

Abstract
TNF receptor‐1 (TNFR1) and TRAIL death receptors preferentially induce pro‐inflammatory or cytotoxic signaling, respectively, via distinct plasma membrane and cytosolic complexes. New studies identifying the pro‐inflammatory factors TRAF2, RIP, and LUBAC in TRAIL death receptor complexes suggest that the latter are more “TNFR1‐like” than anticipated and argue for revision of prevailing models of spatio‐hierarchical TRAIL‐induced signaling complex assembly.
Subject Categories: Autophagy & Cell Death; Post-translational Modifications, Proteolysis & Proteomics
Tumor necrosis factor (TNF) receptor‐1 (TNFR1) and the TNF‐related apoptosis‐inducing ligand (TRAIL) receptors TRAILR1 and TRAILR2 share the characteristic “death domain” (DD) and make use of basically the same set of signaling proteins to induce apoptotic or necroptotic cell death, but also to engage pro‐inflammatory signaling pathways such as the classical NF‐κB pathway. Interestingly, however, while pro‐inflammatory signaling is dominant in the case of TNFR1, cytotoxic activities are most prominent in TRAIL death receptor signaling. These relative preferences appeared to be due to differences in the localization and hierarchy of assembly of signaling complexes triggering cell death and activation of the pro‐inflammatory IκB kinase (IKK) complex (for review see Siegmund et al, 2016). TNFR1 stimulation triggers assembly of a membrane‐localized complex devoid of cytotoxic activities. This “complex I” contains the DD adapter protein TRADD and the serine/threonine kinase RIP, which recruit complexes of TRAF2 and the E3 ubiquitin ligases cIAP1 or cIAP2, as well as the linear ubiquitin chain assembly complex (LUBAC). RIP modification by these E3 ligases with K63‐linked and M1‐linked (linear) ubiquitin chains creates docking sites for the ubiquitin‐binding domain‐containing subunits of the IKK complex and its activator, the TAB 2‐TAK1 complex, and this way ensures robust and very rapid activation of the classical NF‐κB pathway. In contrast, TNFR1‐induced apoptosis and necroptosis are dependent on receptor internalization and arise from secondarily formed receptor‐free cytosolic protein complexes containing RIP, caspase‐8, RIP3, and (in the case of apoptosis) the DD‐containing adaptor protein FADD. Furthermore, cytotoxic TNFR1 signaling typically depends on sensitizing mechanisms and is thus rarely apparent in otherwise unchallenged cells. On the other hand, TRAIL death receptors seem to act directly as the core of a membrane‐associated apoptosis‐inducing signaling complex containing FADD and caspase‐8, while pro‐inflammatory signaling and necroptotic signaling originate from cytosolic complexes additionally containing RIP, RIP3, and TRAF2.
In this issue of The EMBO Journal, new work from the Walczak group (Lafont et al, 2017) now not only demonstrates that the TNFR1 signaling component LUBAC also contributes to TRAIL‐induced NF‐κB signaling, but also finds LUBAC recruitment via TRAF2 and RIP into plasma membrane‐associated TRAIL death receptor signaling complexes, thus making TRAIL death receptors much more “TNFR1‐like”. This notion is further supported by another new study (Henry & Martin, 2017), also reporting TRAIL death receptor recruitment of RIP; while that work did not directly analyze LUBAC recruitment, this event may be inferred based on their observation of recruitment of A20, another functionally relevant ubiquitin‐binding component of the TNFR1 signaling complex with ubiquitination‐modifying activities.
The new work confirms the broad and general relevance of LUBAC for activation of the classical NF‐κB signaling pathway by various stimuli (for review see Sasaki & Iwai, 2015). These observations also match well with the increasing notion of TRAIL death receptors as inducers of inflammation with context‐dependent tumor‐promoting activity (Trauzold et al, 2006; Hoogwater et al, 2010; Hartwig et al, 2017). Indeed, at the first glance the involvement of LUBAC in TRAIL signaling may seem unsurprising given that TRAF2 and RIP had already been implicated in pro‐inflammatory TRAIL signaling (Lin et al, 2000; Varfolomeev et al, 2005). What clearly is unexpected, however, is that TRAF2 and RIP along with LUBAC are not only found in the cytosolic signaling complex, but also in the plasma membrane‐associated TRAIL death receptor signaling complex, which has been the subject of intense research for approximately two decades. It is thus quite remarkable that it took until now to demonstrate robust recruitment of TRAF2, RIP, and TRAF2/RIP‐associated proteins such as LUBAC or A20 to the TRAIL death receptor signaling complex. The interactions of the TRAIL death receptors with TRAF2, RIP, and LUBAC may be less stable or more dynamic than those of TNFR1, and thus more difficult to detect in immunoprecipitation (IP) experiments; indeed, Lafont et al (2017) frequently find significant amounts of TRAF2, RIP, and LUBAC subunits in negative controls of their TRAIL death receptor IPs, something that is not evident in most published reports on TNFR1 IPs. Overall, the new findings of Lafont et al (2017) and Henry and Martin (2017) again highlight the question why TNFR1 and TRAIL death receptors display a relative preference for inflammatory and cytotoxic signaling, respectively.
Since FADD and caspase‐8 are not recruited to TNFR1, the new results at first glance suggest a simplified consideration of TRAIL death receptors as “TNFR1 copies” with the added ability to recruit the apoptosis‐inducing FADD–caspase‐8 dyad, or vice versa the consideration of TNFR1 as a TRAIL death receptor with lost ability to interact with FADD and caspase‐8. However, this would definitely be a misleading oversimplification because FADD and caspase‐8 deficiency abrogates recruitment of TRAF2, RIP, and LUBAC to TRAIL death receptors as well as TRAIL‐induced NF‐κB activation (Henry & Martin, 2017; Lafont et al, 2017), but has no major effect on TNFR1 signaling complex formation and TNF‐induced NF‐κB signaling. Thus, the results of Lafont et al (2017) and Henry and Martin (2017) suggest that the concept of spatially determined hierarchical activation of apoptosis and pro‐inflammatory signaling by TRAIL death receptors might be replaced by a model in which caspase‐8 activation and TRAF2/RIP‐mediated recruitment of IKK activation‐triggering E3 ligases bifurcate at the receptor level (Fig 1). Notably, while the enzymatic activity of caspase‐8 is crucial for TRAIL‐induced apoptosis, it is dispensable (and even inhibitory) for recruitment of TRAF2, RIP, and LUBAC.
Figure 1. Revising models of TRAIL‐induced formation of cell death and NF‐κB‐inducing signaling complexes.
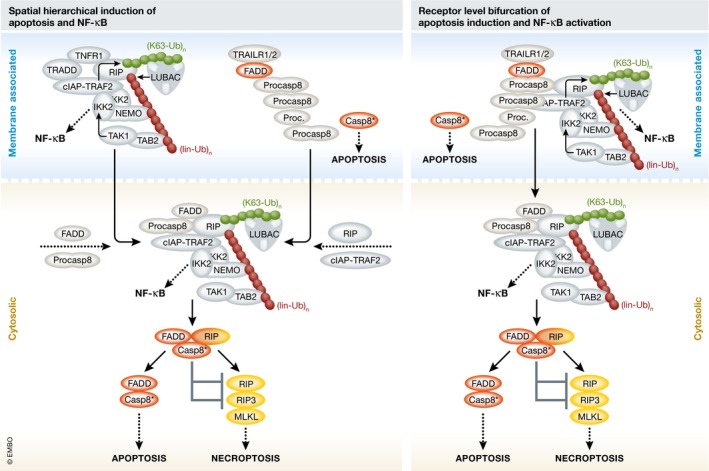
Left, prevalent model based on spatial hierarchy. TRAIL initially stimulates assembly of a membrane‐localized apoptosis‐inducing signaling complex comprising TRAIL death receptors, FADD, and procaspase‐8 filaments. After release into the cytoplasm, the latter two secondarily form an NF‐κB‐activating complex together with RIP and TRAF2. Notably, models of TNFR1 signaling invoke the opposite order of events. Right, revised model with pathway bifurcation at the receptor level. Proteins required for apoptosis induction and NF‐κB activation are concomitantly recruited to the membrane‐localized TRAIL death receptor signaling complex prior to eventual release in the cytoplasm. Although FADD is an essential part of the revised model, it is no obligate component of the cytosolic complex—while FADD‐deficient cells lack cell death induction or NF‐κB activation after TRAIL stimulation, they are still able to activate NF‐κB and necroptosis in response to TNF. The asterisk indicates fully matured heterotetrameric caspase‐8.
FADD has been reported to be, in comparison with caspase‐8, only present in substoichiometric amounts within TRAIL death receptor complexes, serving as a nucleus that promotes filamentous assembly and subsequent trans‐activation of caspase‐8 molecules (Dickens et al, 2012; Fu et al, 2016). Considering the crucial role of caspase‐8 as an adaptor that recruits RIP, TRAF2, and LUBAC to TRAIL death receptors, the question arises whether these molecules then decorate receptor‐associated caspase‐8 filaments, or modify their assembly and/or ability to stimulate caspase‐8 activation. Indeed, Lafont et al (2017) provide evidence that LUBAC restricts TRAIL death receptor‐associated caspase‐8 activation. Since activation of the TRAIL death receptors further results in the appearance of cytosolic complexes containing FADD, caspase‐8, RIP, TRAF2, and LUBAC, it will also be important to clarify whether these complexes are assembled on the expense of TRAIL death receptor‐associated FADD‐nucleated caspase‐8 filaments.
The new studies from Lafont et al (2017) and Henry and Martin (2017) allow us surprising and novel insights into the composition of the TRAIL death receptor signaling complex and put its dual apoptosis‐ and NF‐κB‐stimulating activity into the spotlight. However, these studies also prompt new questions about the mechanisms controlling the quality and quantity of TRAIL activity at the receptor level. This illustrates that even 20 years after the first publications on this topic, we are still far from having a comprehensive picture of the molecular mode of action of TRAIL death receptor signaling complexes, and suggests that there is still much to be discovered.
See also: E Lafont et al (May 2017)
References
- Dickens LS, Boyd RS, Jukes‐Jones R, Hughes MA, Robinson GL, Fairall L, Schwabe JW, Cain K, Macfarlane M (2012) A death effector domain chain DISC model reveals a crucial role for caspase‐8 chain assembly in mediating apoptotic cell death. Mol Cell 47: 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, Srivastava DB, DiMaio F, Penczek PA, Siegel RM, Stacey KJ, Egelman EH, Wu H (2016) Cryo‐EM structure of caspase‐8 tandem DED filament reveals assembly and regulation mechanisms of the death‐inducing signaling complex. Mol Cell 64: 236–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, Taraborrelli L, Draber P, Lafont E, Arce Vargas F, El‐Bahrawy MA, Quezada SA, Walczak H (2017) The TRAIL‐induced cancer secretome promotes a tumor‐supportive immune microenvironment via CCR2. Mol Cell 65: 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CM, Martin SJ (2017) Caspase‐8 acts in a non‐enzymatic role as a scaffold for assembly of a pro‐inflammatory “FADDosome” complex upon TRAIL stimulation. Mol Cell 65: 715–729 [DOI] [PubMed] [Google Scholar]
- Hoogwater FJ, Nijkamp MW, Smakman N, Steller EJ, Emmink BL, Westendorp BF, Raats DA, Sprick MR, Schaefer U, Van Houdt WJ, De Bruijn MT, Schackmann RC, Derksen PW, Medema JP, Walczak H, Borel Rinkes IH, Kranenburg O (2010) Oncogenic K‐Ras turns death receptors into metastasis‐promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 138: 2357–2367 [DOI] [PubMed] [Google Scholar]
- Lafont E, Kantari‐Mimoun C, Draber P, De Miguel D, Hartwig T, Reichert M, Kupka S, Shimizu Y, Taraborrelli L, Spit M, Sprick MR, Walczak H (2017) The linear ubiquitin chain assembly complex regulates TRAIL‐induced gene activation and cell death. EMBO J 36: 1147–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S, Liu ZG (2000) The death domain kinase RIP is essential for TRAIL (Apo2L)‐induced activation of IkappaBkinase and c‐Jun N‐ terminal kinase. Mol Cell Biol 20: 6638–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Iwai K (2015) Roles of linear ubiquitinylation, a crucial regulator of NF‐κB and cell death, in the immune system. Immunol Rev 266: 175–189 [DOI] [PubMed] [Google Scholar]
- Siegmund D, Lang I, Wajant H (2016) Cell death‐independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J doi:10.1111/febs.13968 [DOI] [PubMed] [Google Scholar]
- Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, Emme D, Röder C, Kalthoff H, Wajant H (2006) TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 25: 7434–7439 [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, Ashkenazi A (2005) Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor‐related apoptosis‐inducing ligand. J Biol Chem 280: 40599–40608 [DOI] [PubMed] [Google Scholar]