internal region) failed to detect MTCL1 protein, whereas anti‐LacZ antibody detected expression of a beta‐geo‐fused MTCL1 fragment with a predicted molecular mass of 220 kDa (Fig 1E). Overall, these observations imply that expression of abnormal MTCL1 protein within the cerebellum disrupts motor coordination in mice.
Figure 1. Characterization of Mtcl1 gene‐trap mice.
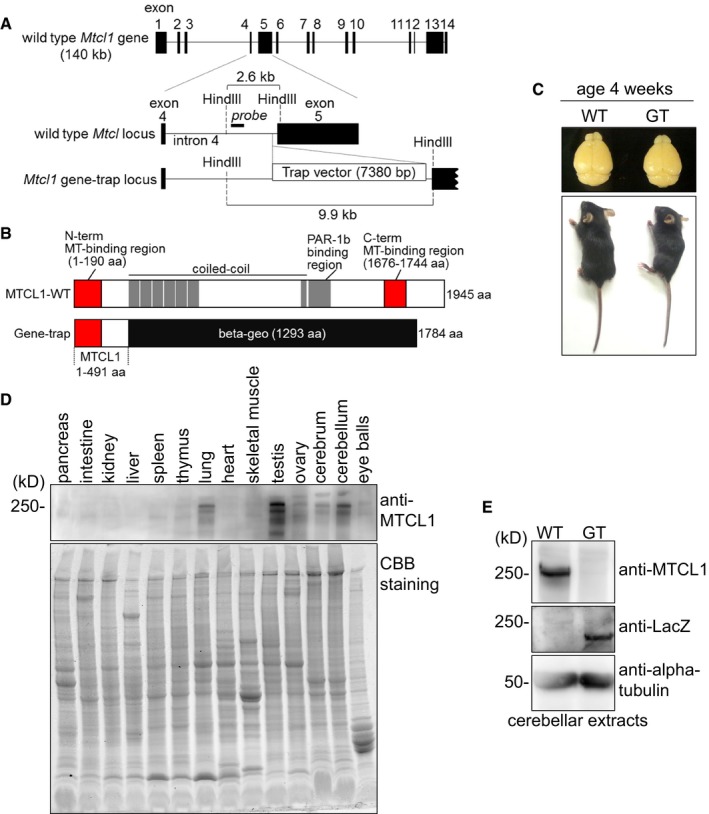
- Schematic of the Mtcl1 allele in mouse. In GT mice, the beta‐geo‐based gene‐trap vector was inserted into intron 4. Black blocks represent exons. For Southern blotting, genomic DNA was digested with HindIII and hybridized with the probe depicted (see also Appendix Fig S1).
- Schematic of MTCL1 WT and mutant proteins.
- Representative pair of WT and GT littermates at 4 weeks of age (lower), with dorsal views of their brains (upper).
- Western blot analysis of various tissue lysates from WT mice at 4 weeks of age using anti‐MTCL1 antibodies (upper). The replica gel was stained with Coomassie brilliant blue to verify protein loading (lower).
- Western blot analysis of cerebellar lysates from WT and GT littermates at 7 weeks of age using anti‐MTCL1, anti‐beta‐galactosidase (LacZ), and anti‐alpha‐tubulin antibodies.
Mtcl1 gene disruption causes cerebellar Purkinje cell degeneration
The brains of GT mice were macroscopically normal (Fig 1C), with a normal layered cerebellar structure (Fig 2A). Therefore, we next examined expression of MTCL1 and the effect of Mtcl1 gene disruption at the microscopic level. Immunostaining of sagittal slices of WT and GT cerebellum showed that MTCL1 is mainly expressed in Purkinje cells, and, to a lesser degree, in granule cells (Fig 2A and B). Further, we found that at 4 weeks of age, Purkinje cells of GT mice frequently showed localized axonal swellings, an early sign of axonal degeneration (Fig 2C, upper right panel, arrows; Coleman, 2005). Ultrastructural analysis of these axonal swellings supported this notion by revealing abnormal accumulation of membranous organelles, a common feature of axonal degeneration (Fig 2D; Coleman, 2005). Partial Purkinje cell loss was also detected in GT mice at 8 months of age, in addition to increased appearance of axonal swellings (Fig 2C, lower right panel, asterisk). Furthermore, weak dilation of dendrites was observed in GT mice (Fig 2E and F), although dendrite density appeared unchanged, and no obvious differences were observed in distribution of synaptic marker proteins (VGLUT1, VGLUT2, and GluRD2; Fig 2G; Hashimoto et al, 2009). These results indicate that disruption of the Mtcl1 gene results in Purkinje cell degeneration, which may cause the abnormal motor coordination.
Figure 2. MTCL1 GT mice exhibit Purkinje cell degeneration.
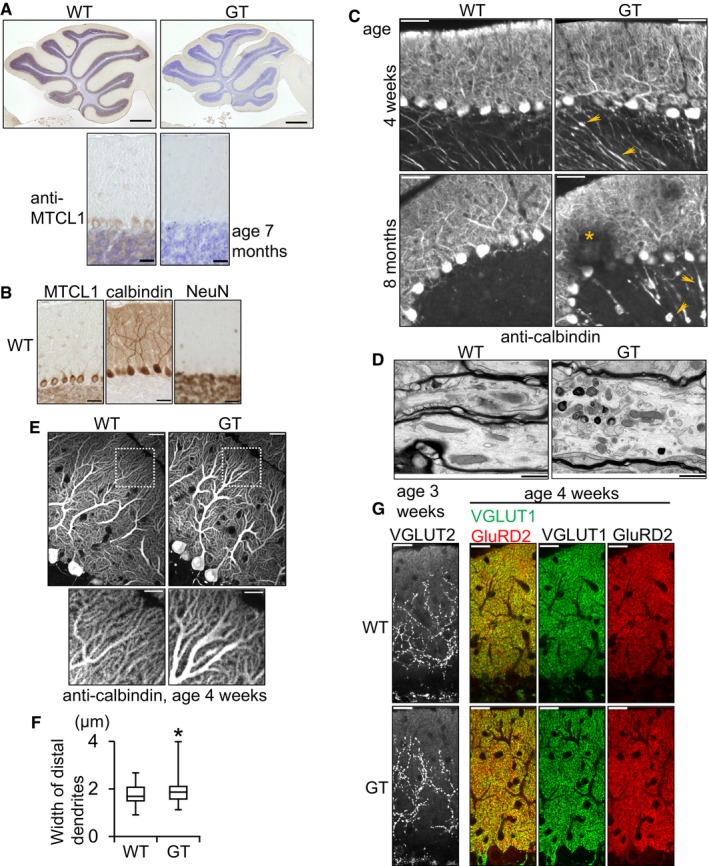
- Cerebellar sections from WT and mutant mice at 7 months of age were immunostained for MTCL1 (brown signal) together with a Nissl counterstain (blue signal).
- Cerebellar sections from WT mouse at 4 weeks of age were immunostained for MTCL1, calbindin, and NeuN. Calbindin and NeuN were used to identify Purkinje cells and granule cells, respectively.
- Cerebellar sections from WT and mutant mice at 4 weeks and 8 months of age were immunostained for calbindin. Arrows indicate dilations and swellings of axons. An asterisk indicates partial Purkinje cell loss.
- Electron micrographs of Purkinje cell axons (surrounded by the myelin sheath) from WT and GT mice at 4 weeks of age.
- Representative calbindin immunostaining of Purkinje cells from WT and GT mice at 4 weeks of age. The boxed regions are magnified and shown in the bottom panels.
- Box plot showing distal dendrite width of Purkinje cells from WT and GT mice at 4 weeks of age, measured 20 μm from the distal edge (WT, n = 59; GT, n = 59). Lines within each box represent medians. Boxes represent the interquartile range from 25th to 75th percentile. Whiskers indicate maximum and minimum values. *P < 0.005, compared with WT (F‐test).
- Cerebellar sections from WT and GT mice were immunostained for VGULT1 and GluRD2 (at 4 weeks of age) or VGLUT2 (at 3 weeks of age).
Disruption of the Mtcl1 gene causes abnormal AIS morphology in Purkinje cells
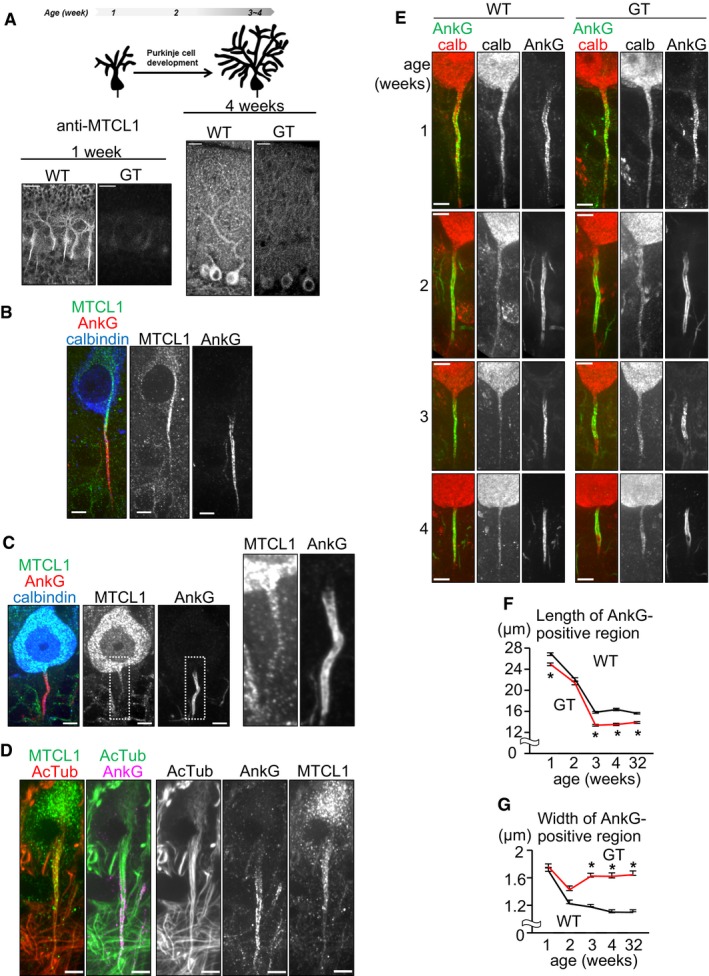
To clarify the role of MTCL1 in Purkinje cells, we next examined the subcellular distribution of MTCL1. By 4 weeks of age, Purkinje cells have completed their postnatal development (Kapfhammer, 2004). At this stage, we found that MTCL1 was evenly distributed within the cell body and stem dendrites (Fig 3A, right). Contrarily, MTCL1 exhibited a characteristic distribution at 1 week of age, demonstrating accumulating needle‐like staining from the perinuclear region to proximal AIS (Fig 3A, left and Fig 3B). Interestingly, the proximal AIS accumulation of ectopically expressed V5‐tagged MTCL1 was clearly detected even at 3 weeks of age when using anti‐V5 antibody, but not the anti‐MTCL1 antibody (Fig 5B). This suggests that MTCL1 localizes to proximal AIS even at the later stage, but the epitope masking hampered clear detection by the anti‐MTCL1 antibody. Consistently, albeit rather weak, the proximal AIS localization was detected by the anti‐MTCL1 antibody for endogenous as well as exogenous MTCL1 (Figs 3C and 5B). Because one of the characteristic features of the AIS is the presence of stable MT bundles, we determined if MTCL1 localizes to these MT bundles. Corresponding with electron microscopy, which first described the presence of AIS MT bundles (Palay et al, 1968), anti‐acetylated tubulin antibody (a stable MT marker) clearly detected MT bundles emerging from the axon hillock and running into the AIS (Fig 3D). As expected, in mice at 1 week of age, MTCL1 colocalized with the proximal part of these MT bundles (Fig 3D). We also found that within the soma, these MT bundles appeared connected to the perinuclear MT network, where MTCL1 is concentrated (see Discussion).
Figure 3. Purkinje cells in GT mice exhibit impaired AnkG localization.
-
ASchematic of postnatal development of murine Purkinje cells (upper). Cerebellar sections from WT mice at 1 and 4 weeks of age were immunostained for MTCL1 (lower).
-
B–DCerebellar sections from WT mice at 1 week (B and D) or 3 weeks (C) of age were immunostained for MTCL1, AnkG, and calbindin (B and C) or acetylated tubulin (AcTub) (D).
-
ECerebellar sections from WT and mutant mice at the indicated age were immunostained for calbindin and AnkG.
-
F, GQuantification of the length (F) and width (G) of AnkG localized regions at the indicated age (WT, n = 112, 84, 124, 66, 120; GT, n = 116, 67, 140, 63, 159, respectively). Data are represented as mean ± SEM. *P < 0.001, compared with WT at each age (Student's t‐test).
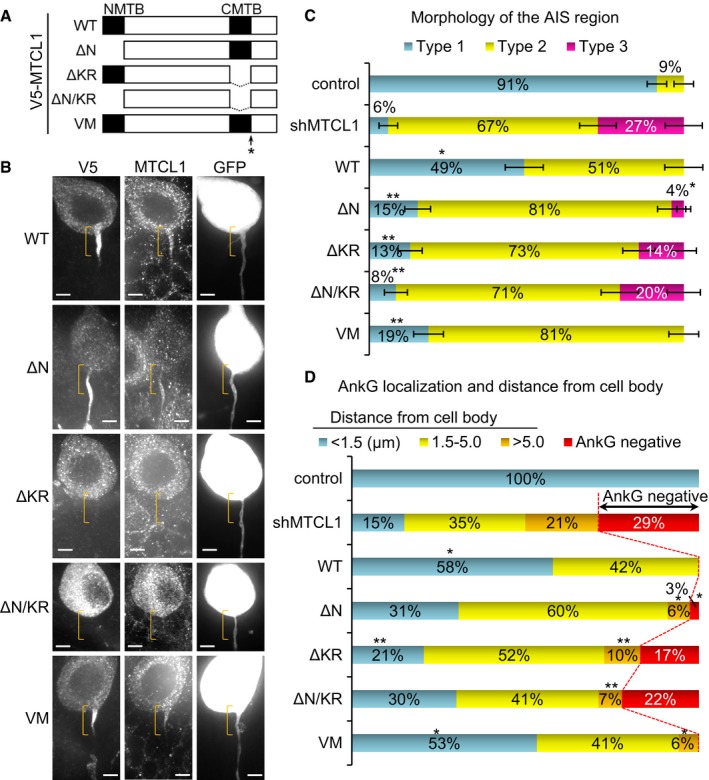
Figure 5. C‐terminal MTBD is required for axonal polarity and AnkG localization.
-
ASchematic of mouse MTCL1 constructs. N‐terminal MTBD (1–190 aa) and C‐terminal MTBD were deleted in ΔN and ΔKR, respectively. ΔN/KR lacks both regions. Val1748 was substituted with Met in VM. All the constructs were tagged with V5 at their N‐termini. V5‐MTCL1 constructs, shMTCL1, and GFP were cloned into a single vector and introduced by IUE (see also Materials and Methods).
-
BGFP‐positive transfected cells were immunostained for V5 and MTCL1. Brackets indicate the proximal AIS region. Scale bars: 5 μm.
-
C, DQuantification of AIS region morphology (C) and AnkG localization (D) (WT, n = 69; ΔN, n = 78; ΔKR, n = 77; ΔN/KR, n = 59; VM, n = 69) (see also Appendix Fig S3). Data represent mean (C and D) ± SEM (C). *P < 0.001, compared with shMTCL1; and **P < 0.001, compared with WT (Fisher's exact test). Note that the control and shMTCL1 data are the same as shown in Fig 4C and D, respectively, because these experiments were performed simultaneously.
The AIS is a specialized region that is critical for action potential generation and maintenance of axonal polarity. Thus, we next examined the possibility that Mtcl1 gene disruption damages Purkinje cells by impairing normal AIS development. In WT mice at 1 week of age, Purkinje cells already show AnkG signal at the proximal axonal region adjacent to the cell body, and this AnkG‐positive region continuously exhibits a slender morphology until 4 weeks of age (Fig 3E, left column). Similarly, AnkG‐positive regions of similar appearance were also observed in GT mice at 1 week of age, but their original morphology was gradually lost, and they became dilated and irregular (Fig 3E, right column). Quantitative analysis of the length and width of these AnkG‐positive regions clearly demonstrated abnormal development of AIS morphology in GT mice. In WT, both the length and width of AnkG‐positive regions progressively became smaller, reaching a plateau around 3 weeks (Fig 3F and G). In contrast, in GT cells, shortening of AnkG‐positive regions accelerated after 2 weeks, reaching a plateau at ~80% of WT length (Fig 3F). The width of AnkG‐positive regions transiently showed a trend toward thinning until 2 weeks, before returning to the original width (Fig 3G). Despite this abnormal AIS development, GT mice exhibited normal colocalization of Nav1.6 with AnkG, and pinceau synapse formation around AnkG localized regions (Appendix Fig S2), suggesting that loss of normal MTCL1 does not severely affect AIS components (Zhou et al, 1998; Ango et al, 2004). We cannot completely exclude the possibility that these morphological AIS changes are not the cause but the result of axonal degeneration at this stage. However, AIS length and width are accompanied by action potential properties such as setting the threshold for firing (Kole & Stuart, 2012; Kuba, 2012), and normal firing is needed for neuronal survival (Wada, 2006). Therefore, these results potentially support the notion that onset of abnormal motor coordination and Purkinje cell degeneration in GT mice are caused by dysregulation of AIS morphology. Since the N‐terminal fragment of MTCL1 expressed in GT mice (Fig 1B) were accumulated at the proximal AIS at 1 week but not at 3 weeks of age, the fragment might compensate the lack of normal MTCL1 at the initial stages (Appendix Fig S1D and E).
MTCL1 depletion in developing Purkinje cells impairs AnkG localization and axonal polarity
To directly determine the cell autonomous function of MTCL1 in Purkinje cells, we next examined the effect of specific MTCL1 depletion in developing Purkinje cells. For this purpose, we introduced expression vectors containing MTCL1 shRNA (shMTCL1) coupled to a GFP expression cassette into nascent Purkinje cells by in utero electroporation (IUE) at embryonic day (E)11.5 (Nishiyama et al, 2012). As expected, MTCL1 expression was specifically reduced in shMTCL1‐transfected cells, but not control shRNA‐transfected cells (Fig 4A, anti‐MTCL1, arrows). GFP fluorescence showed that at 3 weeks of age, shMTCL1‐transfected cells localized in a monolayer together with non‐transfected cells. In addition, they developed highly branched dendrites in the same plane, and a single axon projected into the granule cell layer (Fig 4A, GFP, arrowheads). These observations suggest that MTCL1 knockdown in developing Purkinje cells has little effect on migration, axon formation, and dendrite arborization. However, detailed observations revealed remarkable AIS abnormalities in shMTCL1‐transfected cells. The majority (~91%) of control‐transfected cells showed a stereotypical smooth and slender morphology of the proximal axonal region adjacent to the cell body (hereafter, AIS region) (Fig 4B, Type 1, and Fig 4C). In contrast, only ~6% of shMTCL1‐transfected cells retained this morphology (Fig 4B and C). Instead, the AIS regions of many shMTCL1‐transfected cells (~67%) were irregularly dilated and displayed small protrusions (Fig 4B, Type 2, and Fig 4C). In extreme cases (~27%), not only the AIS region but also the immediate downstream axon and cell body were broadly decorated with multiple protrusions closely resembling dendritic spines (Fig 4B, Type 3, and Fig 4C). Indeed, the immunostaining signal of VGLUT1, a granular cell presynaptic marker, was detected along and close to these protrusions (Fig 4D), suggesting the formation of ectopic dendritic synapses. Because ~89% of Type 3 AIS regions completely lacked AnkG (Fig 4B and Appendix Fig S3A), these results suggest that MTCL1 depletion disrupts axonal polarity by impairing AnkG localization to the AIS region. Notably, even when AnkG signal was detected, MTCL1 knockdown frequently caused detachment of the AnkG‐positive region from the cell body (Fig 4B and E, and Appendix Fig S3B). The length of AnkG‐positive regions in shMTCL1‐transfected cells was also shorter than in control cells (Appendix Fig S3C). In summary, severity of the morphological defect of the AIS region is closely associated with AnkG localization defects. These results suggest that MTCL1 plays an essential role in maintaining AIS structure and function in developing Purkinje cells by regulating AnkG localization to the AIS region.
Figure 4. MTCL1 depletion causes axonal polarity disruption and loss of AnkG localization.
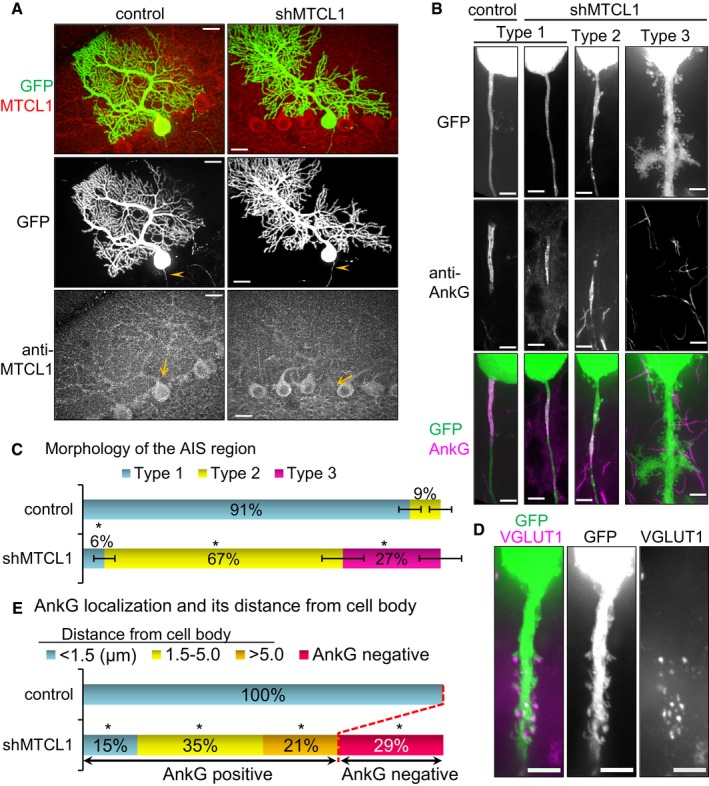
- Cerebellar sections from in utero electroporated mice (control shRNA or MTCL1 shRNA vectors containing GFP expression cassette) at 3 weeks of age were immunostained for MTCL1. Arrows indicate transfected cells. Arrowheads indicate axons of transfected cells.
- Representative images of the three types of AIS regions (GFP panels) and immunostaining for AnkG (middle and bottom panels). Type 1 examples are from control and shMTCL1 cells, and types 2 and 3 are from shMTCL1 cells.
- Quantification of AIS region morphology (control, n = 82; shMTCL1, n = 66). Data represent mean ± SEM. *P < 0.001, compared with control (Fisher's exact test).
- Representative Type 3 AIS region of MTCL1 shRNA‐transfected Purkinje cells immunostained for VGLUT1.
- Quantification of AnkG localization at the AIS region (control, n = 82; shMTCL1, n = 66) (see also Appendix Fig S3). Data represent mean ± SEM. *P < 0.001, compared with control (Fisher's exact test).
C‐terminal MTBD is required for AnkG localization and axonal polarity in Purkinje cells
We observed transient localization of MTCL1 to AIS MT bundles in early postnatal Purkinje cells (Fig 3D). In addition, recent studies have suggested a link between AIS MT bundles and AnkG localization (Leterrier et al, 2011; Jones et al, 2014; van Beuningen et al, 2015). Taken together, our data strongly suggest that MTCL1 maintains AnkG localization by promoting formation of stable MT bundles via MT‐regulating activity. To validate this, we next tested the necessity of the MT‐regulating activity of MTCL1 in promoting AIS development. For this purpose, we prepared a V5‐tagged, RNAi‐resistant MTCL1 construct and its deletion mutants lacking N‐terminal and/or C‐terminal MTBD (Fig 5A) and examined their rescue capability on the MTCL1 knockdown effect. To ensure simultaneous expression of MTCL1 shRNA, GFP, and one of the MTCL1 constructs in a single cell, all of the corresponding DNA fragments were cloned into a single vector and introduced by IUE (see Materials and Methods).
Subcellular localization of each V5‐MTCL1 construct under suppression of endogenous MTCL1 expression is shown (Fig 5B). In Purkinje cells at 3 weeks of age, preferential concentration of ectopic V5‐MTCL1 WT was observed at the axon hillock and proximal part of the AIS region (Fig 5B, brackets). The MTCL1 mutant lacking N‐terminal MTBD (ΔN) still localized to the AIS region, but exhibited a broader distribution throughout the entire AIS region. In addition, this mutant was excluded from the axon hillock and proximal end of the axon (Fig 5B). In contrast, mutants lacking C‐terminal MTBD (ΔKR and ΔN/KR) lost the ability to localize to the AIS region and distribute evenly within the cell body (Fig 5B). These results indicate that C‐terminal MTBD is essential for MTCL1 localization to the AIS region, whereas N‐terminal MTBD plays a modifying but not indispensable localization role. As already mentioned, localization of V5‐MTCL1‐WT to the hillock and proximal part of the AIS region was clearly detected when using anti‐V5 antibody, but not anti‐MTCL1 antibody, whose epitope is located at the central region of MTCL1 (Fig 5B; T. Satake and A. Suzuki, our unpublished observations). This may imply that not only both MTBDs, but also the central region (including the Par‐1b binding region; Fig 1B), is involved in MTCL1 localization to the hillock and proximal AIS.
We also examined the rescue activity of each mutant on morphology and AnkG localization to the AIS region (Fig 5C and D). V5‐MTCL1 WT expression completely abolished Type 3 AIS (lacking AnkG localization) and significantly increased cells with normal Type 1 AIS (Fig 5C and D). Detachment of AnkG‐positive regions from the cell body was also largely suppressed (Fig 5D and Appendix Fig S3D). Likewise, V5‐MTCL1 ΔN expression reduced the number of Type 3 AIS (Fig 5C and D), although it did not show enough activity to increase Type 1 AIS similar to WT levels (Fig 5C). On the other hand, V5‐MTCL1 ΔKR and ΔN/KR had no substantial activity in reducing Type 3 AIS (Fig 5C and D). All mutants suppressed detachment of AnkG‐positive regions from the cell body, but with significantly weaker activity than WT (Fig 5D and Appendix Fig S3D). These results indicate that: (i) C‐terminal MTBD is indispensable for AnkG localization to the AIS region and subsequent maintenance of axonal polarity; and (ii) N‐terminal MTBD is not required for AnkG localization, but contributes to normal morphology of the AIS region. As for reduced length of AnkG‐positive regions, even expression of V5‐MTCL1 WT did not exhibit a rescue effect (Appendix Fig S3E), suggesting that an appropriate amount of MTCL1 and/or the presence of other splicing variant(s) of MTCL1 are required to regulate this aspect of AIS development.
MTCL1 dysfunction causes disorganization of stable MT bundles within the AIS
To confirm the importance of the MT‐regulating activity of MTCL1 in AIS development, we examined AIS MT arrays in cells lacking normal MTCL1 expression. In GT mice, Purkinje cells developed apparently normal AIS MT bundles at 1 week of age (Appendix Fig S4A). However, at 4 weeks of age, when abnormal AIS morphology is prominent, AIS MT bundles became looser compared with WT mice and split toward the distal region (Fig 6A, arrowheads). Similar disorganization of AIS MT bundles was also observed at 3 weeks of age in shMTCL1‐transfected Purkinje cells (Fig 6B). These results were supported by the ultrastructural analysis shown in Fig 6C and D. Consistent with the immunofluorescence data, MTs extended from the axon hillock, gathered into bundles, and were then funneled into the proximal AIS in both WT and GT cells (Fig 6C, arrowheads). In the distal AIS, continuous MTs were readily observed beneath the plasma membrane in single longitudinal sections of WT cells (Fig 6D, left panel, arrowheads). However, such MTs were rarely observed in corresponding sections for GT cells (Fig 6D‐i and ii, arrowheads). In these cells, short MTs were sometimes observed, but they disappeared shortly after, and the contiguous part of MTs reappeared in neighboring sections (Fig 6D‐i and ii, arrowhead with asterisk). This suggests that AIS MT bundles are distorted in GT mice. Taken together, these observations are consistent with the notion that the MT‐regulating activity of MTCL1 is required for formation of straight AIS MT bundles running parallel to the longitudinal axonal axis.
Figure 6. MT bundles in the AIS region are disorganized in Purkinje cells of GT mice and shMTCL1‐transfected cells.
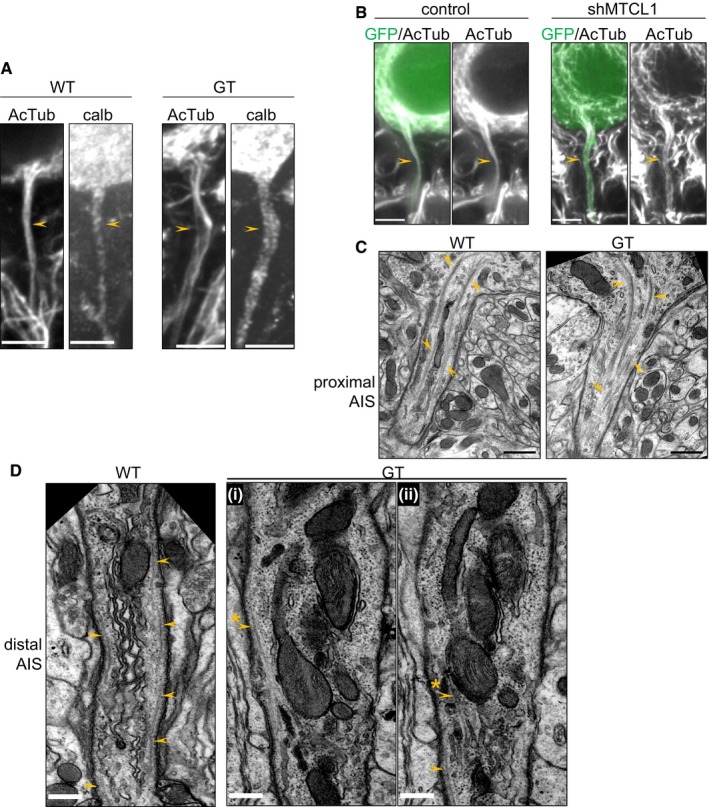
-
ACerebellar sections from WT and GT mice at 4 weeks of age were immunostained for acetylated tubulin (AcTub) and calbindin. Arrowheads represent the AIS region.
-
BControl or shMTCL1‐transfected Purkinje cells were immunostained for AcTub. Arrowheads represent the AIS region.
-
C, DElectron micrographs of the AIS region in Purkinje cells from WT and mutant mice at 4 weeks of age. Processes that extend from the bottom of the Purkinje cell body were recognized as the proximal AIS region (C), and those associated with the beginning of myelination were recognized as the distal AIS region (D) (see also Appendix Fig S4B). Arrowheads indicate MTs. Image (D‐ii) is the adjacent section of (D‐i). Arrowheads with asterisks represent continuous MTs between adjacent sections.
Conditional knockout of MTCL1 in Purkinje cells also results in abnormal motor coordination with gradual AIS impairment
Finally, we analyzed Mtcl1 conditional knockout (cKO) mice (Appendix Fig S1F), in which the Mtcl1 gene is disrupted under control of Cre recombinase driven by a Purkinje cell‐specific promoter, Purkinje cell protein 2 (Pcp2). Consistent with the fact that Pcp2‐Cre is activated around 1 week of age (Barski et al, 2000), MTCL1 expression was observed in Purkinje cells at 1 week, but had significantly reduced at 3 weeks and 2 months of age (Fig 7A). Accordingly, cKO mice began to exhibit abnormal motor coordination at 3–4 weeks of age (Movie EV2), indicating that specific reduction of MTCL1 expression in Purkinje cells is sufficient to cause abnormal motor coordination.
Figure 7. Purkinje cell‐specific Mtcl1 gene disruption after initial AnkG localization impairs correct developmental alteration of AnkG localization length.
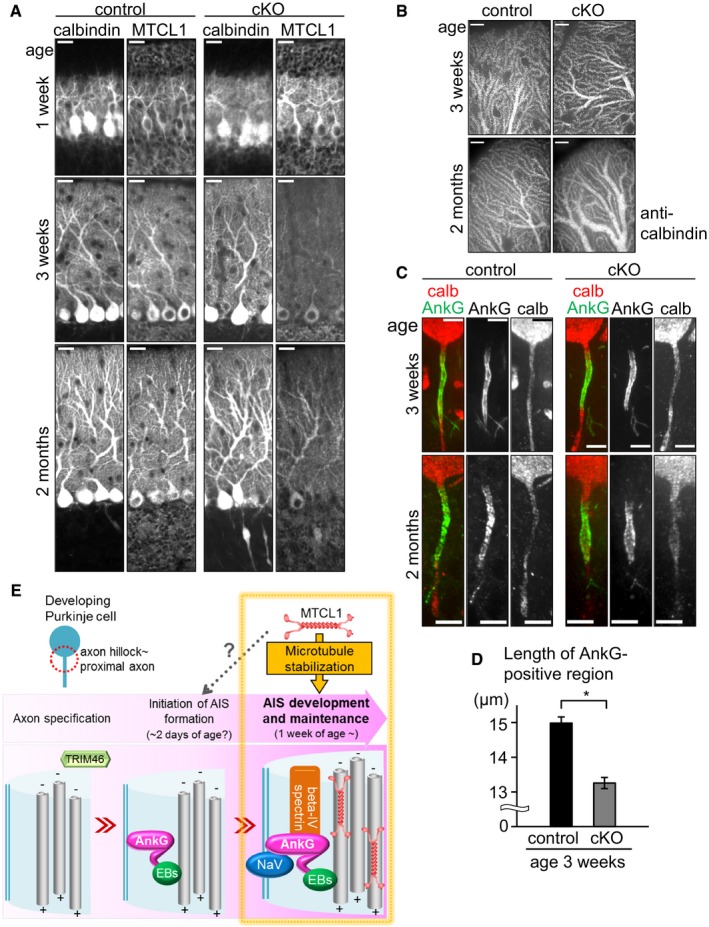
- Cerebellar sections from control and cKO mice at 1 and 3 weeks, and 2 months of age were immunostained for calbindin and MTCL1.
- Representative distal dendrites of Purkinje cells in control and cKO mice.
- Cerebellar sections of control and cKO mice at 3 weeks of age were immunostained for calbindin and AnkG.
- Measurement of the length of AnkG localized regions in control and cKO mice at 3 weeks of age (control, n = 42; shMTCL1, n = 45). Data represent mean ± SEM. *P < 0.001, compared with control (Student's t‐test).
- A model for the role of MTCL1 in AIS development and maintenance in Purkinje cells.
Similar to GT mice, Purkinje cells in cKO mice showed axonal swellings, dilation of distal dendrites, and irregular AIS morphology. However, these defects were detected at 2 months of age (Fig 7A–C), and not obvious at 3 weeks of age when mice start to show abnormal motor coordination (Fig 7A–C). The only defect detected at 3 weeks of age was a substantial reduction of the length of AnkG‐positive regions compared with control mice (Fig 7C and D). As previously observed, AnkG‐positive regions had already appeared in Purkinje cells at 1 week of age, and gradually completed their morphological development (Fig 3D–F). Therefore, the above results suggest that gradual loss of MTCL1 expression after initial AnkG localization affects postnatal AIS development and ultimately causes gross disorder and degeneration of Purkinje cells. Because cKO mice show clear abnormal motor coordination at 3–4 weeks of age, our present results suggest that subtle disturbances of the AIS at this age critically impair Purkinje cell function and cerebellar motor coordination activity.
C‐terminal MTBD point mutation detected in spinocerebellar ataxia patients
Spinocerebellar ataxia (SCA) is a clinically and genetically heterogeneous group of neurological disorders primarily characterized by imbalance, progressive gait and limb ataxia, and dysarthria (Klockgether, 2008; Sun et al, 2016). To examine the possible involvement of MTCL1 gene mutations in relevant human diseases, we performed targeted resequencing of MTCL1 in 167 patients with SCA, as well as 13 patients with multiple system atrophy and one with cerebellar cortical atrophy, diseases that can both also manifest ataxia (see Appendix Supplementary Methods). We identified two SCA patients in one family (Fig 8A) with a MTCL1 variant in which evolutionarily conserved Val1435 within the C‐terminal MTBD was changed to a methionine in a heterozygous state (Fig 8B and C). This V1435M variation was predicted to be pathogenic by three in silico prediction tools, and not registered in ~68,000 normal exomes data in an appropriate website and our in‐house 575 Japanese exomes data (Table 1). In addition, one patient (II‐2) was negative for the nucleotide repeat expansions associated with SCA1, 2, 3, 6, 7, 12, and 17, and dentatorubral–pallidoluysian atrophy. Notably, substitution of Val1435 to Met in human MTCL1 significantly reduced MT stabilization activity of the C‐terminal MTBD fragment in vitro (Fig 8D). Furthermore, mouse MTCL1 containing the corresponding mutation (V1748M) showed substantially lower activity than WT in restoring abnormal AIS region morphology in MTCL1‐depleted Purkinje cells (Fig 5). Taken together, these results suggest the possibility that this MTCL1 mutation is involved in pathogenesis of SCA or related human diseases.
Figure 8. Point mutation in C‐terminal MTBD in spinocerebellar ataxia patients.
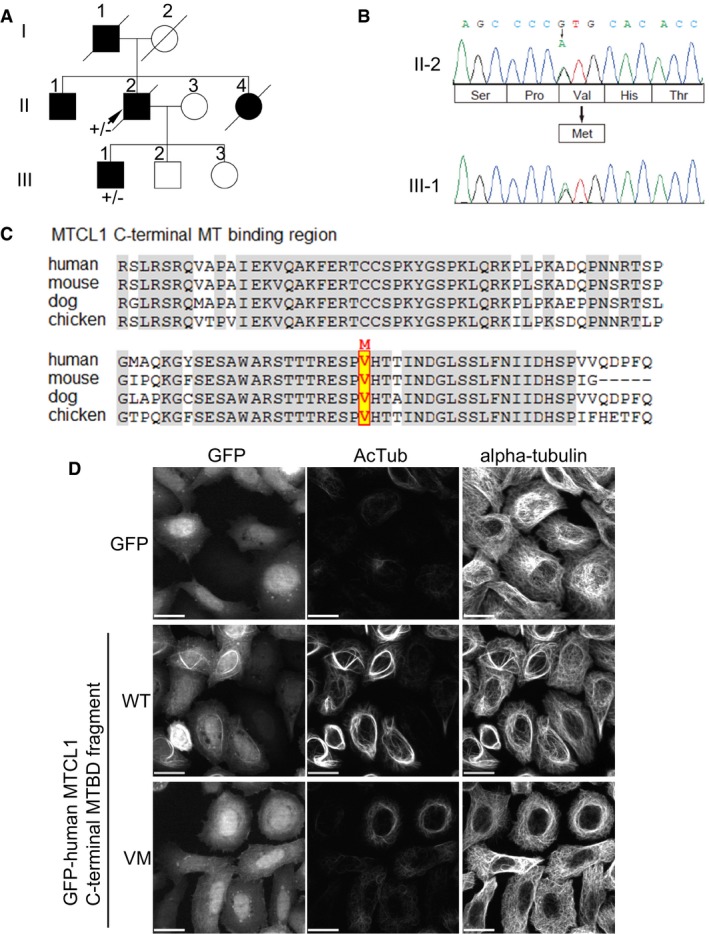
- Familial pedigree of a patient with a MTCL1 variant. II‐2 was subjected to targeted resequencing of MTCL1, as well as whole‐exome sequencing (arrow). Boxes indicate males and circles indicate females. Black indicates affected individuals.
- Electropherogram of c.4303G>A (p.Val1435Met) in MTCL1. II‐2 and III‐1 both carry this variant in a heterozygous state.
- Amino acid conservation of C‐terminal MTBD in MTCL1 between the species is indicated. Valine corresponding to V1435 in human MTCL1 is highlighted.
- The fragment corresponding to C‐terminal MTBD of MTCL1 was expressed in HeLa‐K cells as a GFP‐fusion protein, and its MT‐stabilizing and bundling activity examined. MT stabilization was estimated by signal intensity of acetylated tubulin (AcTub) staining. Scale bars: 20 μm.
Table 1.
Pathogenic prediction of the p.V1435M mutation
| Computational prediction of pathogenicity on p.V1435Ma | Allele frequency in control exome databaseb | ||||
|---|---|---|---|---|---|
| PolyPhen2 (HumVar) | SIFT | MutationTaster | HGVD | ESP6500 | ExAc (all) |
| 1 (pathogenic) | 0 (pathogenic) | Disease causing (score 0.999) | 0 | 0 | 0 |
The V1435M mutation was predicted to be damaging by the following web tools: PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and MutationTaster (http://www.mutationtaster.org/).
Allele frequencies in the following databases are shown: Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and Exome Aggregation Consortium Browser (http://exac.broadinstitute.org/). The number of control exomes showing V1435M variation is reported.
Discussion
In this study, we have shown that MTCL1 is highly expressed in cerebellar Purkinje cells, and disruption of the Mtcl1 gene results in abnormal motor coordination and degeneration of Purkinje cells without any macroscopic brain defects. We also show that acute knockdown of MTCL1 in nascent developing Purkinje cells severely impairs AnkG localization to the AIS region. These findings are consistent with our observation that Mtcl1 mutant mice show defects in normal postnatal development of AIS morphology as the earliest sign of Purkinje cell abnormalities. Taken together, our results demonstrate that MTCL1 plays a critical role in cerebellar function by regulating AIS development in Purkinje cells.
MTCL1 regulates AnkG localization and axonal polarity via MT stabilization activity
Here, we demonstrate that normal AnkG localization to the proximal axon of Purkinje cells requires the C‐terminal MTBD of MTCL1, which has a characteristic MT‐stabilizing activity (Sato et al, 2014). We reveal here that MTCL1 localizes to the proximal region of AIS MT bundles from the early postnatal stage and these AIS MT bundles are disorganized in Purkinje cells lacking normal MTCL1 expression (Fig 6). Considering that MT stabilization is frequently coupled with formation of tight MT bundles (Chapin et al, 1991; MacRae, 1992), these results indicate that MTCL1 plays an essential role in AnkG localization by stabilizing MTs (Fig 7E). This is consistent with the finding that AIS MT bundles are highly stabilized (Tapia et al, 2010; Sanchez‐Ponce et al, 2011). Then, how does the MTCL1‐mediated formation of stabilized MT bundles maintain AnkG localization to the proximal axon? An important clue for this question was provided by a recent study demonstrating that the C‐terminal tail domain of 480AnkG, the longest AnkG isoform, specifically interacts with EB1 or 3, which accumulate on AIS MT bundles, and this interaction is essential for normal AnkG localization at the proximal axon (Freal et al, 2016; Fig 7E). Considering that EB proteins preferentially bind to GTP‐MTs than to GDP‐MTs (Dimitrov et al, 2008; Zanic et al, 2009; Maurer et al, 2011; Nakata et al, 2011), MTCL1 may indirectly facilitate EB1/3 recruitment to AIS MT bundles by increasing the abundance of GTP‐MT lattice which is more stable than GDP‐MT lattice.
In mice, AnkG localization to the proximal axon is observed in the majority of Purkinje cells (82.7%) 2 days after birth (T. Satake and A. Suzuki, our unpublished observations). As we did not examine the effect of MTCL1 depletion at this stage, our present results do not show an indispensable role for MTCL1 in initial targeting of AnkG to the proximal axon. The role of MTCL1 at this stage should be clarified in future studies (Fig 7E).
Role of MT cross‐linking activity of MTCL1
MTCL1 has another MTBD within its N‐terminus, which does not stabilize MTs by itself, but instead cross‐links them with the aid of the adjacent coiled‐coil motifs (Fig 1B; Sato et al, 2013). Here, we demonstrate that this region is not essential for AnkG localization, but is required for appropriately localizing the AnkG‐positive region adjacent to the cell body. Interestingly, N‐terminal MTBD is required for MTCL1 localization to the axon hillock and proximal axonal end, but not to the AIS region. These results suggest that N‐terminal MTBD‐mediated MT cross‐linking occurs not in the AIS but in the cell body, where AIS MT bundles originate, and this MT cross‐linking is essential for AIS positioning close to the soma.
With regard to MT cross‐linking factors in the AIS, a strong candidate was recently reported that localizes to the proximal axon partly overlapping the AIS, and induces MT bundles in vitro with a strikingly similar appearance to AIS MT bundles. Interestingly, this protein, tripartite motif containing 46 (TRIM46), is required for neuronal polarity and axon specification by promoting formation of parallel MT arrays with their plus‐end exposed (van Beuningen et al, 2015). In contrast, MTCL1 is not required for axon specification in Purkinje cells because its depletion in nascent Purkinje cells has little effect on axon formation and overall cell morphology (Fig 4A; Miyata et al, 2010). Because initial AnkG localization to the future AIS region occurs after axon specification (Jenkins & Bennett, 2001; Hedstrom et al, 2007; Galiano et al, 2012; Kevenaar & Hoogenraad, 2015), this suggests the intriguing possibility that TRIM46 works first in formation of uniformly orientated MT arrays for axon specification and subsequent initiation of AIS formation, while in the later stages, MTCL1 works in the stabilization of MT bundles to maintain AnkG localization at the proximal axon (Fig 7E).
MTCL1 is expressed not only in Purkinje cells, but also in other neuronal cells (Appendix Fig S5). Thus, it will be intriguing to determine whether MTCL1 plays a general role in AIS development in other neurons. From our examination, abnormalities of AnkG localization and AIS morphology were not detected in cortical and hippocampal neurons in GT mice at 3 weeks of age (Appendix Fig S5). This suggests that MTCL1 mainly plays a role in AIS morphology in Purkinje cells. The MTCL1 paralog (registered as SOGA) may compensate for MTCL1 dysfunction in other neuronal types. Alternatively, characteristic features of Purkinje cells (e.g., their size) may necessitate stronger support from stabilized MTs to stably maintain the AIS.
The possible role of MTCL1 in selective vesicle transport
The AIS is proposed to function as a selective filter for cytoplasmic organelles, which is essential for establishing axonal polarity. Therefore, striking loss of axonal polarity in MTCL1‐knockdown Purkinje cells may be sufficiently explained by a defective AIS lacking AnkG localization. Nevertheless, a recent study proposed that polarized sorting of cytoplasmic organelles in neurons occurs at a region more proximal than the AIS, which is located at the axon hillock, and named the “pre‐axonal exclusion zone (PAEZ)” (Farias et al, 2015). In this zone, organelles are suggested to selectively attach to MT motors, each of which is appropriately targeted to the correct destination. Notably, AIS MT bundles extend from the axon hillock at which MTCL1 shows colocalization with these bundles (Fig 3C). Considering that one of the axonal MT motors, kinesin‐1, preferentially interacts with stabilized MTs (Hammond et al, 2010; Nakata et al, 2011), the intriguing possibility is raised that MTCL1 regulates neuronal polarity not only by promoting AIS development but also by producing specific MT tracks that show preferential interaction with certain motors. In this regard, it is noteworthy that MTCL1 interacts with the Golgi membrane in cultured cells and works at cross‐linking and stabilizing Golgi‐derived MTs to facilitate Golgi‐ribbon formation and polarized vesicle transport (Sato et al, 2014). Interestingly, it has been shown that the cross‐linked MT bundles are extended from Golgi area of cell body to the AIS in goldfish Purkinje cells (Matsumura & Kohno, 1991). Consequently, one intriguing question that remains to be solved in the near future is the relationship between AIS MTs and Golgi‐derived MTs, both of which are highly cross‐linked and stabilized.
MTCL1 plays a crucial function in motor coordination
Purkinje cells in MTCL1 GT and cKO mice exhibit milder AIS phenotypes than MTCL1 knockdown cells acutely induced at E11.5 by IUE: Specifically, they show neither loss of AnkG localization nor detachment of the AnkG‐positive region from the cell body. However, Purkinje cells in both mice strains commonly exhibit an abnormal AIS morphology as the earliest defect, which is associated with disorganization of AIS MT bundles in GT mice. This indicates that reduced MTCL1 activity for developing stabilized AIS MT bundles differentially results in distinct AIS phenotypes depending on the different context of each experimental system. In genetically modified mice, disorganized AIS MTs may retain the ability to maintain AIS localization of AnkG, but not to functionally support postnatal development of a slender AIS region of an appropriate length and width. The MTCL1 paralog (registered as SOGA) may compensate for loss of MTCL1 function in mutant mice to some extent because it shows MT stabilization activity and cerebellar expression similar to MTCL1 (Sato et al, 2013) (A. Suzuki and M. Miki, our unpublished observations). In any case, what is most striking is that MTCL1 cKO mice exhibit abnormal motor coordination at 3–4 weeks of age, while Purkinje cells only show changes in length of the AnkG‐positive region at this stage. We cannot completely exclude the possibility that other signs of abnormalities were missed in our analysis. However, our results suggest that slight disturbance to MTCL1‐mediated AIS development is highly crucial for cerebellar function, which in turn is important for motor coordination.
Additionally, we identified a rare variation of the evolutionarily conserved Val1435 to Met MTCL1 mutation in SCA patients, which significantly affects MT‐stabilizing and bundling activity of C‐terminal MTBD in vitro. As for in vivo MTCL1 function in Purkinje cells, the V1435M variant was more functional than deletion mutants lacking the MT‐binding region. Nevertheless, the variant exhibited significantly lower activity than WT for normal morphological development of the AIS region in Purkinje cells. Therefore, our results suggest that MTCL1 dysfunction is involved in the symptoms of the affected patient, at least as one important genetic effect. We believe that further work on MTCL1 will provide novel insight into the underlying mechanisms of this human neurodegenerative disease.
Materials and Methods
Animals
All mice used in this study were maintained and handled in accordance with the Institutional Animal Care and Use Committees at Yokohama City University, Medical Life Science and RIKEN Kobe Branch. Embryonic stem (ES) cells from Mtcl1 gene‐trap mice (AT0556 generated by the Sanger Institute Gene Trap Resource, Cambridge, UK) were obtained from the Mutant Mouse Regional Resource Center (MMRRC, Davis, CA, USA). For the conditional knockout strategy, exon 1 in the Mtcl1 locus was flanked by two loxP sites, which were excised upon Cre recombinase excision. Further details are found in the Appendix Supplementary Methods. Homozygous Mtcl1 gene‐trap mice were always compared with wild‐type littermates, and Mtcl1 (flox/flox) with Pcp2‐Cre (cKO) mice were compared with Mtcl1 (flox/flox) without Pcp2‐Cre (control) littermates. Both male and female mice were used equally in all experiments.
Immunostaining of mouse cerebellar sections
Mouse cerebellar sections were immunostained and imaged. A detailed description of the procedures can be found in the Appendix Supplementary Methods. Acquired images were processed using ImageJ software (NIH, Bethesda, MD, USA) to appropriately adjust the brightness and contrast.
Antibodies
For Western blotting, the following antibodies were used: anti‐MTCL1 rabbit pAb (1:250, W19; Santa Cruz, Dallas, TX, USA), anti‐beta‐galactosidase rabbit pAb (1:500, ab616; Abcam, Cambridge, UK), anti‐alpha‐tubulin mouse mAb (1:5,000, DM1A; Sigma‐Aldrich, St. Louis, MO, USA).
For immunostaining, the following antibodies were used: anti‐MTCL1 rabbit pAb (1:250, W19; Santa Cruz), anti‐calbindin rabbit pAb (1:1,000, AB1778), and anti‐NeuN (1:500, MAB377) (Merck Millipore, Darmstadt, Germany); anti‐calbindin mouse mAb (1:1,000, C9848), anti‐MAP2 mouse mAb (1:500, M9942), and anti‐acetylated tubulin mouse mAb (1:1,000, T6793) (Sigma‐Aldrich); anti‐AnkG mouse mAb (1:500, N106/36) and anti‐Nav1.6 mouse mAb TC‐sup (1:5, K87A/10) (NeuroMab, Davis, CA, USA); anti‐GluRD2 rabbit pAb (1:1,000, Rb‐Af1200), anti‐VGLUT1 rabbit pAb (1:1,000, Rb‐Af500), anti‐VGLUT2 rabbit pAb (1:1,000, Rb‐Af720), and anti‐VGAT rabbit pAb (1:1,000, Rb‐Af500) (Frontier Institute, Hokkaido, Japan); and anti‐V5 mouse mAb (1:1,000, R960CUS; Thermo Fisher Scientific, Waltham, MA, USA).
Mouse tissue lysate preparation and Western blotting
Equal protein amounts (25 μg) of various tissue lysates were separated by SDS–PAGE and analyzed by Western blotting, according to standard protocols. A detailed description of the procedures can be found in the Appendix Supplementary Methods.
In utero electroporation (IUE)
Pregnant ICR mice (SLC, Shizuoka, Japan) at E11.5 were used for IUE. Further details can be found in the Appendix Supplementary Methods.
Short hairpin RNA (shRNA) expression vectors
Expression vectors for control shRNA (5′‐GAATATAGAAGAAGACTAG‐3′) and mouse MTCL1 shRNA (5′‐GAATTCCATAGCAACGACT‐3′) were constructed. A detailed description of the procedures can be found in the Appendix Supplementary Methods.
Expression vectors
For rescue experiments using V5‐tagged mouse MTCL1 constructs (V5‐MTCL1/pCAG‐GS) (Sato et al, 2013), five silent mutations were introduced into the corresponding MTCL1 shRNA target site to generate the resulting sequence: 5′‐GAATTCCACTCCAATGATT‐3′. Underlined nucleotides represent the mutated sites. Next, DNA fragments containing a gfp/neo cassette and shRNA expression cassette from the pEB‐Super‐gfp vector (Sato et al, 2013) were cloned into the AvrII site of the V5‐MTCL1/pCAG‐GS vector.
Transmission electron microscopy examination
A detailed description of the procedures can be found in the Appendix Supplementary Methods.
Target resequencing of MTCL1 in SCA patients
Experimental protocols were approved by the Institutional Review Board of Yokohama City University School of Medicine. Written informed consent was obtained from all the patients or their parents. A detailed description of the procedures can be found in the Appendix Supplementary Methods.
Author contributions
TS performed the main parts of the experiments and wrote the manuscript. KYa generated GT mice and performed the first analysis of abnormal motor coordination. KH performed the first immunohistochemical analysis. SM, HD, and NM performed exome analysis. MT‐N performed electron microscopy. YF and GS generated flox mice. EM, YHT, and MY provided advice on in utero electroporation procedures. KYo and HY collected and evaluated the clinical data. AS supervised the research and wrote manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Review Process File
Acknowledgements
The authors thank Hirokazu Hirai and Akira Iizuka for comments; Sandy Chen for her skilled technical assistance; and Aya Ogino for her assistance in the early days of the study. This research was supported by KAKENHI (to A.S.) (Grant Numbers 21116004 and 15K15069), and the Special Coordination Fund for Promoting Science and Technology “Creation and Innovation Centers for Advanced Interdisciplinary” (to A.S.) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, and partly supported by a grant for 2013 Strategic Research Promotion (No. IR2504) of Yokohama City University, Japan (to A.S.). The research was also partially supported by The Naito Foundation (to A.S.).
The EMBO Journal (2017) 36: 1227–1242
References
- Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ (2004) Ankyrin‐based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell 119: 257–272 [DOI] [PubMed] [Google Scholar]
- Baas PW, Deitch JS, Black MM, Banker GA (1988) Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci USA 85: 8335–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, Meyer M (2000) Cre recombinase expression in cerebellar Purkinje cells. Genesis 28: 93–98 [PubMed] [Google Scholar]
- van Beuningen SF, Will L, Harterink M, Chazeau A, van Battum EY, Frias CP, Franker MA, Katrukha EA, Stucchi R, Vocking K, Antunes AT, Slenders L, Doulkeridou S, Sillevis Smitt P, Altelaar AF, Post JA, Akhmanova A, Pasterkamp RJ, Kapitein LC, de Graaff E et al (2015) TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88: 1208–1226 [DOI] [PubMed] [Google Scholar]
- Chapin SJ, Bulinski JC, Gundersen GG (1991) Microtubule bundling in cells. Nature 349: 24 [DOI] [PubMed] [Google Scholar]
- Coleman M (2005) Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 6: 889–898 [DOI] [PubMed] [Google Scholar]
- Dimitrov A, Quesnoit M, Moutel S, Cantaloube I, Pous C, Perez F (2008) Detection of GTP‐tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science 322: 1353–1356 [DOI] [PubMed] [Google Scholar]
- Farias GG, Guardia CM, Britt DJ, Guo X, Bonifacino JS (2015) Sorting of dendritic and axonal vesicles at the pre‐axonal exclusion zone. Cell Rep 13: 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freal A, Fassier C, Le Bras B, Bullier E, De Gois S, Hazan J, Hoogenraad CC, Couraud F (2016) Cooperative interactions between 480 kDa Ankyrin‐G and EB proteins assemble the axon initial segment. J Neurosci 36: 4421–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN (2012) A distal axonal cytoskeleton forms an intra‐axonal boundary that controls axon initial segment assembly. Cell 149: 1125–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J (2010) Building and maintaining the axon initial segment. Curr Opin Neurobiol 20: 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ (2010) Posttranslational modifications of tubulin and the polarized transport of kinesin‐1 in neurons. Mol Biol Cell 21: 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Yoshida T, Sakimura K, Mishina M, Watanabe M, Kano M (2009) Influence of parallel fiber‐Purkinje cell synapse formation on postnatal development of climbing fiber‐Purkinje cell synapses in the cerebellum. Neuroscience 162: 601–611 [DOI] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN (2007) Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol 178: 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN (2008) AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol 183: 635–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2001) Ankyrin‐G coordinates assembly of the spectrin‐based membrane skeleton, voltage‐gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155: 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, Korobova F, Svitkina T (2014) Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J Cell Biol 205: 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapfhammer JP (2004) Cellular and molecular control of dendritic growth and development of cerebellar Purkinje cells. Prog Histochem Cytochem 39: 131–182 [DOI] [PubMed] [Google Scholar]
- Kevenaar JT, Hoogenraad CC (2015) The axonal cytoskeleton: from organization to function. Front Mol Neurosci 8: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T (2008) The clinical diagnosis of autosomal dominant spinocerebellar ataxias. Cerebellum 7: 101–105 [DOI] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ (2008) Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci 11: 178–186 [DOI] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ (2012) Signal processing in the axon initial segment. Neuron 73: 235–247 [DOI] [PubMed] [Google Scholar]
- Komada M, Soriano P (2002) [Beta]IV‐spectrin regulates sodium channel clustering through ankyrin‐G at axon initial segments and nodes of Ranvier. J Cell Biol 156: 337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H (2012) Structural tuning and plasticity of the axon initial segment in auditory neurons. J Physiol 590: 5571–5579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers M, van de Willige D, Freal A, Chazeau A, Franker MA, Hofenk J, Rodrigues RJ, Kapitein LC, Akhmanova A, Jaarsma D, Hoogenraad CC (2016) Dynein regulator NDEL1 controls polarized cargo transport at the axon initial segment. Neuron 89: 461–471 [DOI] [PubMed] [Google Scholar]
- Leterrier C, Vacher H, Fache MP, d'Ortoli SA, Castets F, Autillo‐Touati A, Dargent B (2011) End‐binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc Natl Acad Sci USA 108: 8826–8831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Dargent B (2014) No Pasaran! Role of the axon initial segment in the regulation of protein transport and the maintenance of axonal identity. Semin Cell Dev Biol 27: 44–51 [DOI] [PubMed] [Google Scholar]
- MacRae TH (1992) Microtubule organization by cross‐linking and bundling proteins. Biochim Biophys Acta 1160: 145–155 [DOI] [PubMed] [Google Scholar]
- Matsumura A, Kohno K (1991) Microtubule bundles in fish cerebellar Purkinje cells. Anat Embryol (Berl) 183: 105–110 [DOI] [PubMed] [Google Scholar]
- Maurer SP, Bieling P, Cope J, Hoenger A, Surrey T (2011) GTPgammaS microtubules mimic the growing microtubule end structure recognized by end‐binding proteins (EBs). Proc Natl Acad Sci USA 108: 3988–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Ono Y, Okamoto M, Masaoka M, Sakakibara A, Kawaguchi A, Hashimoto M, Ogawa M (2010) Migration, early axonogenesis, and Reelin‐dependent layer‐forming behavior of early/posterior‐born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev 5: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Niwa S, Okada Y, Perez F, Hirokawa N (2011) Preferential binding of a kinesin‐1 motor to GTP‐tubulin‐rich microtubules underlies polarized vesicle transport. J Cell Biol 194: 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Hayashi Y, Nomura T, Miura E, Kakegawa W, Yuzaki M (2012) Selective and regulated gene expression in murine Purkinje cells by in utero electroporation. Eur J Neurosci 36: 2867–2876 [DOI] [PubMed] [Google Scholar]
- Palay SL, Sotelo C, Peters A, Orkand PM (1968) The axon hillock and the initial segment. J Cell Biol 38: 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Proskauer CC, Kaiserman‐Abramof IR (1968) The small pyramidal neuron of the rat cerebral cortex. The axon hillock and initial segment. J Cell Biol 39: 604–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN (2010) The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci 11: 552–562 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Ponce D, Munoz A, Garrido JJ (2011) Casein kinase 2 and microtubules control axon initial segment formation. Mol Cell Neurosci 46: 222–234 [DOI] [PubMed] [Google Scholar]
- Sato Y, Akitsu M, Amano Y, Yamashita K, Ide M, Shimada K, Yamashita A, Hirano H, Arakawa N, Maki T, Hayashi I, Ohno S, Suzuki A (2013) The novel PAR‐1‐binding protein MTCL1 has crucial roles in organizing microtubules in polarizing epithelial cells. J Cell Sci 126: 4671–4683 [DOI] [PubMed] [Google Scholar]
- Sato Y, Hayashi K, Amano Y, Takahashi M, Yonemura S, Hayashi I, Hirose H, Ohno S, Suzuki A (2014) MTCL1 crosslinks and stabilizes non‐centrosomal microtubules on the Golgi membrane. Nat Commun 5: 5266 [DOI] [PubMed] [Google Scholar]
- Sobotzik JM, Sie JM, Politi C, Del Turco D, Bennett V, Deller T, Schultz C (2009) AnkyrinG is required to maintain axo‐dendritic polarity in vivo . Proc Natl Acad Sci USA 106: 17564–17569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM (2009) A selective filter for cytoplasmic transport at the axon initial segment. Cell 136: 1148–1160 [DOI] [PubMed] [Google Scholar]
- Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, Johnson GV, Brady ST (2013) Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron 78: 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YM, Lu C, Wu ZY (2016) Spinocerebellar ataxia: relationship between phenotype and genotype – a review. Clin Genet 90: 305–314 [DOI] [PubMed] [Google Scholar]
- Tapia M, Wandosell F, Garrido JJ (2010) Impaired function of HDAC6 slows down axonal growth and interferes with axon initial segment development. PLoS ONE 5: e12908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A (2006) Roles of voltage‐dependent sodium channels in neuronal development, pain, and neurodegeneration. J Pharmacol Sci 102: 253–268 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Al‐Bassam S, Miyazaki Y, Wandless TJ, Webster P, Arnold DB (2012) Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins. Cell Rep 2: 1546–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ogawa Y, Hedstrom KL, Rasband MN (2007) betaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol 176: 509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW, van Beuningen SF, Cunha‐Ferreira I, Cloin BM, van Battum EY, Will L, Schatzle P, Tas RP, van Krugten J, Katrukha EA, Jiang K, Wulf PS, Mikhaylova M, Harterink M, Pasterkamp RJ, Akhmanova A, Kapitein LC, Hoogenraad CC (2014) Microtubule minus‐end binding protein CAMSAP2 controls axon specification and dendrite development. Neuron 82: 1058–1073 [DOI] [PubMed] [Google Scholar]
- Zanic M, Stear JH, Hyman AA, Howard J (2009) EB1 recognizes the nucleotide state of tubulin in the microtubule lattice. PLoS ONE 4: e7585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V (1998) AnkyrinG is required for clustering of voltage‐gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143: 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta B, Desmazieres A, Rinaldi A, Tait S, Sherman DL, Nolan MF, Brophy PJ (2011) A critical role for Neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron 69: 945–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Movie EV2
Review Process File