

Key Clinical Message
Alveolar capillary dysplasia (ACD) is a rare condition with variable presentation and clinical course. Clinicians should consider this diagnosis in neonates presenting with nonlethal congenital gastrointestinal malformation, a period of well‐being after birth then unremitting hypoxemia and refractory pulmonary hypertension. Lung biopsy and FOXF1 gene testing may help in diagnosis.
Keywords: Alveolar capillary dysplasia, FOXF1 gene, gastrointestinal malformation, misaligned pulmonary vein, pulmonary hypertension
Introduction
Alveolar capillary dysplasia (ACD) is a fatal developmental anomaly resulting in “misalignment” of the pulmonary veins (MPV) and abnormal intrapulmonary shunting 1. Most patients with ACD have multiple associated anomalies, predominantly of the gastrointestinal tract, but cardiovascular, urogenital, and musculoskeletal system involvement has also been described 2. Infants with ACD generally become critically unwell in the first few days of life with pulmonary hypertension, but presentation in later life has also been reported 3, 4, 5. Currently available therapies are mostly ineffective in reversing hypoxemic respiratory failure. However, a few studies have suggested some therapeutic benefits of pulmonary vasodilators with a single case report of a long‐term survivor 6.
The precise etiology of ACD remains unclear, but recent literature suggests that FOXF1 gene mutations are present in some cases 7, 8. In this report, we describe the clinical outcomes of three infants that illustrate the extensive clinical spectrum and pathophysiology of this disorder. All three infants succumbed to respiratory failure and in two, a pathogenic heterozygous FOXF1 gene mutation was detected. We compare our cases to reports in the literature, especially in atypical cases and provide a comprehensive update on this condition.
The Cases
Case 1
A female infant, the second child of a healthy nonconsanguineous Caucasian couple with a previous term healthy infant, was born to a mother with a history of depression and mild temporal lobe epilepsy who was treated with escitalopram in early pregnancy. Prenatal ultrasound scans at 19 weeks showed a fetal right‐sided abdomino‐pelvic cystic mass, initially thought to be ovarian in origin and which increased in size on sequential scans. The infant was born by a spontaneous vaginal delivery at 38+5 weeks’ gestation with a birthweight of 3340 g. Her Apgar score at 5 min was 9. The infant was admitted to the nursery for further management and investigation of the intra‐abdominal lesion and at admission, the infant's pulse oximetry (Spo2) readings were consistently >95%. An abdominal X‐ray showed a gas filled bowel loop in the right abdomen with a paucity of distal bowel gas and normal lung fields.
At 12 h of age, the infant developed hypoxemia requiring supplemental oxygen. She deteriorated rapidly and required mechanical ventilation by 22 h of age. Pulmonary hypertension was confirmed on 2D‐echocardiography with a structurally normal heart. Therapy was quickly escalated to high‐frequency ventilation, inhaled nitric oxide up to 20 ppm and inotropic support, including milrinone. Preductal SpO2 remained persistently below 90% after intubation, and there was no response to exogenous surfactant. She continued to deteriorate with profound hypoxemia, hypercapnia, metabolic acidosis, and hypotension. Extracorporeal membrane oxygenation (ECMO) was considered and discussed with the parents, but in view of the critical situation, a palliative care plan was instituted and the infant died at 47 h of age 9.
At autopsy, lung microscopy showed diffuse ACD with MPV (Figure 1). The alveolar septa were thickened with several thin walled capillaries that were not opposed to the epithelium. Dilated veins accompanying the arteries and bronchi in the bronchovascular bundles were noted. Electron microscopy showed normal surfactant bodies. A duodenal volvulus and an aganglionic distal colon with the transition zone placed about 20 cm from the anorectal junction were also present. Sequence analysis of a DNA sample identified a novel heterozygous FOXF1 nonsense mutation in exon 1(c.668C>A); p.Ser223Ter 7.
Figure 1.
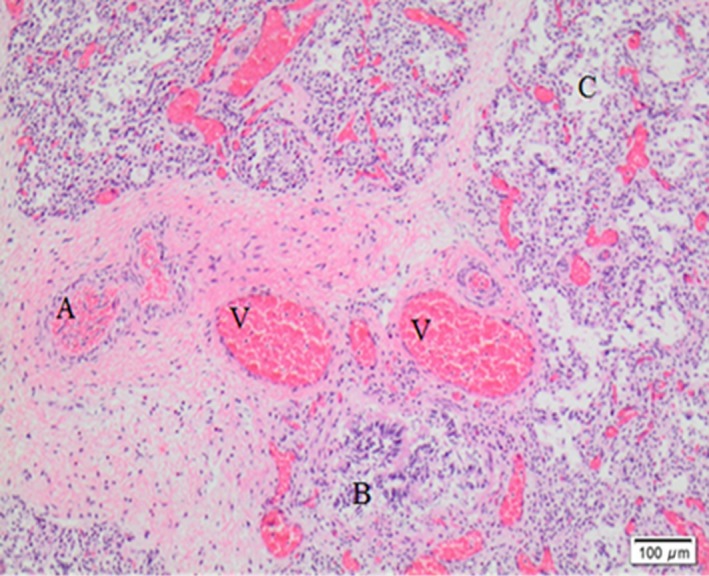
Lung tissue histology – abnormally located veins adjacent to arteries and bronchioles. Dilated capillaries are not in contact with the alveolar epithelium. A, Arteries; V, veins; B, bronchioles; C, alveolar space between thickened alveolar septa.
Case 2
This infant was the second child of a mother with polycystic ovarian syndrome. Her other child was born at term and was well. The pregnancy was complicated by gestational diabetes mellitus. Duodenal atresia was suspected on an 18‐week ultrasound scan and polyhydramnios subsequently developed. She was delivered by elective cesarean section at 38 weeks with a birthweight of 3470 g and 5‐min Apgar score of 9. The first postnatal abdominal X‐ray was suggestive of duodenal atresia, and an imperforate anus was noted on clinical examination. Cardiorespiratory status was normal, and initial SpO2 readings were consistently >95%.
On day 2, she was electively intubated and placed on mechanical ventilation in preparation for abdominal surgery. Congenital adhesions causing duodenal obstruction were noted, and a sigmoid colostomy was formed. She was extubated the next day and continued for 15 days without respiratory difficulties. On day 15, a second laparotomy was performed for a duodenoplasty and Bishop‐Kerr jejunostomy as the infant had persistent feeding difficulties secondary to a dysfunctional and dilated duodenum. She was then extubated uneventfully again to room air on day 17.
On day 22, she developed a urinary tract infection (UTI) from E. coli and required supplemental oxygen by high flow nasal cannula until day 30. On day 41, she became profoundly unwell with suspected necrotising enterocolitis. High flow nasal oxygen (FiO2 0.3) was recommenced with empiric antibiotics (meropenem and vancomycin) but respiratory deterioration continued and she required mechanical ventilation on day 43. Chest X‐ray showed persistent bilateral perihilar opacities and hyperinflated lung fields. There was no response to exogenous surfactant. High‐frequency ventilation and nitric oxide were commenced for pulmonary hypertension (confirmed on 2D‐echocardiography, which also showed a small secundum atrial septal defect). Empirical antifungal treatment was given with minimal response. She developed bilateral pneumothoraces requiring treatment with chest drains. Hypoxemia remained persistent, and the infant passed away on day 48 despite intensive care.
At autopsy, lung microscopy revealed patchy ACD with MPV (Figure 2). Abnormally sited and patchily‐distributed pulmonary veins were seen. However, some pulmonary veins were also appropriately positioned within the interalveolar septa. The alveolar septa were thickened with several thin walled nonapposed capillaries. Cytomegalovirus (CMV) infection without an inflammatory response in the lungs was diagnosed by identification of viral inclusions and confirmation on immunohistochemical staining and molecular testing. Sequence analysis of a DNA sample identified a novel “de novo” heterozygous FOXF1 missense mutation at exon 1(c.260G>T); p.Gly87Val.
Figure 2.
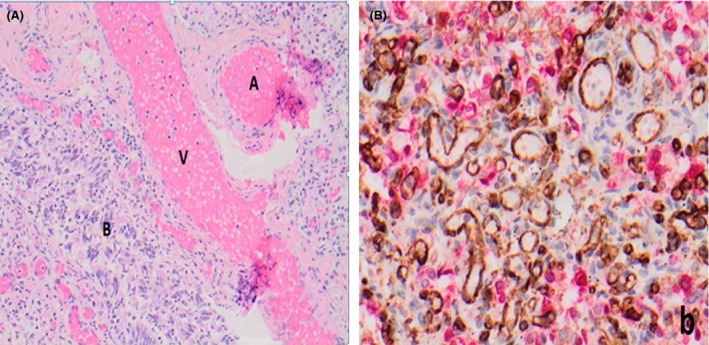
(A) Lung tissue histology – abnormally located veins adjacent to arteries and bronchioles. A, Arteries; V, veins; B, bronchioles. (B) Immunohistochemically dual stained for CD31 (brown, endothelial marker) and cytokeratin (red; epithelial cell marker) demonstrating variability in arrangement and size of alveolar capillaries.
Case 3
This male infant was the first child of a healthy nonconsanguineous Caucasian couple, who had previously had six miscarriages. Prenatal ultrasound scans showed a fetal omphalocele and duodenal atresia. This pregnancy was complicated by polyhydramnios. Amnio‐reductions were performed twice for maternal comfort. Preterm labor and rupture of membranes occurred at 34 weeks’ gestation. She proceeded to cesarean section in view of a nonreassuring fetal trace and clinical chorioamnionitis at 36+6 weeks’ gestation. Antenatal steroids and antibiotics were given prior to delivery.
The infant had a small omphalocele and a birthweight of 2985 g. His Apgar score at 5 min was 9. The cord blood gas was normal, and there was meconium staining of the amniotic fluid. From birth, he required supplemental oxygen to keep SpO2 >90%. A small left‐sided spontaneous pneumothorax was noted on X‐ray and managed conservatively. An abdominal X‐ray showed a “double bubble” sign suggestive of duodenal atresia. Antibiotic therapy (penicillin, gentamicin, and metronidazole) was started as prophylaxis.
The infant subsequently deteriorated at 24 h of age. He remained persistently hypoxemic despite mechanical ventilation, 100% oxygen, exogenous surfactant, inhaled nitric oxide (pulmonary hypertension was confirmed on echocardiography with a structurally normal heart), high‐frequency ventilation, and inotropic support. In view of the critical situation, a palliative care plan was discussed with parents and instituted. The infant died at 45 h of age.
At autopsy, lung microscopy showed generalized ACD (Figure 3) and MPV. There was also evidence of meconium aspiration with numerous keratinocytes within alveolar spaces. Duodenal atresia with a mesenteric band causing malrotation of ascending and mid‐transverse colon was also noted. The ileum, cecum, and appendix were located within the omphalocele which showed autolytic changes but were otherwise unremarkable. Genetic studies were not undertaken.
Figure 3.
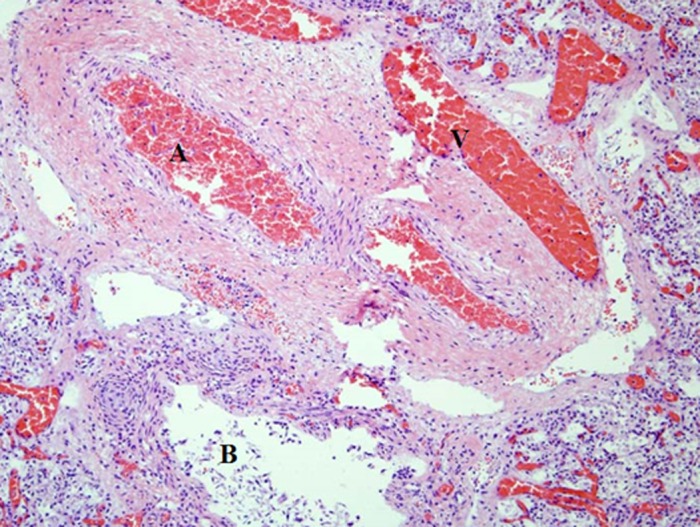
Lung tissue histology – abnormally located vein adjacent to arteries and bronchioles. Dilated capillaries are not in contact with the alveolar epithelium. A, Arteries; V, veins; B, bronchioles.
Discussion
Alveolar capillary dysplasia was first described in 1981 and is almost invariably lethal 1. The exact incidence of the disease is unclear as post mortems or lung biopsies are not routinely performed in deceased infants. In confirmed cases of ACD, affected infants are usually born at term with normal Apgar scores 2. Symptom progression depends on the extent of lung involvement. Most patients present within 24 h of life with respiratory distress and hypoxemia but as illustrated by case two and other patients, presentation may be delayed and can be as late as 7 months of age if pulmonary involvement is patchy 5 (Table 1).
Table 1.
Review of literature on ACD with relevant gastrointestinal malformation (intestinal malrotation, HSCR) and FOX gene mutation
| Case report/Case series | Age of onset | Age of survival | Sex | Gastrointestinal malformation | Histopathological finding | Genetic testing |
|---|---|---|---|---|---|---|
|
Boggs et al. (1994) 4
Abdallah (1993) 10 Reported the case of sibling |
Day 1 5 week |
Day 14 5 weeks and 2 days |
Male Sister |
Nil reported |
Generalized ACD Patchy ACD |
Not tested |
|
Sen et al. (2004) 1
Case series of 30 cases |
One case of onset Day 1 Rest had onset in first 2 days of life |
Survival till day 113
Died in neonatal period |
Female Overall up to 56% were Females |
HSCR, Intestinal malrotation |
Generalized ACD Generalized ACD |
BMPR2, EMAPII gene tested but no sequence change was noted |
| Michalsky et al. (2005) 27 | Day 1 | 7 days | Female | Omphalocele | Generalized ACD | Not tested |
| Shehata et al. (2005) 14 | Day 1 | 3 months | Male | Meckel's diverticulum | Generalized ACD | Trisomy 21 |
| Shankar et al. (2006) 13 | 7 weeks | 4 months | Female | HSCR disease |
1st lung biopsy inconclusive 2nd lung biopsy showed Patchy ACD |
Not tested |
| Danhaive et al. (2008) 12 | At Birth | 8 months | Sex not reported | Nil reported | Generalized ACD | ABCA3 mutation |
| Ahmed et al. (2008) 11 | 7 month of age | 7 months and 3 weeks | Female | Nil reported | Patchy to generalized ACD with few normally apposed capillaries | Not tested. |
|
Antano et al. (2006) 18
3 cases reported |
Day 2 | Up to 2 weeks of age | 2 males and 1 female |
Intestinal malrotation HSCR |
Generalized ACD | Not tested |
|
Stankiewicz et al. (2009) 7
6 cases with FOX genes mutation in 16q24.1 cluster (FOXF1, FOXC2 and FOXL1) and ACD |
Day 1 |
Day 1 Day 40 Day 15 Day 25 Day 20 Day 13 |
Female Male Female Female Female Female |
Intestinal malrotation, annular pancreas, duodenal stenosis | Generalized ACD | FOXF1 mutation |
| YY Chee et al. (2010) 25 | Day 1 | Day 2 | Male | Omphalocele and intestinal malrotation | Generalized ACD | Not tested |
| Yu et al. (2010) 26 | Day 1 | Day 3 | Male | Intestinal malrotation | Generalized ACD | FOXF1, FOXC2, FOXL1, IRF8, MTHFSD deletion |
| Yoshihiko Kodama et al. (2012) 5 | Day 1 | About 8 months (237 days) | Female | Intestinal malrotation, DORV | Generalized ACD | Not tested |
|
Sen et al. (2013) 8
53 cases. These included the cases retrieved retrospectively which possibly included some of previously reported cases |
Age of onset was not reported | 4 cases of survival beyond neonatal period‐ up to 50–120 days |
3 Females and 1 male Among longer survivals Overall there were up to 60% Female |
8 cases with intestinal malrotation, one case of HSCR with imperforate anus and intestinal malrotation | Nature of ACD on histopathology not stated in individual patient | FOXF1 code sequencing with 37 (70%) cases showing mutations including nonsense, missense and frameshift mutation and upstream mutations |
| Yukie Ito et al. (2014) 6 | 3 months of age | 36 months | Male | Nil reported | Patchy ACD | Frameshift mutation c.899Tdel, p.L300RfsX79, in the first exon of FOXF1 |
| Current case series |
Day 1 Day 22 Day 1 |
Day 3 Day 48 Day 2 |
Female Female Male |
HSCR, duodenal volvulus Imperforate anus with congenital adhesions Duodenal atresia, intestinal malrotation omphalocele |
Generalized ACD Patchy ACD Generalized ACD |
De novo FOXF1 nonsense mutation at exon 1 (c.668C>A)
FOXF1 missense mutation at exon 1(c.260G>T) parental testing not available Not tested |
Only cases of ACD and MPV confirmed on lung histology are included with long‐term survivors are in bold. ACD, alveolar capillary dysplasia; MPV, misaligned pulmonary veins; HSCR, Hirschsprung's disease.
Most infants with ACD die from hypoxemic respiratory failure within the first 2 weeks of life. There are eight cases in which long‐term survival beyond the neonatal period have been described and one infant survived for 36 months 4, 5, 6, 10, 11, 12, 13, 14. In this case series, two infants with generalized lung involvement presented within the first 2 days of life and died the day after. In the infant with patchy lung involvement from our cases, respiratory symptoms arose at 3 weeks of age and death occurred much later, at 3 months of age.
The pathophysiology of ACD is not fully defined, and the mechanism to explain its delayed presentation is far more unclear. The findings of Melly et al. 15 suggest that variations in disease severity and timing of presentation correspond to alterations in capillary density and contact with the alveolar epithelium or to better lobular development and patchy involvement 4, 16, 17. Case 3 in this series would support this proposition, where a later clinical presentation was accompanied histologically by alveolar capillaries appropriately juxtaposed in many areas to the alveolar epithelium.
Histological findings have only been characterized from symptomatic cases, and the evolution of ACD in asymptomatic patients has not been ascertained in humans. Triggers of acute deterioration signaling the onset of fulminant disease also need clarification. These triggers may initiate constriction of hypertrophied pulmonary arteries leading to pulmonary hypertensive crises 16 and may include various infections, such as viral upper respiratory tract infections or as in case 2, a urine tract infection.
Initial diagnosis may be difficult as common investigations such as X‐rays are often noninformative. Currently, histopathology is the gold standard for the diagnosis, but lung tissue may also be difficult to obtain ante‐mortem if there is no surgical expertise or if the infant is very unwell. Furthermore, selective lung biopsy may not capture patchy involvement 13. Typical histological findings include evidence of misaligned pulmonary veins (anomalous distended pulmonary veins within the bronchovascular bundles instead of the interlobular septa) representing the intrapulmonary shunts, paucity of capillaries proximal to the alveolar epithelium, medial thickening of small pulmonary arteries (reflecting pulmonary hypertension), and immature alveolar development 15.
An index of suspicion of ACD should be entertained if there are extrapulmonary anomalies in an infant with deteriorating respiratory status as these have been noted in up to 80% of infants with ACD 18. Gastrointestinal malformations, such as Hirschsprung disease and omphalocele, are rare but a recognized association with ACD (Table 1). These associations suggest that the molecular pathophysiological factors underlying ACD may be affected by a common genetic pathway, for example, an embryonic mesenchymal migration defect 19.
Mutations of the FOXF1 gene causing ACD were first described in 2009 7 and have been detected in up to 55–70% of ACD cases 8, 20 (Table 1). The FOXF1 gene is located at chromosome 16q24.1, and the gene consists of two exons, a DNA binding “Forkhead box” domain (DBD), and the cell‐type‐specific activation domain coded by exon 1 and the general activation domain coded by exon 2. Most (90%) cases are sporadic and are a result of de novo FOXF1 mutations. Rare familial recurrences are described on the basis of a maternal mutation on the paternal imprinted allele, which is not expressed. ACD occurs when this mutated allele is maternally transmitted 7, 8. Therefore, females may be carriers of a FOXF1 mutation on their paternal allele and have no clinical features. This is a very important group to identify as the recurrence risk is high (50%). In contrast, recurrence risk in de novo cases is ~1% based on the possibility of gonadal mosaicism in one of the parents.
FOXF1 is involved in mesenchymal – epithelial interactions during development of the gastrointestinal tract and lungs as it is expressed in splanchnic mesenchyme closely opposed to endoderm 20. It was proposed that FOXF1 point mutations were associated with bowel malrotation (as in one of our cases), while FOXF1 microdeletions appear to be linked with hypoplastic left heart syndrome and gastrointestinal atresias caused by involvement of adjacent genes FOXC2 and FOXL17. Cases with mutations in upstream regulators of FOXF1 are also reported, showing variable lung involvement and severity of disease 21, 22. In our patients (Case 1 and 2), a FOXF1 nonsense mutation at exon 1(c.668C>A);p.Ser223Ter and exon 1(c.260G>T); p.Gly87Val were found, which has not been previously reported, adding further to the mutation and phenotypic spectrum of FOXF1‐related disorders. The availability of faster turnaround times for gene testing may allow a genetic diagnosis to support timely clinical decision making.
The clinical presentation of ACD is similar to any infant presenting with pulmonary hypertension, but the response to therapy is often minimal or transient 23, 24. This serves as an initial diagnostic clue. Lung recruitment strategies, such a high‐frequency ventilation or surfactant, are often the first step in clinical practice in view of severe hypoxemia. There are reports of transient improvement with pulmonary vasodilators, but this is usually futile23. Other reports have demonstrated a similar pattern of response to inhaled or intravenous prostacyclin including epoprostenol at doses up to 120 ng/kg/h 5. Extracorporeal membrane oxygenation (ECMO) may temporize death 24, but lung transplantation is currently the only theoretically curative option but no successful cases have so far been reported and no data on long‐term neurodevelopmental outcome and life expectancy are available.
There are emerging data of longer survival of some patients with less severe disease which could help clinicians triage patients for potential lung transplantation. A single long‐term survivor is reported in literature who recovered from his pulmonary hypertension crisis by a combination therapy of inhaled NO, epoprostenol, milrinone, and oral pulmonary vasodilators. The mild phenotype of this case can be explained by unique lung CT and histopathological findings 6 (Table 1).
In any infant with severe hypoxemic respiratory failure, a broad differential diagnosis, especially of treatable conditions, must be considered. These include severe sepsis, surfactant protein deficiency, congenital infections, and other congenital lung malformations that may be amenable to surgery. Infection screens including blood cultures for bacterial and fungal infections and blood tests (PCR) for herpes simplex, toxoplasma, and cytomegalovirus virus DNA must also be performed and were negative in all three of our cases.
In conclusion, the combination of an infant presenting with escalating and intractable respiratory failure and a possible nonpulmonary, gastrointestinal malformation, should alert clinicians to the possibility of ACD after exclusion of other treatable conditions. A lung biopsy is the current gold standard of diagnosis for this disease, but genetic analysis (chromosome microarray looking for 16q24.1 deletion and FOXF1 sequencing and microdeletion screening) may offer rapid confirmation and counseling for future pregnancies.
Conflict of Interest
None declared.
Authorship
DG: Prepared first draft of manuscript, reviewed cases, and literature. JLO: Supervised Dr Goel, revised manuscript, and approved final manuscript to be published. KL: Assisted Dr Goel with data interpretation and approved final manuscript to be published. MW: Revised and approved final manuscript to be published. AWS: Assisted Dr Goel with data acquisition and interpretation and approved final manuscript to be published. DM: Assisted Dr Goel with data acquisition and interpretation and approved final manuscript to be published. AJG: Performed the autopsy on two of the presented cases. Established the diagnosis of ACD/MPV in these cases. Revised manuscript and approved final manuscript to be published. CL: Performed the autopsy on one of the presented cases and established the diagnosis of ACD/MPV, supervised Dr Goel, revised manuscript, and approved final manuscript to be published.
Acknowledgments
We thank the department of Anatomical Pathology, SEALS, Prince of Wales Hospital, NSW, for the technical support. We also thank Dr Megan Dishop (Children's Hospitals and Clinics, Minneapolis, MN) for her histo‐pathological review of Case 2.
References
- 1. Sen, P. , Thakur N., Stockton D. W., Langston C., and Bejjani B. A.. 2004. Expanding the phenotype of alveolar capillary dysplasia (ACD). J Pediatr 145:646–651. [DOI] [PubMed] [Google Scholar]
- 2. Ng, P. C. , Lewindon P. J., Siu Y. K., To K. F., and Wong W.. 1995. Congenital misalignment of pulmonary vessels: an unusual syndrome associated with PPHN. Acta Pædiatr. 84:349–353. [DOI] [PubMed] [Google Scholar]
- 3. Bishop, N. B. , Stankiewicz P., and Steinhorn R. H.. 2011. Alveolar capillary dysplasia. Am J Respir Crit Care Med. 184:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boggs, S. , Harris M. C., Hoffman D. J., Goel R., McDonald‐McGinn D., Langston C., et al. 1994. Misalignment of pulmonary veins with alveolar capillary dysplasia: affected siblings and variable phenotypic expression. J Pediatr. 124:125–128. [DOI] [PubMed] [Google Scholar]
- 5. Kodama, Y. , Tao K., Ishida F., Kawakami T., Tsuchiya K., Ishida K., et al. 2012. Long survival of congenital alveolar capillary dysplasia patient with NO inhalation and epoprostenol: effect of sildenafil, beraprost and bosentan. Pediatrics International: Official Journal of the Japan Pediatric Society 54:923–926. [DOI] [PubMed] [Google Scholar]
- 6. Ito, Y. , Akimoto T., Cho K., Yamada M., Tanino M., Dobata T., et al. 2015. A late presenter and long‐term survivor of alveolar capillary dysplasia with misalignment of the pulmonary veins. Eur.J.Pediatr. 174:1123–1126. [DOI] [PubMed] [Google Scholar]
- 7. Stankiewicz, P. , Sen P., Bhatt S. S., Storer M., Xia Z., Bejjani B. A., et al. Genomic and Genic Deletions of the FOX Gene Cluster on 16q24.1 and Inactivating Mutations of FOXF1 Cause Alveolar Capillary Dysplasia and Other Malformations. Am.J.Hum.Genet. 84:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sen, P. , Yang Y., Navarro C., Silva I., Szafranski P., Kolodziejska K. E., et al. 2013. Novel FOXF1 mutations in sporadic and familial cases of alveolar capillary dysplasia with misaligned pulmonary veins imply a role for its DNA Binding Domain. Hum.Mutat. 34:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goel, D. , Oei J. L., Shand A. W., Mowat D., and Loo C.. 2016. FOXF1 gene mutation in alveolar capillary dysplasia associated with Hirschsprung's disease and clinical review. J Paediatr Child Health. 52:787–788. [DOI] [PubMed] [Google Scholar]
- 10. Abdallah, H. I. , Karmazin N., and Marks L. A.. 1993. Late presentation of misalignment of lung vessels with alveolar capillary dysplasia. Crit Care Med. 21:628–630. [DOI] [PubMed] [Google Scholar]
- 11. Ahmed, S. , Ackerman V., Faught P., and Langston C.. 2008. Profound hypoxemia and pulmonary hypertension in a 7‐month‐old infant: late presentation of alveolar capillary dysplasia. Pediatr Crit Care Med. 9:e43–e46. [DOI] [PubMed] [Google Scholar]
- 12. Danhaive, O. , Peca D., and Boldrini R.. 2008. ABCA3 mutation and pulmonary hypertension: A link with alveolar capillary dysplasia? J. Pediatr. 152:891–892. [DOI] [PubMed] [Google Scholar]
- 13. Shankar, V. , Haque A., Johnson J., and Pietsch J.. 2006. Late presentation of alveolar capillary dysplasia in an infant. Pediatr Crit Care Med. 7:177–179. [DOI] [PubMed] [Google Scholar]
- 14. Shehata, B. M. , and Abramowsky C. R.. 2005. Alveolar Capillary Dysplasia in an Infant With Trisomy 21. Pediatr.Dev.Pathol. 8:696–700. [DOI] [PubMed] [Google Scholar]
- 15. Melly, L. , Sebire N. J., Malone M., and Nicholson A. G.. 2008. Capillary apposition and density in the diagnosis of alveolar capillary dysplasia. Histopathology 53:450–457. [DOI] [PubMed] [Google Scholar]
- 16. Cater, G. , Thibeault D. W., Beatty E. C. Jr, Kilbride H. W., and Huntrakoon M.. 1989. Misalignment of lung vessels and alveolar capillary dysplasia: a cause of persistent pulmonary hypertension. J.Pediatr. 114:293–300. [DOI] [PubMed] [Google Scholar]
- 17. Butler, M. W. , Ursell P. C., Wung J. T., and Stolar C. H.. 1992. Misalignment of pulmonary veins with alveolar capillary dysplasia as a cause of persistent neonatal pulmonary hypertension. Pediatr Res. 31:303. [Google Scholar]
- 18. Antao, B. , Samuel M., Kiely E., Spitz L., and Malone M.. 2006. Congenital alveolar capillary dysplasia and associated gastrointestinal anomalies. Fetal Pediatr.Pathol. 25:137–145. [DOI] [PubMed] [Google Scholar]
- 19. Sirkin, W. , O'Hare B. P., Cox P. N., Perrin D., Cutz E., et al. 1997. Alveolar capillary dysplasia: lung biopsy diagnosis, nitric oxide responsiveness, and bronchial generation count. Pediatr.Pathol.Lab.Med. 17:125–132. [PubMed] [Google Scholar]
- 20. Castilla‐Fernandez, Y. , Copons‐Fernandez C., Jordan‐Lucas R., Linde‐Sillo A., Valenzuela‐Palafoll I., et al. 2013. Alveolar capillary dysplasia with misalignment of pulmonary [corrected] veins: concordance between pathological and molecular diagnosis. J.Perinatol. 33:401–403. [DOI] [PubMed] [Google Scholar]
- 21. Szafranski, P. , Yang Y., Nelson M. U., Bizzarro M. J., Morotti R. A., Langston C., et al. 2013. Novel FOXF1 deep intronic deletion causes lethal lung developmental disorder, alveolar capillary dysplasia with misalignment of pulmonary veins. Hum.Mutat. 34:1467–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parris, T. , Nik A. M., Kotecha S., Langston C., Helou K., Platt C., et al. 2013. Inversion upstream of FOXF1 in a case of lethal alveolar capillary dysplasia with misalignment of pulmonary veins. Am.J.Med.Genet.A. 161:764–770. [DOI] [PubMed] [Google Scholar]
- 23. Steinhorn, R. H. , Cox P. N., Fineman J. R., Finer N. N., Rosenberg E. M., Silver M. M., et al. 1997. Inhaled nitric oxide enhances oxygenation but not survival in infants with alveolar capillary dysplasia. J.Pediatr. 130:417–422. [DOI] [PubMed] [Google Scholar]
- 24. Al‐Hathlol, K. , Phillips S., Seshia M. K., Casiro O., Alvaro R. E., and Rigatto H.. 2000. Alveolar capillary dysplasia. Report of a case of prolonged life without extracorporeal membrane oxygenation (ECMO) and review of the literature. Early Human Dev. 57:85–94. [DOI] [PubMed] [Google Scholar]
- 25. Chee, Y. Y. , Chim S., Beh P., and Wong K. Y.. 2010. Alveolar capillary dysplasia associated with ompahlocoele. Hong Kong J.Pediatr. 15:243–246. [Google Scholar]
- 26. Yu, S. , Shao L., Kilbride H., and Zwick D. L.. 2010. Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am.J.Med.Genet.A. 152a:1257–1262. [DOI] [PubMed] [Google Scholar]
- 27. Michalsky, M. P. , Arca M. J., Groenman F., Hammond S., Tibboel D., and Caniano D. A.. 2005. Alveolar capillary dysplasia: a logical approach to a fatal disease. J.Pediatr.Surg. 40:1100–1105. [DOI] [PubMed] [Google Scholar]