

Abstract
Control of energy homeostasis and metabolism is achieved by integrating numerous pathways, and miRNAs are involved in this process by regulating expression of multiple target genes. However, relatively little is known about the posttranscriptional processing of miRNAs and a potential role for the precursors they derive from. Here, we demonstrate that mature miRNA‐22 is more abundant in muscle from male mice relative to females and that this enables sex‐specific regulation of muscular lipid metabolism and body weight by repressing estrogen receptor alpha (ERα) expression. We found that the ERα adjusts its own activity by preventing processing of miR‐22 via direct binding to a conserved ERα‐binding element within the primary miR‐22 precursor. Mutation of the ERα binding site within the pri‐miR‐22 in vivo eliminates sex‐specific differences in miR‐22 expression. We reason that the resulting tissue selective negative feedback regulation is essential to establish sex‐specific differences in muscle metabolism and body weight development.
Keywords: ERα, miR‐22, miRNA processing, muscle lipid metabolism, sexual dimorphism
Subject Categories: Metabolism, RNA Biology
Introduction
Metabolic activity is regulated by complex interactions to control energy homeostasis and to support different metabolic needs of specific cell types. Many of these processes are hormonally controlled, and estrogen is known to be one of the key players of metabolic homeostasis as indicated by metabolic changes during menopause or by studies involving genetically engineered animals. Deficiency in estrogen signaling is associated with metabolic dysfunction and increased body weight, and at the level of the organism, this occurs by affecting food intake and changing energy expenditure. However, different tissues, including the central nervous system, skeletal muscle, adipose tissue, and immune cells, respond to estrogen signaling either by inducing systemwide changes or by activation of cell‐autonomous processes (Mauvais‐Jarvis et al, 2013). Skeletal muscle constitutes a large proportion of the body mass and contributes profoundly to energy expenditure at rest and activity (Rolfe & Brown, 1997; Baskin et al, 2015). Estrogen supplementation in males stimulates lipid catabolic effects in muscle (Hamadeh et al, 2005; Salehzadeh et al, 2011), which are mediated by estrogen receptors such as the estrogen receptors alpha and beta (ERα/β). Physiological responses require a balance between ERα and ERβ, but ERα is assumed to be the major effector of estrogen signaling (Barros & Gustafsson, 2011). Most of the actions of ERα are achieved at the transcriptional level via estrogen response elements (EREs), but ERα also exerts important functions via alternative, non‐classical routes (Park et al, 2011). Loss of ERα leads to metabolic imbalance resulting in obesity (Heine et al, 2000; Hewitt et al, 2010). Tissue‐specific mutations of ERα have helped decipher the contribution from different tissues in ERα‐dependent regulation of energy homeostasis, but numerous questions are still open (Ribas et al, 2011; Xu et al, 2011). Specifically in female skeletal muscle, ERα signaling is essential to maintain mitochondrial function and loss of ERα in female muscle leads to increased adiposity (Ribas et al, 2016); however, the molecular machinery that allows sex‐specific control of metabolic responses is not completely understood.
The diversity and importance of non‐protein coding RNAs transcribed from the genome have long been overlooked, and we are just beginning to understand the multiplicity of mechanisms by which ncRNAs regulate gene expression (Rinn & Chang, 2012). Non‐coding RNAs may be categorized according to their function or size, which permits distinction of long non‐coding RNAs (lncRNAs) and small ncRNAs such as miRNAs. lncRNAs are defined as non‐coding RNA molecules of more than 200 bases, are usually encoded by more than one exon, and are often polyadenylated (Derrien et al, 2012). miRNAs are small ncRNAs that are transcribed as larger precursors and are processed to mature ~20‐nt miRNAs. Processing occurs in two consecutive steps (executed by RNase III enzymes) where Drosha cleaves the primary miRNA precursor (pri‐miR) releasing the pre‐miRNA, which is then subsequently exported to the cytoplasm and cleaved by Dicer, forming the mature miRNA form that is loaded on an Argonaute protein to repress target mRNAs (Bernstein et al, 2001; Gregory et al, 2004; Lee et al, 2004; Ha & Kim, 2014). In addition, it is known that RNA‐binding proteins like hnRNP A1 (Guil & Caceres, 2007), KSRP (Trabucchi et al, 2009), or ADAR1 (Yang et al, 2006) can selectively modulate biogenesis of specific miRNAs via interaction with the precursor forms.
Moreover, processing of precursor miRNAs might be modulated by various signaling pathways like SMAD (Davis et al, 2008), p53 (Suzuki et al, 2009), and potentially also by estrogen signaling (Katchy et al, 2012; Klinge, 2012). Effects of miRNAs are exerted by posttranscriptional repression, either reducing mRNA stability and/or inhibiting translation. The partial complementarity of miRNAs to target molecules allows miRNAs to regulate large sets of target genes (Selbach et al, 2008; Guo et al, 2010; Huntzinger & Izaurralde, 2011). Since the biological activity of miRNAs depends on the cellular context, that is, the relative abundance of interacting molecules and turnover or biological activity of miRNA targets in a specific cell type, it is necessary to study the function of miRNAs under physiological conditions in vivo (Boettger & Braun, 2012).
In this study, we describe the regulatory interactions between the miR‐22 precursor, mature miRNA miR‐22, and ERα occurring at the posttranscriptional level, which restrict ERα abundance specifically in male skeletal muscles. We found high concentration of mature miR‐22 in male muscle, and after deletion of miR‐22 in mutant animals, we observed increased expression of the miR‐22 target ERα in male muscle. Increased expression of ERα stimulates fatty acid oxidation (FAO) in muscle, causing a reduction in body weight and WAT stores in male animals, that was reversed by muscle‐specific ablation of ERα. Further analysis revealed that ERα represses processing of the pri‐miR‐22—preferentially in females—by binding to a conserved ERα binding element within the pri‐miR‐22. Accordingly, we found that inactivation of ERα or mutation of the ERα‐binding site in pri‐miR‐22 abolishes the repression of miR‐22 processing. We postulate that miR‐22 and ERα form a regulatory feedback loop, which assures high miR‐22 concentrations and limits ERα abundance in male muscles, thereby restricting FAO.
Results
Sex‐specific differences in miR‐22 abundance regulate body weight in male mice
miRNA‐22 is detected in several tissues of the mouse with the strongest expression seen in skeletal muscle and the heart (Fig 1A). Northern blot analysis confirmed this result revealing high concentrations of miR‐22 in heart and skeletal muscle (Fig EV1A) but only low levels in other tissues. Similarly, we detected strong expression of miR‐22 in human differentiating myoblasts but low expression in other cell lines (Fig EV1B). Muscles of male mice showed significantly higher concentrations of mature miR‐22 compared to female muscles (Fig 1B). In contrast, the pri‐miR‐22 was more abundant in female muscle tissue (Fig 1C), resulting in higher miR‐22/pri‐miR‐22 ratios in male compared to female muscles (Fig 1D). So far, miR‐22 has been associated with different types of cancer and was shown to regulate the cardiac stress response in cardiomyocytes (Gurha et al, 2013; Huang et al, 2013), but a role in the regulation of metabolic processes was not known. Therefore, we genetically inactivated miR‐22 in mice to investigate the role of miR‐22 for the regulation of sex‐specific differences and the metabolism (Fig EV1C and D). Inactivation of miR‐22 was achieved by replacing the miR‐22 coding exon with an IRES‐lacZ‐neomycin resistance cassette, which allowed us to monitor the transcriptional activity of the locus encoding pri‐miR‐22 as a single spliced transcript by β‐galactosidase staining. A survey of different tissues uncovered strong β‐galactosidase activity in the heart and skeletal muscle (Fig 1E–G), but no staining was evident in adipose tissue, liver, kidney, or in pancreas including islets of Langerhans validating our Northern blot and qRT–PCR analysis. Heterozygous as well as homozygous miR‐22 mutant mice were born according to the expected Mendelian ratios, developed normally, were fertile, and showed no changes in survival compared to littermate controls (Fig EV1E and F). However, analysis of body weight revealed an attenuated gain of weight in homozygous male miR‐22 mutants compared to WT animals, which was not observed in female animals (Fig 2A). The reduced body weight was not associated with a reduced size of the animals as determined by tibia length measurements of adult animals (Fig 2B). MRI‐based analysis of body composition (Fig 2C and D) indicated unchanged muscle volume (Fig 2E) but a reduced amount of white adipose tissue (WAT; Fig 2F), which corresponded to reduced serum levels of the fat cell‐derived hormone leptin (Fig EV2A) and reduced cross‐sectional area of fat cells in miR‐22−/− (KO) vs. miR‐22+/+ (WT) animals (Fig EV2B). No evidence for reduced food intake, increased locomotor activity, or changes in the respiratory exchange quotient at day or night was found (Fig EV2C–H). Blood glucose concentration and plasma triglyceride levels were unchanged in mice fed ad libitum as well as in fasted animals. Free fatty acids were unchanged in serum of mice fed ad libitum, however were observed reduced free fatty acids in fasted miR‐22−/− (KO) vs. miR‐22+/+ (WT) animals again in accordance with reduced WAT mass in mutant animals (Fig EV2I–K).
Figure 1. miR‐22 is predominantly expressed in male striated muscle.
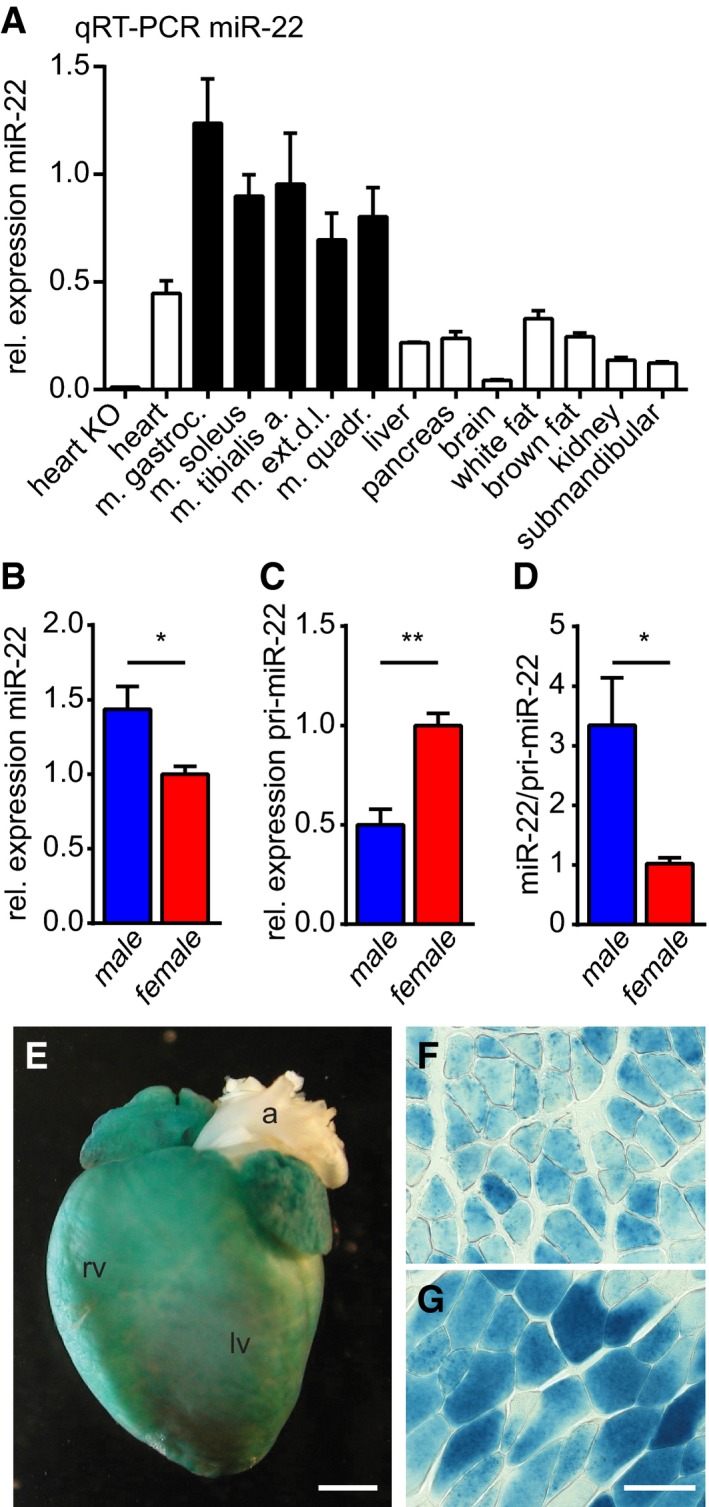
-
AqRT–PCR analysis of miR‐22 expression in mouse tissues indicates high expression of miR‐22 in all striated muscle tissues. qRT–PCR using miR‐22−/− mutant tissue proves the specificity of the assay. These data were confirmed by Northern blot analysis (Fig EV1A), and similarly, miR‐22 expression is induced in differentiating human myoblasts and is much higher than in other established human cell lines (Fig EV1B).
-
B–DHigher amounts of miR‐22 are found in muscle of male compared to female mice (B). In contrast, the pri‐miR‐22 is enriched in female muscle (C) and this results in a higher miR‐22/pri‐miR‐22 ratio in male animals (D; B–D: male, n = 5 pools of three mice; female, n = 5 pools of three mice).
-
E–GX‐Gal staining indicates β‐galactosidase activity under control of the miR‐22 locus in tissues isolated from miR‐22−/− animals. We detected activity of the locus in the adult heart (E; rv: right ventricle, lv: left ventricle, a: aorta) and in cross sections of skeletal muscles like M. soleus (F) or M. tibialis anterior (G). In line with low detection of miR‐22 in qRT–PCR and Northern blot analysis (Fig EV1A), we detected no β‐galactosidase activity in white or brown adipose tissue, liver, or pancreas. The scale bar in (E) corresponds to 2 mm; the scale bar in (G) corresponds to 50 μm in (F) and (G).
Figure EV1. Inactivation of mmu‐miR‐22.
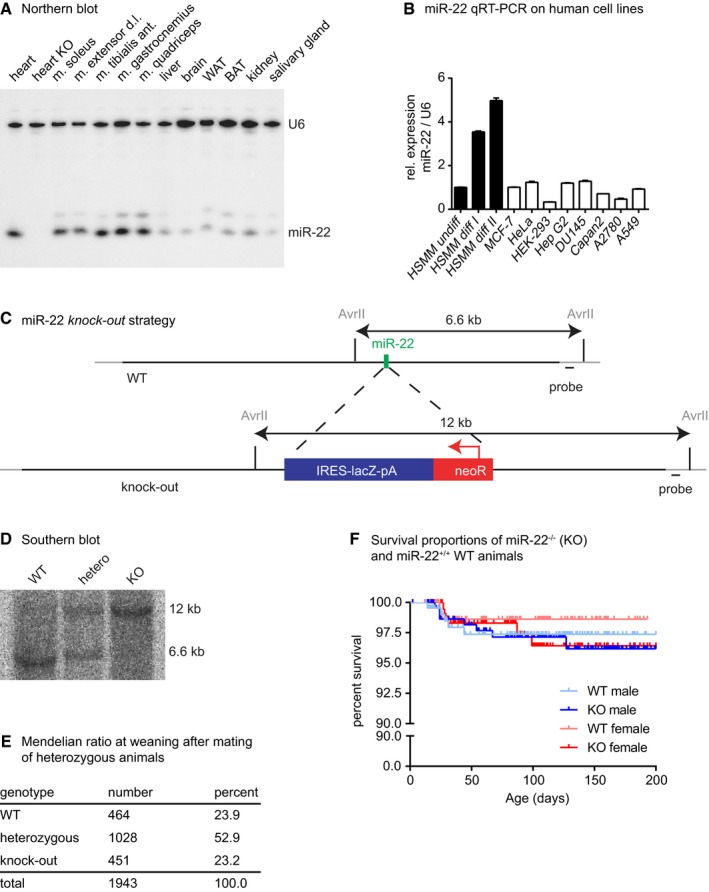
- Northern blot analysis of miR‐22 expression reveals predominant detection of miR‐22 in striated muscle tissue. Loss of the miR‐22 signal in miR‐22−/− (KO) heart proves loss of miR‐22 in KO mice and indicates specificity of the signal detected in the Northern blot.
- miR‐22 is detected in different human cell lines. MiR‐22 expression is low in established human cell lines and in proliferating primary human myoblasts, but is increased in differentiating human myoblasts (HSMM, human skeletal muscle cells and myoblasts; MCF‐7, breast cancer cell line; HeLa, cervical cancer cell line; HEK‐293, human embryonic kidney cell line; Hep G2, human liver cancer cell line; DU145, human prostate cancer cell line; Capan2, pancreatic adenocarcinoma cell line; A2780, human ovary cancer cell line; A549, human lung carcinoma cell line). Data are presented as mean ± SEM (n = 3 technical replicates).
- The miR‐22‐containing region was deleted by replacing a 282‐bp genomic fragment by an IRES‐lacZ‐pA‐PGK‐neoR cassette using GalK‐mediated recombination of a BAC containing genomic DNA covering the miR‐22 locus. This deletion removes the complete exon 2 of the miR‐22‐containing RNA. Exon 1 and exon 3 of the miR‐22 precursor RNA remain intact.
- Southern blot analysis of AvrII‐digested genomic DNA of WT, heterozygous and knock‐out animals confirms the targeting of the miR‐22 locus.
- miR‐22−/− (KO) mice were born at the expected Mendelian ratio and survived the first 2 weeks after birth as demonstrated by genotyping at the time of weaning.
- Survival of miR‐22−/− (KO) male or female mice was monitored for 200 days and was not significantly different from miR‐22+/+ (WT) mice according to the log‐rank (Mantel–Cox) test and the Gehan–Breslow–Wilcoxon test.
Figure 2. miR‐22 is required for normal body weight development in male mice.

-
AGenomic deletion of miR‐22 leads to reduced body weight of mutant male mice (miR‐22+/+, WT, n ≥ 15; miR‐22−/−, KO, n ≥ 17; weight is significantly (*–***) decreased for all time points after 5th week, two‐tailed unpaired t‐test); female animals are not affected (WT, n ≥ 8; KO, n ≥ 8). Data are presented as mean ± SEM.
-
BReduction in body weight in male miR‐22 knock‐out mice is not due to a general reduction in body size as indicated by the unchanged tibia length in adult male animals (WT, n = 20; miR‐22 KO, n = 22; two‐tailed unpaired t‐test). Data are presented as mean ± SEM.
-
C–FBody composition was analyzed by MRI and an ImageJ plug‐in was used to distinguish and color code fat (light blue) and muscle tissue (brown) at consecutive coronary levels of the mouse body (C, D). This classification of fat and muscle tissue is used to quantify the muscle and fat volume of the mouse from ankle (an) to the inguinal lymph node (iLN) in miR‐22+/+ (WT, n = 21) and congenital miR‐22−/− (KO, n = 27) mice. No change in muscle volume is observed (E); however, adipose tissue volume is significantly reduced (F) (P < 0.05, two‐tailed unpaired t‐test). Data are presented as mean ± SEM.
-
G–KGene set enrichment analysis (GSEA) was performed using Affymetrix microarray transcriptome data to identify significantly enriched gene sets in miR‐22 knock‐out skeletal muscle (n = 5 biological replicates) compared to WT skeletal muscle (n = 5 biological replicates). In the C3 miR‐motive gene set collection, the miR‐22 gene set was the only significantly enriched gene set (G). In the C5‐gene ontology gene set collection, only gene sets related to lipid metabolism (H–K) were identified to be significantly enriched (ES: enrichment score; NES: normalized enrichment score; FWER: familywise error rate, FWER control exerts a conservative control over false discovery associated with multiple hypothesis testing).
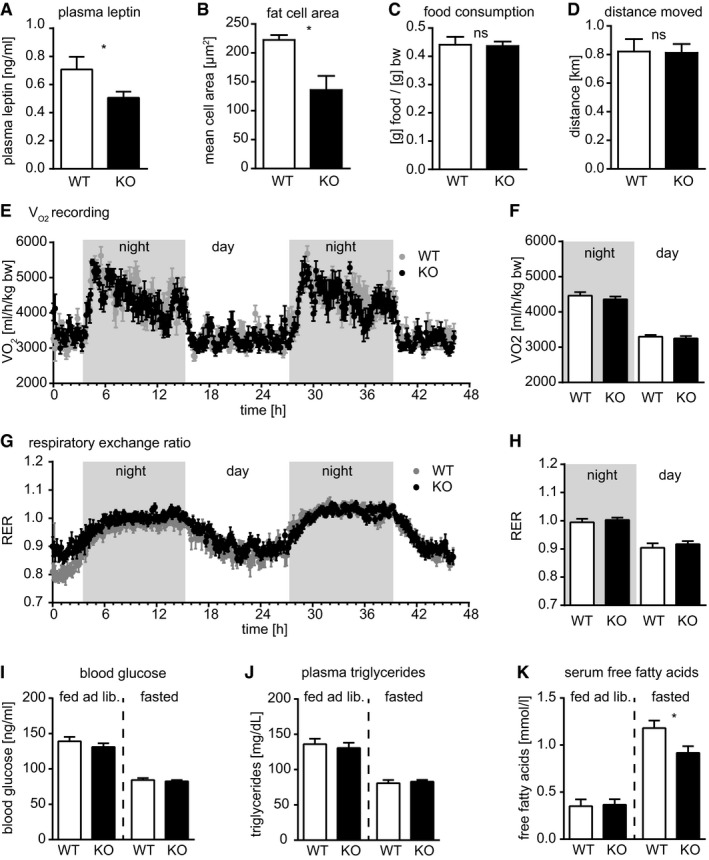
Figure EV2. miR‐22 affects WAT mass and adipocyte diameter in male mice.
-
AIn accordance with reduced white fat mass (Fig 2C–F), we detect reduced plasma leptin levels in the male miR‐22−/− (KO) mice (WT, n = 12; miR‐22 KO, n = 13).
-
BThe reduced fat mass correlates with reduced cross‐sectional area of subcutaneous fat cells in male miR‐22−/− (KO) animals (WT, n = 3; KO, n = 3). Calculation of adipocyte number from cross‐sectional area of adipocytes assuming spherical adipocytes and WAT volume as determined by MRI indicates that the number of adipocytes is not decreased in miR‐22 KO mice.
-
C–HNo differences between miR‐22+/+ (WT) and miR‐22−/− (KO) mice were detected in Phenomaster metabolic cages for (C) food consumption or (D) distance moved. Similarly, we detected no differences in oxygen consumption (E; F, averaged values for day and night) or carbon dioxide production resulting in no differences in respiratory exchange ratio (G; H, averaged values for day and night). Data were recorded for 48 h (WT, n = 8; KO, n = 10).
-
I–KNo differences in blood glucose concentration were detected between miR‐22+/+ (WT) and miR‐22−/− (KO) mice fed ad libitum (WT, n = 13; KO, n = 12), nor in fasted animals (WT, n = 17; KO, n = 16). No differences in plasma triglycerides concentration were detected between miR‐22+/+ (WT) and miR‐22−/− (KO) mice fed ad libitum (WT, n = 12; KO, n = 12), nor in fasted animals (WT, n = 9; KO, n = 10). The concentration of free fatty acids was unchanged in serum of mice fed ad libitum (miR‐22+/+, WT, n = 10; miR‐22−/− KO, n = 10); however, free fatty acids were significantly reduced in fasted miR‐22−/− (KO, n = 12) compared to fasted miR‐22+/+ (WT, n = 10).
MiR‐22 controls lipid metabolism in skeletal muscles
Since miR‐22 expression is highest in skeletal muscle, we reasoned that miR‐22 controls metabolic processes in the musculature, which might be responsible for the observed weight changes and the reduced amount of WAT in miR‐22 mutants. Transcriptome analysis of muscle tissue identified 91 transcripts that changed significantly (> 1.5 fold) between male WT and miR‐22 mutant animals. Among these genes were key regulators of metabolic pathways including Agpat2 (Cortes et al, 2009; Subauste et al, 2012), Cidea (Gong et al, 2009) (upregulated) as well as ACSS1/AceCS2 (Sakakibara et al, 2009), Egln3/Phd3 (Luo et al, 2011), Ostn/Musclin (Subbotina et al, 2015), Sln (Bal et al, 2012), and Crebl2 (Ma et al, 2011) (downregulated). Unbiased analysis of gene expression changes by Gene Set Enrichment Analysis (GSEA) (Subramanian et al, 2005) using the C3‐motiv gene set of the molecular signatures database (http://software.broadinstitute.org/gsea/msigdb) revealed significant enrichment of potential miR‐22 targets (Fig 2G). A similar analysis using the C5‐gene ontology gene set revealed strong enrichment of gene sets related to lipid metabolic processes (Fig 2H–K). In stark contrast, analysis of transcriptional changes in WT and miR‐22 mutant WAT did not reveal enrichment of the C5‐gene ontology gene sets although 39 significantly regulated (> 1.5 fold) transcripts were identified after inactivation of miR‐22, suggesting that miR‐22 does not directly control a distinct program in WAT. Similarly, no molecular changes indicating browning of WAT (Rodriguez et al, 2016) were detected (Table EV1), and in skeletal muscle, we detected no changes in expression of cytokines potentially targeting the metabolic activity of WAT (Table EV2). Taken together, our results indicate that miR‐22 primarily regulates lipid metabolism of skeletal muscle, which affects energy balance of the animals and thus WAT volume, a hypothesis that is also supported by the high expression of miR‐22 in skeletal muscle but low expression in WAT.
Transcriptome profiling may not reveal all potential miRNA targets; however, one of the identified potential primary miR‐22 targets was estrogen receptor alpha (Esr1; ERα), which has a known impact on metabolic processes (Pandey & Picard, 2009; Ribas et al, 2016). In line with the strong expression of miR‐22 in skeletal muscles, we detected upregulation of ERα protein in miR‐22 mutant skeletal muscle (Fig 3A and B, and Appendix Fig S1A), but not in white adipose tissue (WAT) or the liver of miR‐22 mutants (Fig 3B, and Appendix Fig S1B and C). Intriguingly, the concentration of ERα was only increased in skeletal muscles of male but not female miR‐22 mutants (Fig 3C), which fits well to the lower concentrations of mature miR‐22 seen in female mice and the lack of weight reduction in female miR‐22 mutants. Luciferase reporter assays demonstrated the ability of miR‐22 binding sites in the 3′ UTRs of ERα (Fig 3D) to mediate repression of protein synthesis in proliferating and differentiating C2C12 myoblasts (Fig 3E). Since miRNAs often modulate expression of several direct targets, we additionally analyzed the expression of Cav3, another potential direct target of miR‐22 identified here. Cav3 expression was significantly increased in male but not in female miR‐22 mutant skeletal muscles (Fig EV3). Although Cav3 is involved in trafficking of proteins and deletion of Cav3 impairs uptake of glucose into cells and increases WAT mass (Capozza et al, 2005), transgenic overexpression of Cav3 does not induce metabolic changes (Galbiati et al, 2000), which excludes increased Cav3 expression as a dominant cause of reduced WAT mass in miR‐22 mutants.
Figure 3. Molecular consequences of loss of miR‐22 in muscle tissue.

-
A, BThe direct miR‐22 target protein ERα is upregulated in muscle tissue of miR‐22 (KO) male mice, but not in fat tissue (muscle: WT, n = 5; miR‐22 KO, n = 5; epididymal fat: WT, n = 4; miR‐22 KO, n = 4).
-
CERα is not significantly regulated in muscle tissue of female mice (WT, n = 5; miR‐22 KO, n = 5). Original Western blot data are included in Appendix Fig S1. ERα/GAPDH ratios were normalized to the mean of WT values.
-
D, ELuciferase reporter assays in C2C12 proliferating myoblasts or differentiating myocytes demonstrate that the WT miR‐22 target site originating from the 3′ UTRs of ERα mRNA represses the activity of luciferase upon transfection of pre‐miR‐22 (miR‐22), but not upon transfection of scrambled control miRNA (scr) and this is not observed with a mutant version (mut) of the target sites. Myocytes express miR‐22 endogenously, and this results in additional repression of luciferase under control of WT target site compared to mutant target site in myocytes (n = 3 independent transfections each).
Figure EV3. Cav3 is a direct molecular target of miR‐22.

-
A, BCav3 is a direct target of miR‐22 with a miR‐22 recognition sequence (A) in the 3′ UTR of Cav3 mRNA. The miR‐22 recognition site in the 3′ UTR of Cav3 mediates repression of a luciferase reporter in proliferating and differentiating C2C12 myoblasts. Note lower activity of luciferase under control of the WT miR‐22 binding site compared to luciferase with mutant miR‐22 site in differentiating compared to proliferating C2C12 cells, most likely due to low expression of miR‐22 in proliferating myocytes and increased expression of miR‐22 in differentiating C2C12 cells (n = 3 independent transfections each).
-
C–GOriginal data (C–E) and statistical evaluation (F, G) reveal dynamic control of Cav3 protein expression. The miR‐22 target protein Cav3 is upregulated in gastrocnemius muscle (C, F) of male miR‐22−/− mice (KO, n = 5) compared to muscle of congenic miR‐22+/+ mice (WT, n = 5). However, Cav3 is not significantly regulated in gastrocnemius muscle of female mice (D, F; WT, n = 7; miR‐22 KO, n = 6). In epididymal WAT of KO mice Cav3 protein expression is decreased; however, Cav3 abundance is increased in epididymal WAT of miR‐22−/−/lox3ERαfl/fl/MCK‐Cre pos mice (E, G; WT, n = 5; miR‐22 KO, n = 5, MCK dKO, n = 5). This indicates that in male muscle Cav3 is upregulated due to loss of miR‐22‐mediated repression in muscle of miR‐22 KO mice. This repression is not as obvious in female muscle and not detected in male WAT, probably due to reduced expression of miR‐22 in WAT and female muscle. Moreover, Cav3 protein expression in WAT correlates with changes in WAT volume; thus, expression in WAT might be dominated by physiological changes occurring in WAT in the different models.
MiR‐22 regulates metabolism in male skeletal muscles by repression of ERα
ERα regulates metabolic activity of tissues at several levels and has a critical role for the control of lipid metabolism in skeletal muscles (Mauvais‐Jarvis et al, 2013; Cavalcanti‐de‐Albuquerque et al, 2014; Ribas et al, 2016). Since we detected an enrichment of lipid metabolic processes in miR‐22 mutant male muscles by GSEA, we asked whether the increased levels of ERα in muscles of male miR‐22 mutants account for the observed sex‐specific differences. Therefore, we generated miR‐22/ERα compound mutants using the previously described Ex3αER allele (Fig 4A), which does not alter the body weight in males whereas female Ex3αER become obese (Hewitt et al, 2010). Deletion of ERα in miR‐22 mutants abrogated body weight differences caused by loss of miR‐22 (Fig 4B and Appendix Fig S2A), indicating that upregulation of ERα could be instrumental for the miR‐22 mutant phenotype. In addition, loss of ERα rescued the expression of several molecular markers related to metabolic changes in muscles of miR‐22/ERα compound mutants (Fig 4C and Appendix Fig S3A–C). Next, we generated miR‐22 KO/lox3ERαfl/fl/MCK‐Crepos compound mutant mice, in which the third exon (lox3ERα) of ERα is specifically deleted by Cre recombination in striated muscle using the MCK‐Cre strain (Bruning et al, 1998) (Fig 4A). Muscle‐specific inactivation of ERα abolished the body weight differences between miR‐22 KO/lox3ERαfl/fl/MCK‐Crepos mice and miR‐22 WT/lox3ERαfl/fl/MCK‐Crepos control mice (Fig 4B and Appendix Fig S2B) excluding potential effects in other tissues, which might be caused by the germline mutation of ERα.
Figure 4. Repression of ERα signaling rescues the miR‐22 phenotype.
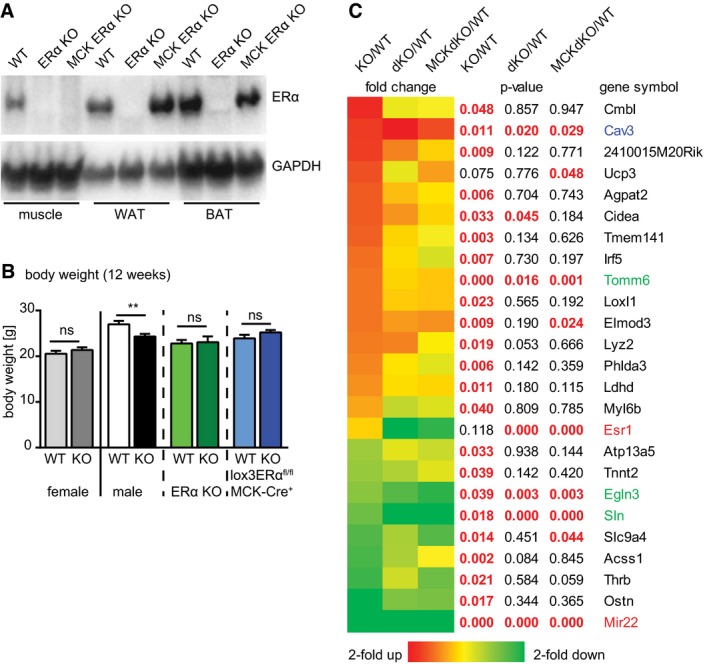
- Deletion of the 3rd exon of the ERα gene leads to loss of ERα protein in all examined tissues of constitutive ERα KO (ERα−/−, Ex3αER−/−) animals. In MCK‐Cre positive, homozygous lox3ERαfl/fl mice (MCK ERα KO) ERα expression is lost in muscle, but maintained in white or brown adipose tissue (WAT/BAT).
- Deletion of miR‐22 does not change body weight of female miR‐22−/− (KO) mice compared to congenic female miR‐22+/+ (WT) controls; however, body weight is significantly reduced in male miR‐22−/− (KO) mice compared to the congenic miR‐22+/+ (WT, see Fig 2A). Additional germline‐ (ERα KO, ERα−/−) or muscle‐specific deletion of ERα (lox3ERαfl/fl/MCK‐Crepos) eliminates the body weight differences between male miR‐22+/+ (WT) and male miR‐22−/− (KO) mutant animals (miR‐22+/+ female, n = 9; miR‐22−/− female, n = 9; miR‐22+/+, n = 18, miR‐22−/−, n = 22; miR‐22+/+/ERα−/−, n = 12; miR‐22−/−/ERα−/−, n = 8; miR‐22+/+/lox3ERαfl/fl/MCK‐Crepos, n = 10; miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos, n = 10). Time course of BW development is depicted in Appendix Fig S2. Data are presented as mean ± SEM. **P < 0.01, two‐tailed unpaired t‐test.
- Deletion of ERα abolishes molecular changes observed in muscle tissue of miR‐22−/− (KO) mice. Several markers associated with metabolic regulation return to WT levels in miR‐22 knock‐out mice with additional germline‐ (dKO, miR‐22−/−/ERα−/−) or muscle‐specific deletion of ERα (MCK dKO, miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos). Although ERα accounts for several of the observed molecular changes, there are also ERα‐independent mechanisms, indicated by independent upregulation of the primary miR‐22 target Cav3 (blue) and regulation of other metabolically important genes (green). Transcriptome data were confirmed by qRT–PCR (Appendix Fig S3A–E) or by Western blot analysis (Appendix Fig S4A–C).
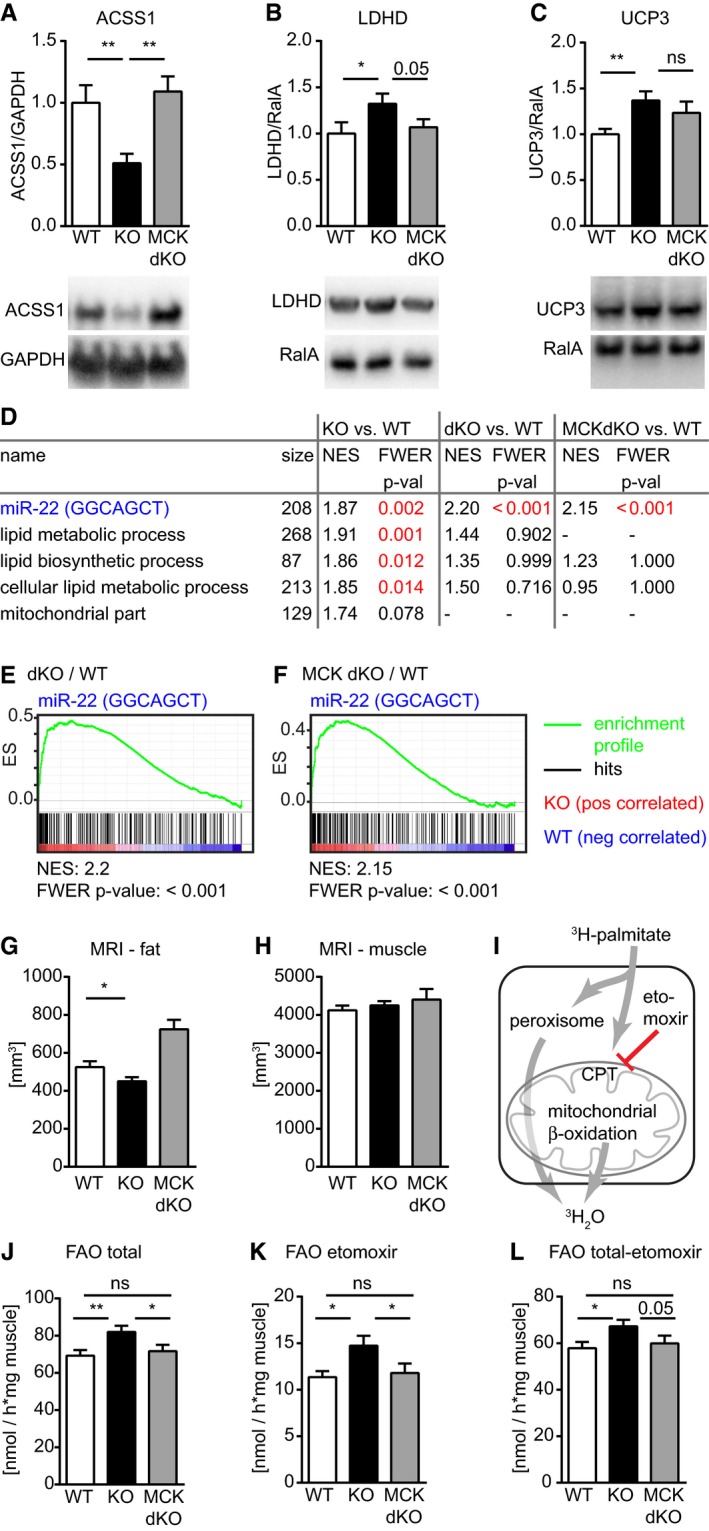
The expression of several molecular markers that were changed in muscle of the miR‐22 KO returned to WT levels after muscle‐specific inactivation of ERα in miR‐22 dKOs (Fig 4A). Sixty‐two of the 91 significantly changed (> 1.5 fold) genes in miR‐22 mutants were normalized including ACSS1, Agpat2, and musclin (Fig 4C), which was further validated by qRT–PCR (Appendix Fig S3A–C) and Western blot analysis (Fig 5A–C and Appendix Fig S4). Hence, in germline and muscle‐specific ERα/miR‐22 dKOs, we did not observe an enrichment of gene sets related to lipid metabolism anymore (Fig 5D), while the enrichment of miR‐22 target genes was maintained as expected (Fig 5E and F). This confirms our assumption that increased ERα signaling due to loss of miR‐22 is responsible for altered lipid metabolism in male muscles. It needs to be mentioned, however, that some aspects of metabolic dysregulation (Figs 4C and 5C, and Appendix Fig S3D and E) were not reversed by inactivation of ERα in miR‐22 KOs, indicating that not all effects of miR‐22 are mediated via ERα. To exclude possible impact of changes in muscle fiber type composition on metabolic properties in our animal models, we analyzed fiber type composition and mitochondrial content in WT, miR‐22 KO, and miR‐22 KO/lox3ERαfl/fl/MCK‐Crepos mice and detected no significant changes (Appendix Fig S5).
Figure 5. Repression of ERα signaling rescues the molecular signatures of the miR‐22 phenotype.
-
A–CWestern blot analysis confirms regulation of molecular markers of metabolic changes in skeletal muscle tissue of miR‐22−/− animals (KO, n = 5) compared to muscle of congenic miR‐22+/+ (WT, n = 5) tissue and the at least partial rescue of these changes in muscle of miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos (MCK dKO, n = 5; n = 4 was used for KO and MCK dKO in C; see also Appendix Fig S4). Ratios of signals for protein of interest and respective protein used for normalization were normalized to the mean of WT values.
-
D–FUnbiased gene set enrichment analysis (GSEA) of transciptomics data confirms highly significant enrichment of the miR‐22 target gene set in miR‐22−/− animals (KO, n = 5) compared to miR‐22+/+ (WT, n = 5; KO vs. WT), in miR‐22−/−/ERα−/− (dKO, n = 4) compared to WT (dKO vs. WT) and in miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos (n = 4) compared to WT (MCK dKO vs. WT). In contrast, we do not observe enrichment of lipid metabolic, cellular lipid metabolic or lipid biosynthetic processes after additional germline or muscle‐specific deletion of ERα. All gene sets with FWER P‐value < 0.05 enriched in KO animals vs. WT are shown.
-
G, HMRI analysis of body composition demonstrates that the striated muscle‐specific deletion of ERα leads to increased body fat content in miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos (MCK dKO, n = 14) that is reduced in miR‐22−/− (KO; n = 27) animals compared to miR‐22+/+ (WT, n = 21) animals, indicating that the amount of ERα in striated muscle directly influences the body fat content. The muscle volume is not changed upon modulation of ERα concentration.
-
I–LIsolated skeletal muscle was incubated in medium containing 3H‐labeled palmitate to determine fatty acid oxidation (FAO). CPT1 was blocked by Etomoxir to discriminate mitochondrial oxidation of palmitate from other processes resulting in oxidation of palmitate (I). FAO is increased in muscle tissue isolated from miR‐22−/− mice (KO, n = 21) compared to muscle isolated from congenic miR‐22+/+ (WT, n = 14) animals, but returns to WT levels in muscle of miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos (MCK dKO, n = 13). This is observed for total FAO (J), for extramitochondrial (K) as well as for mitochondrial FAO (L).
Closer analysis of body composition using MRI revealed that deletion of ERα in muscles of miR‐22−/− (KO) animals affected WAT (Fig 5G) but not muscle mass (Fig 5H). The increased concentration of ERα in miR‐22 mutants correlated with reduced WAT volume, whereas deletion of ERα in miR‐22 mutant muscles increased WAT volume (Fig 5G). The inverse correlation of ERα expression in male skeletal muscle with WAT volume corroborates the profound impact of muscle metabolism on WAT volume.
To prove the effect of miR‐22 and the contribution of ERα on muscle fatty acid catabolism, we directly analyzed FAO of muscle tissue isolated from WT, miR‐22 KO as well as from miR‐22 KO/lox3ERαfl/fl/MCK‐Crepos compound mutant mice. Isolated muscles were incubated in medium containing 3H‐labeled palmitate. To distinguish between mitochondrial β‐oxidation and extramitochondrial lipid metabolism, we used etomoxir to block carnitine palmitoyltransferase‐1 (CPT1)‐mediated uptake of fatty acids into mitochondria (Fig 5I). We observed a surge of FAO in muscle isolated from miR‐22 KO mice by 18% compared to congenic WT controls (Fig 5J). This increase was due to both increased peroxisomal metabolism of fatty acids (Fig 5K) and increased mitochondrial β‐oxidation (Fig 5L). FAO returned to WT levels after deletion of ERα in muscles of miR‐22 KO/lox3ERαfl/fl/MCK‐Crepos compound mutant mice (Fig 5J–L) indicating that ERα directly regulates FAO in muscles in a miR‐22‐dependent manner.
ERα binds to motif in the miR‐22 precursor to prevent maturation of miR‐22
So far, our results indicated that miR‐22 specifically represses ERα expression in male muscles to prevent exaggerated lipid metabolism leading to reduced WAT stores and body weight. However, a critical question remained: Which process is responsible for limiting the effects of ERα signaling in male muscle without impairing ERα signaling in females? The answer might lay in the reduced concentration of mature miR‐22 in females compared to males. We reasoned that the differential regulation of miR‐22 could occur at the posttranscriptional level, with repressed miR‐22 processing in females, since the pri‐miR‐22 is much more abundant in females than in males while the distribution of mature miR‐22 shows the exact opposite pattern (Fig 1B–D). The pri‐miR‐22 is transcribed as a spliced long non‐coding RNA that consists of three exons (miR22hg; ENSMUST00000149940; Fig 6A), in which the second exon contains the sequence of the mature miR‐22 molecule. We noticed that the first exon of the pri‐miR‐22 RNA encompasses a conserved sequence that is highly reminiscent of the known consensus ERα DNA ERE binding site (Fig 6B; http://jaspar.genereg.net) (Sandelin et al, 2004; Welboren et al, 2009). Bioinformatic analysis using the RNAfold algorithm (Lorenz et al, 2011) predicted that this sequence element is part of a stemlike double‐stranded structure (Fig 6C and E). Sequences of long non‐coding RNAs are in general not well conserved between species (Derrien et al, 2012), but the sequence constituting the double‐stranded ERE‐like motif in the pri‐miR‐22 shows only little changes among different mammalian species (Fig 6D). Moreover, RNAfold predicts that the stem‐loop‐like structural elements at the ERE‐ and at the miR‐22 sequences are the regions of lowest entropy in mouse and human pri‐miR‐22 secondary structures (Fig 6E and F). Analysis of miR‐22 expression in ERα mutant mice revealed a profound influence of ERα on the abundance of mature miR‐22. Compared to WT animals, we observed a higher expression of mature miR‐22 especially in female ERα mutants, and loss of ERα completely abolished the differences in expression of mature miR‐22 between male and female animals (Fig 6G). Moreover, we observed a concomitant decrease in pri‐miR‐22 levels and loss of the male/female differences in pri‐miRNA abundance in ERα mutants (Fig 6H) indicating that sex‐associated differences in miR‐22 processing depend on the presence of ERα. To investigate whether ERα suppresses miR‐22 processing by direct sequence‐specific binding to the ERE‐like sequence element in pri‐miR‐22 or by other indirect mechanisms, we performed RNA immunoprecipitation (RIP) experiments. We used WT pri‐miR‐22, a pri‐miR‐22 with mutation in the putative ERα binding motif in the first exon, and a pri‐miR‐22 control with mutations of the putative ERα binding motif and mutation of the sequence corresponding to the mature miR‐22. Expression of these constructs in HEK293 cells together with flag‐tagged ERα (Badia et al, 2009) followed by immunoprecipitation using an anti‐FLAG antibody allowed efficient recovery of the ERα protein and of WT pri‐miR‐22 (Fig 7A). In contrast, GAPDH mRNA was not precipitated demonstrating specificity of the pulldown of pri‐miR‐22 by ERα. Importantly, mutation of the potential ERα binding motif abolished the interaction between pri‐miR‐22 and ERα indicating that the physical association of pri‐miR‐22 and ERα depends on the specific ERα binding motif in the first exon of pri‐miR‐22 (Fig 7A). To provide direct evidence that the ERα‐binding site in pri‐miR‐22 is instrumental for differential processing of miR‐22 in male and female animals in vivo, we generated a mutant mouse strain, in which the ERα binding site in pri‐miR‐22 was replaced with a loxP sequence (Fig 7B). Analysis of mutant miR‐22d/d mice revealed equal expression levels of mature miR‐22 (Fig 7C) and of mutated pri‐miR‐22 in male and female animals (Fig 7D) corroborating that the ERα binding motif in pri‐miR‐22 indeed is required for sex‐specific processing of miR‐22 in vivo.
Figure 6. ERα regulates maturation of miR‐22.

-
A–CTranscription of the miR‐22 locus gives rise to a spliced precursor RNA that encodes the mature miR‐22 in the second exon. The first exon of the pri‐miR‐22 contains a sequence element that strongly resembles the ERα DNA binding site (B, ERE) known from ChIP experiments. (C) The RNAfold algorithm predicts this element to be part of a stemlike double‐stranded structure.
-
DThis sequence element and the corresponding antisense sequence, together predicted to form a double‐stranded structure, are highly conserved in mammals.
-
E, FSecondary structure of the first 500 bases of the mouse (E, ENSMUST00000149940) and human (F, ENST00000571595) pri‐miR‐22 were predicted using RNAfold, and centroid secondary structures are depicted with color‐coded local entropies. In both, mouse and humans, stem‐loop structures for the ERE‐like structure and the stem‐loop structures of miR‐22 were predicted with low local entropy, whereas other predicted structural features with predicted low local entropy are not conserved.
-
G, HDeletion of ERα leads to increased abundance of mature miR‐22 in skeletal muscle and abolishes the differences in miR‐22 abundance between male and female animals (G). Moreover, loss of ERα decreases the abundance of the miR‐22 precursor RNA and again loss of the differences between male and female is observed (H) (ERα+/+, WT male, n = 13; ERα+/+, WT female, n = 11; ERα−/−, KO male, n = 11; ERα−/−, KO female, n = 14). This indicates an ERα‐dependent repression of miR‐22 maturation from pri‐miR‐22 that is especially pronounced in female animals. *P < 0.05, ***P < 0.001, one‐tailed unpaired t‐test. Data were normalized to U6 (G) or Rplp0 (H), respectively, and WT female values were used as reference. Data are presented as mean ± SEM.
Figure 7. ERα directly binds to pri‐miR‐22.

-
ARNA immunoprecipitation using HEK293 cells transfected with ERα‐FLAG and pri‐miR‐22 variants indicate an interaction between the WT pri‐miR‐22 (WT), but not with mutants containing a mutated ERE site (mut) or with a control construct with mutated ERE and miR‐22 replaced by miR‐1 sequences (con).
-
B–DIn vivo mutation of the identified ERα binding element (ERE, see Fig 6A) in the first exon of the miR‐22 host gene to a loxP site (miR‐22d/d; B) abolished the differences in the abundance of mature miR‐22 (C; miR‐22d/d male, n = 22; miR‐22d/d female, n = 18; two‐tailed unpaired t‐test) and of pri‐miR‐22 between male and female animals (D, miR‐22d/d male, n = 23; miR‐22d/d female, n = 17; two‐tailed unpaired t‐test). Data were normalized to U6 (C) or Rplp0 (D), respectively, and WT female values were used as reference. Data are presented as mean ± SEM.
-
EIn summary, miR‐22 is expressed predominantly in striated muscle tissue, and at the posttranscriptional level, ERα represses the processing of miR‐22 host RNA to mature miR‐22 by direct binding to an ERE‐like sequence in the miRNA host RNA. This results in higher abundance of mature miR‐22 in male compared to female muscle. Accordingly, complete disruption of ERα increases mature miR‐22 and decreases the pri‐miR‐22 concentration (blue arrows). The miRNA miR‐22 represses in vivo the activity of the direct target ERα especially in male muscle. Repressed ERα abundance in wild‐type male muscle tissue is essential to restrict the effects of ERα on the lipid metabolic genes and on fatty oxidation in male muscle tissue. Deletion of miR‐22 leads to increase in ERα and fatty acid oxidation and subsequently to decrease WAT and body weight in male animals (red arrows); these effects can be eliminated by additional muscle‐specific deletion of ERα (green arrows).
Discussion
Our study revealed that a posttranscriptional regulatory feedback loop between pri‐miR‐22, mature miR‐22, and ERα is crucial for the control of male muscle lipid metabolism and permits sex‐specific regulation of ERα expression in skeletal muscles. We showed that mature miR‐22 is present at higher concentrations in male than in female skeletal muscles while the miR‐22 precursor shows the exact opposite pattern. Analysis of miR‐22 expression in ERα mutants demonstrated that ERα is essential for the sex‐specific differences and regulates the amount of mature miR‐22 at the posttranscriptional level by controlling processing of the primary transcript. Deletion of ERα results in increased mature miR‐22 especially in female muscle and concomitant decrease in the abundance of pri‐miR‐22. This indicates that in female, ERα represses the processing of pri‐miR‐22 to mature miR‐22, whereas in male muscle ERα does not affect the release of miR‐22 from pri‐miR‐22. Identification of a conserved ERE‐like sequence element in the pri‐miR‐22 suggested that ERα might bind directly to the miR‐22 precursor RNA thereby modulating miRNA processing. RNA‐IP experiments using WT and mutant versions of miR‐22 precursor prove that ERα can bind the ERE‐like sequence contained in the RNA molecule and mutation of the sequence abolishes this binding. Mutation of the ERα binding sequence in vivo leads to the loss of the sex‐specific differences in the abundance of miR‐22 and pri‐miR‐22 demonstrating that the identified ERα binding sequence element is indeed important for sex‐specific differences in miR‐22 and pri‐miR‐22 abundance in vivo. Thus, the combined evidence from analysis of ERα KO mice, miR‐22d/d mutants, and RIP experiments suggests that ERα attenuates maturation of miR‐22 in female tissues by direct binding to the pri‐miR‐22 via a specific ERE‐like sequence element, whereas lower activity of ERα in male tissues is further repressed by the unimpaired release of the ERα repressor miR‐22. While it seems clear that direct binding of ERα to pri‐miR‐22 represses miR‐22 release, it is less evident whether reduced ERα activity in male preventing inhibition of pri‐miR‐22 processing is due to lower estrogen activity in male or whether additional male‐specific modulators of ERα signaling limit ERα activity. Most likely the function of ERα on miR‐22 processing depends on interaction of ERα with additional protein complexes as well as on subcellular localization of these protein complexes; however, the current study did not address these molecular interactions in greater detail.
Our study disclosed that miR‐22 specifically suppresses ERα expression in male skeletal muscles and thereby modulates lipid metabolism including suppression of FAO. So far, the mechanisms that restrict ERα expression in males were unknown, although there is clearly a need to control ERα signaling in males, since estrogen is not only present as a circulating hormone in females but is synthetized in males from testosterone at extragonadal sites (Jones et al, 2000). Estrogen acts as a paracrine factor and abolished estrogen release affects male metabolism resulting in increased body weight (Simpson et al, 2005). Unbiased analysis of transcriptional changes in muscle of miR‐22 mutants by GSEA revealed enrichment of potential miR‐22 target genes and enrichment of gene sets associated with lipid metabolic processes. Among the few significantly regulated potential miR‐22 target genes, we identified ERα as the most important effector of miR‐22 on lipid metabolism. Additional germline and muscle‐specific deletion of ERα in miR‐22 mutant mice normalized lipid metabolic genes and returned FAO in skeletal muscles to WT levels, thereby rescuing the metabolic phenotype of miR‐22 mutants.
The impact of ERα on the metabolism has been studied in various settings and in great detail. It is well known that ERα affects energy input and expenditure (Heine et al, 2000; Ribas et al, 2010). Germline and tissue‐specific deletion of ERα have helped to decipher effects of ERα in the discrete tissues (Ribas et al, 2011; Xu et al, 2011). In female muscle tissue, ERα is important for FAO (Mauvais‐Jarvis et al, 2013; Ribas et al, 2016), which corresponds to our observation that increased expression of ERα caused by loss of miR‐22 changes the expression of lipid metabolic genes. Accordingly, metabolic changes in male miR‐22 mutants were rescued by deletion of ERα, which agrees with known sex‐specific differences in skeletal muscle lipid handling resulting in higher β‐oxidation capacity in female compared to male skeletal muscle (Cavalcanti‐de‐Albuquerque et al, 2014; Lundsgaard & Kiens, 2014). We therefore concluded that the miR‐22–ERα regulatory feedback loop contributes to sex‐specific differences by negative regulation of the lipid metabolic genes specifically in male skeletal muscles. We argue that tissue specificity is achieved by low expression of miR‐22 in other tissues, thereby restricting the identified mechanism to skeletal muscles.
miR‐22 does not only directly target ERα but also other genes involved in the regulation of the metabolism, which makes it difficult to distinguish primary from overlaying secondary effects. However, analysis of miR‐22/ERα compound mutants proved to be a useful tool to distinguish specific effects of the primary miR‐22 target ERα from those depending on other miR‐22 activities. We found that despite normalization of numerous aberrantly expressed genes after muscle‐specific inactivation of ERα in miR‐22 mutants some metabolically important genes like UCP3 (Aguer et al, 2013), Egln3/PHD3 (Luo et al, 2011), or Sln (Bal et al, 2012) remained dysregulated. In contrast, Cav3, which contains a miR‐22 target sequence and which has been described to cause metabolic effects under certain physiological conditions (Capozza et al, 2005), was downregulated in WAT of miR‐22 mutants but increased in miR‐22/ERα compound mutants. Thus, expression of Cav3 in WAT primarily correlates with WAT volume and not with miR‐22 expression, which rules out an important role of Cav3 for mediating miR‐22 effects in WAT. The complex regulation uncovered by our analysis is in agreement with the theory that miRNAs integrate different signaling cascades to target and adjust related biological processes (Bartel, 2009; Small & Olson, 2011). We reason that miRNA‐dependent regulatory mechanisms do not always act in a simple synergistic manner but might evoke complementary or antagonistic effects that eventually confer complexity and robustness to regulatory cascades.
In our study, we have demonstrated that repression of ERα by miR‐22 controls lipid metabolism in male skeletal muscles allowing acquisition of WAT depots and proper gain of body weight in males. We have defined a posttranscriptional regulatory feedback loop that restricts ERα signaling in male but not in female skeletal muscles. The ERα‐miR‐22 feedback loop provides a mechanistic model that explains some sex‐specific differences in muscle lipid metabolism and body weight regulation. Future studies will further improve our understanding of estrogen signaling and the multifaceted consequences, which result from therapeutic application of estrogen.
Materials and Methods
Animal experiments
All animal experiments were in accordance with German animal protection laws and were approved by the local governmental animal protection committee (Regierungspraesidium Darmstadt, Hessen, Germany).
Mouse models
A 129S7AB2.2 BAC clone (bMQ‐420K21) was used for recombination in SW102 cells (Warming et al, 2005) to replace the sequence coding for mature miR‐22 with an AscI‐flanked galK cassette that was amplified with homologous flanking sequences (CTGAAAGTCAGCTAGAGCCAGGTTTGGATGGCCTCTGGGTCGCTGGCCCTGTCACCCAG; ATTCCCCAATTTATAGACAGTGCCTTTAACCTAAATCTTTCACTGGGCCAGTGGGGGTAA). The mutated genomic sequence was recombined into a pKO targeting vector containing a DTA expression cassette. Gap repair resulted with miR‐22 locus genomic sequence from 5′ AACCCTTAAACTCCAGGTGTGGTGG to 3′ GCCTTGTTGGACCGGGAATTCTAGG. The galK cassette was replaced by a loxP‐flanked IRES‐lacZ‐neoR cassette using AscI. MPI‐II (129SV) embryonic stem cells (Voss et al, 1997) were used for homologous recombination, and chimeric animals were generated using standard procedures. Chimeras were mated to C57BL/6, and animals used for analysis were backcrossed to C57BL/6 for > 5 generations. miR‐22−/− (KO) and miR‐22+/+ (WT)‐control animals were obtained by interbreeding of heterozygous animals. AvrII‐digested genomic DNA and a 3′ probe amplified by PCR (TGAGTTGGACATGCTCCCTTACTAC, TGTTGCTGGAGGTTTGCTGTGAGG; 249 bp) were used to determine the genotype. ERα−/− (ERαKO; Ex3αER KO) and ERα conditional mutant mice (floxed exon 3 of ERα, lox3ERαfl/fl) were provided by Sylvia C. Hewitt & Kenneth S. Korach, NIEHS/NIH (Hewitt et al, 2010). To generate a muscle‐specific knock‐out of ERα, lox3ERα mice were mated to MCK‐Cre mice (Bruning et al, 1998). For mutation of the ERα binding site in the first exon of pri‐miR‐22 in vivo, the WT genomic DNA was recovered from the BAC bMQ‐420K21 as described above. A PCR fragment using primers (GGAGTAAGCAGAGGTGCTGGCGCCCCCGAGTGGGTGTGGcgcgCCAGCAATGGGCGGTGATTG; GGAGCCCAGCGATGACTAACCCAGG) was digested with KasI and inserted into the KasI digested WT genomic DNA fragment to replace the ERα binding site by an AscI recognition site. A neomycin resistance cassette was inserted into the AscI site. Embryonic stem cells were analyzed by Southern blot (BamHI digest, 5′ probe: TCCAAAGCACTTGGTTAATAACTTGC, TTGCTGGTCTGGCCTGTGTATGAG; WT: 16.5 kb, mutant: 8.7 kb). Chimeric mice were mated to Cre‐deleter mice (Su et al, 2002) to delete the selection cassette, resulting in mice with an AscI site‐flanked loxP site replacing the potential ERα binding site in the first exon of miR‐22 host gene. These mice were backcrossed to obtain Cre‐negative mice with homozygous mutation of (miR‐22d/d) of the potential ERα binding site. This mutation of the first exon was identified by PCR (GTTGGTAGGCGTGGCTTAGAGAAGCTC, ACCGGCTCCAGTCTCCGATCAGCTGG; WT: 218 bp, miR‐22d/d: 268 bp). Mice were kept in IVC with ad libitum access to water and food (Altromin #1314).
The food intake, activity, and respiratory exchange quotient of mice were determined in metabolic cages of the phenomaster system (TSE‐systems). The mice were kept in type 2 cages with food and water ad libitum. The experiment was performed inside the animal facility with habitual day and night cycles, temperature and humidity. Animals were accustomed to the new cages for 2 days before the measurements were started. The measurement parameters were detected every 10 min for 48 h. Blood was collected with or without prior 16‐h fasting by retro‐orbital puncture or decapitation of the mice. Blood glucose concentration was determined using Accu‐Check Sensor (Roche). Commercially available kits were used according to the suppliers instructions to quantify leptin (ELISA; Millipore, EZML‐82K), triglycerides (GPO method; Biolabo, #80019), and free fatty acids (colorimetric; Abcam, ab6541).
Cell culture
MCF‐7, HeLa, HEK‐293, Hep G2, Capan2, and A549 cells were obtained from ATCC, DU145 cells were from DSMZ, and A2780 cells were from Sigma. Cells were cultured in DMEM containing 10% FCS. Human skeletal muscle myoblasts (HSMM; Lonza) were grown in skeletal muscle cell growth medium (Lonza; SKGM2 bullet kit CC‐3245), and 80% confluent cells were differentiated in differentiation medium (DMEM/Ham's F12, 2% horse serum). All cells were tested mycoplasma negative.
MRI measurements
Mice were measured under volatile isoflurane (1.5–2.0% in oxygen and air with a flow rate of 1.0 l/min) anesthesia; the body temperature was maintained 37°C by a thermostatically regulated water flow system during the entire imaging protocol. MRI experiments were performed on a 7.0 T superconducting magnet (Bruker Biospin, Pharmascan, 70/16, 16 cm; Ettlingen, Germany) equipped with an actively shielded imaging gradient field of 300 mT/m. The frequency for the 1H isotope is 300.33 MHz. A 60‐mm inner diameter linear polarized 1H volume resonator (Bruker Biospin) was used for RF pulse transmission and signal reception. A coronal MSME‐(Multi‐Slice‐Multi‐Echo) spin‐echo sequence with an echo time TE = 8.6 ms, repetition time TR = 453 ms, a field of view FOV = 7 × 7 cm², matrix size MTX = 512 × 256, and a slice thickness of 1 mm was recorded. For volumetric quantification of fat and muscle tissue, we used an in‐house developed ImageJ plugin. A list of anatomically defined landmarks are set by the operator and used to derive tissue‐specific signal intensity thresholds and to define the region of interest for intensity‐sensitive region growing segmentation. The resulting tissue voxel volumes inside the region of interest is counted and returned as cubic millimeters for each tissue.
Transcriptome analysis
Total RNA was extracted from tissue or cultured cells using the TRIzol (Life Technologies) method. For Northern blot analysis, 3 μg total RNA was separated by a 15% polyacrylamide–urea gel (Invitrogen) and blotted on a Hybond XL (Amersham) membrane using semi‐dry blotting (1.5 mA/cm², 30 min). The probe was labeled with γ‐32P‐ATP in using PNK (NEB). The probe was purified using spin columns (Bio‐Rad, 732‐6221) and hybridized to the membrane overnight at 30°C. The blots were washed with 2× SSC containing 0.1% SDS, and signals were detected with a Bass‐2500 reader. Affymetrix GeneChip Mouse Gene 1.0 ST arrays were used for transcriptome analysis of soleus muscle and epididymal white adipose tissue according to the manufacturer's instructions (muscle: miR‐22+/+, n = 5; miR‐22−/−, n = 5, miR‐22−/−/ERα−/−, n = 4; miR‐22−/−/lox3ERαfl/fl/MCK‐Crepos, n = 4; WAT: miR‐22+/+, n = 3; miR‐22−/−, n = 3). Data were analyzed using the RMA algorithm and DNAStar Arraystar 12 software. Calculation of the median was used as averaging method, fold changes between groups were calculated, and the moderated t‐test was used to determine statistical significance of the observed changes in gene expression (Smyth, 2004). For unbiased identification of regulated biological processes, Gene Set Enrichment Analysis (GSEA; http://www.broadinstitute.org/gsea/index.jsp) was performed (Subramanian et al, 2005). Expression values of all genes represented on the MoGene 1.0 Affymetrix array were imported into the software and compared with appropriate genes sets of the Molecular Signature Database (MSigDB v5.1; release 13‐January‐2016). We used the miRNA target gene sets (C3; MIR—microRNA targets; sets of genes that contain a specific potential microRNA binding motif in the 3′‐UTR) and GO gene sets (C5; gene sets are named by GO term and contain genes annotated by that term) using default parameters but limited the analysis to gene sets with at least 75 genes for a robust analysis and set the permutation type to gene_set according to the GSEA instructions. For analysis of expression of individual mRNAs and mature miRNA expression, Applied Biosystems Taqman qRT–PCR kits were used. Total RNA, isolated from soleus muscle if not otherwise stated, was reverse transcribed using Superscript (Invitrogen) with a mix of oligo dT and random primers. For miRNA detection using the Applied Biosystems Taqman System, the assay‐specific RT reactions were used. Taqman analysis was performed in duplex reactions with FAM‐labeled probes for the genes of interest (miR‐22, RT/TM000398; Acss1, mm00475647_m1; Crebl2, Mm00624585_m1; Egln3, Mm00472200_m1; Ostn, Mm00813799_m1; Sln, Mm00481536_m1; Tomm6, mm00481240_m1) and VIC‐labeled controls (U6, custom 001973; Gapdh, 4352339E) for normalization in a StepOnePlus real‐time PCR system. For quantification of pri‐mir‐22 KAPA SYBR® FAST qPCR Kits with primers to detect pri‐mir‐22 (GCTGCCAGTTGAAGAACTGTTGCCCT/gcaactcgagCTTCCGGATGATAGGCAAAGCTGC) were used. Pri‐miR‐22 signals were normalized to Rplp0 (ribosomal protein, large, P0; Arbp)‐Expression (CCGCAGGGGCAGCAGTGGT/AAGCGCGTCCTGGCATTGTCT). Experiments were done with technical triplicates for each sample, and RQ values were calculated using StepOne V2 Software.
Western blot analysis
Protein lysates (10–30 μg) were resolved on 4–12% SDS–PAGE gradient gels and transferred onto nitrocellulose membranes (GE). The following antibodies were used according to manufacturer's instructions: ACSS1 (Proteintech 17138‐1‐AP), Cav3 (BD Biosciences 610420), ERα (AbD Serotec MCA1799T); GAPHD (CST 2118), LDHD (Sigma SAB2103557), RalA (BD R23520), UCP3 (Abcam ab3477) with corresponding HRP‐conjugated secondary antibodies (Pierce 1858413/5; eBioscience 18‐8877) using chemiluminescence (Femto‐Kit, Pierce) with a Versadoc Imaging System. Soleus (Acss1, LDHD, UCP3) or gastrocnemius (ERα, Cav3) muscle was used for Western blot analysis. Signal intensity was quantified using Quantity One or Image Lab 5.0 software.
Histological analysis
Skeletal muscle fiber types were identified as previously described (Sandona et al, 2012) using concentrated monoclonal antibodies BA‐D5, SC‐71, BF‐F3 (developed by S. Schiaffino, obtained from DSHB) in combination with AffiniPure goat anti‐mouse IgG, Fcγ subclass 2b—DyLightTM405 (111‐475‐207), goat anti‐mouse IgG, Fcγ subclass 1—Alexa Fluor® 488 (115‐545‐205), and goat anti‐mouse IgM μ chain—Alexa Fluor® 594 (115‐585‐075; Jackson ImmunoResearch). Tissues were snap‐frozen on liquid nitrogen, 10 μm cryosections were mounted on Superfrost slides. Tissue sections were dried at RT, fixed with 4% PFA/0.1% sodium deoxycholate/0.02% NP‐40/PBS for 5 min, washed three times with PBS, and blocked in 2% FCS, 0.5% NP‐40/PBS for 1 h. Sections were incubated with primary antibodies (1:100) overnight at 4°C, washed three times with PBS and were than incubated with secondary antibodies (1:300), washed three times with PBS and were embedded in Fluoromount. For succinate dehydrogenase activity staining, cryosections were dried at RT and stained for 1.5 h at RT with 0.02 M KH2PO4, 0.076 M Na2HPO4, 0.1 M sodium succinate, 0.02% w/v nitroblue tetrazolium. X‐Gal staining of tissues was performed as previously described (Boettger et al, 2009), and corresponding WT (miR‐22+/+) tissues were used as controls.
Luciferase reporter assay
Luciferase reporter assays were performed as previously described (Wystub et al, 2013). Briefly, miRNA target sites were cloned quadruplicate into the pmirGLO Dual‐Luciferase Vector (Promega E1330) using oligonucleotides (ERα binding site: GGAAAATTATTCAGGGCAGCTAAT; ERα mutated site: GGAAAATTATTCAGGGACTAGTAT; Cav3 WT binding site: TGAGGGTTCCTTAAGAGGCAGCTATG; Cav3 mutated binding site: TGAGGGTTCCTTAAGACCGACCAATG). Reporter vectors were cotransfected with miRIDIAN microRNA mimics miR‐22 (GE Dharmacon) into C2C12 cells (ATCC) cultured in proliferation medium (DMEM, 10% FBS, 2 mM glutamine, 1× penicillin/streptomycin). Proliferating C2C12 cells were lysed for luciferase assays 24 h after transfection; for analysis of luciferase activities in differentiating C2C12 cells, transfected cells were transferred to differentiation medium (DMEM, 2% horse serum) 24 h after transfection and differentiating myotubes were lysed 72 h after transfection. Firefly luciferase and Renilla activity were determined using the Dual‐Luciferase Reporter Assay (Promega). Firefly luciferase was normalized to Renilla activities. C2C12 cells were tested mycoplasma negative.
Fatty acid oxidation assay
Fatty acid oxidation of skeletal muscle tissue was determined as described previously (Moon & Rhead, 1987) with modifications (Schoors et al, 2015). In brief, to determine FAO soleus muscles were isolated from mice. One muscle was incubated in medium (DMEM, 10% FBS) supplemented with 60 μM 9,10‐3H palmitate (PerkinElmer; 30 Ci/mmol) for 6 h at 37°C/5% CO2. The second corresponding muscle of each mouse was incubated in similar medium additionally containing 100 μM etomoxir (Sigma E1905) to block mitochondrial uptake of fatty acids by carnitine palmitoyltransferase‐1 (CPT‐1). After incubation, supernatant was transferred into vials sealed with rubber stoppers and containing hanging wells with pre‐wetted Whatman paper. 3H2O labeled water was allowed to equilibrate between medium and hanging wells by incubation of vials at 37°C for 48 h. Labeled water captured in the hanging wells was determined using LSC‐cocktail (Ultima Gold™, PerkinElmer) and a PerkinElmer Tri‐Carb 2810 TR scintillation analyzer. Standard values were used to calculate the amount of metabolized palmitate. Data were normalized to the tissue weight; weight of isolated muscles did not differ between experimental groups.
RNA immunoprecipitation
Variants of the cDNA of the miR‐22hg (AK008813, Fantom2 clone ID: C030026K10) were cloned into a pIRES2‐EGFP (Clontech)‐based vector with the IRES2‐EGFP removed by NotI/BamHI digest and religation. The WT miR‐22hg cDNA was amplified using oligonucleotides (TB914: gcatgctagcTGGCGCCCCCGAGTGGGTGTGGTCACGTTGC, TB916: atgcgaattcGGACAGTCTCATATAGCCCAGGCTG). The potential ERα binding site was mutated by amplification using mutated oligonucleotides (TB915: gcatgctagcTGGCGCCCCCGAGTGGGTGTtcagACGTacatCCAGCAATGGGCGGTGATTGGCCCTG, TB916). We further mutated the miR‐22 stem‐loop sequence to a miR‐1 stem‐loop sequence by a two‐step PCR protocol using oligonucleotides (TB915, TB919: TAGCTTAGCAGGTTCATATGGGCATATAAAGAAGTATGTACTGCGGCTCAGCCAGGTGAGGGTGTGGGAGGCACAGGTTCCTGATG and TB916, TB920: ATATGAACCTGCTAAGCTATGGAATGTAAAGAAGTATGTATTGCCCTCTGCCCCTGGCTTCGTGGAGGAAGAGGAGAAGCAGCAGC). Subsequently, we used these PCR products as templates to amplify a control pri‐miR with mutated ERα binding site and miR‐1 stem‐loop sequence using oligonucleotides (TB915, TB916). PCR products were inserted in the vector using NheI/EcoRI restriction enzyme sites. All PCRs were performed using Phusion DNA polymerase (NEB); the accuracy of the constructs was confirmed by sequencing.
RNA immunoprecipitation followed published protocols (Moran et al, 2012) with the following modifications. Six 15‐cm plates of HEK293 cells were co‐transfected with the plasmids expressing variants of the miR‐22hg and with a plasmid expressing the Flag‐tagged ERα (Badia et al, 2009) using the TurboFect transfection reagent (Thermo Scientific). Approximately 1.6 × 108 cells were harvested by trypsinization, washed twice with PBS, and were crosslinked with 0.1% f.c. formaldehyde (Sigma) for 10 min. Reaction was quenched using glycine 125 mM f.c. Cells were washed twice with PBS and resuspended in 2.4 ml RIPA‐buffer containing RNasin (Promega, N2511; 1:100) and protease inhibitors (Roche, 11697498001 1 tablet/10 ml). Cells were disrupted using a Covaris‐Sonicator (S220; 1 min, 100 W peak power, 200 cycle/burst, 1‐ml tubes), and the lysate was centrifuged 10 min at 2,600 g at 4°C. The respective supernatants were incubated overnight at 4°C with 50 μl magnetic beads (Thermo, Dynabeads Protein A; 10001D) preincubated with 8 μg antibody (IgG: Invitrogen #10400C; anti‐Flag: Sigma#F1804). Following the incubation, beads were washed three times with RIPA‐buffer, a 1/10 aliquot was removed for Western blot analysis, the beads were further washed three times with high‐salt RIPA‐buffer (1 M NaCl, 4 M urea), and once with PBS. Beads were incubated in proteinase K for 30 min at 42°C and 4 h at 65°C (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 10 mM DTT, 1% SDS, 100 μg/ml proteinase K). RNA was isolated using the TRIzol method, combined with an RNeasy (Qiagen) clean‐up step. RNA was incubated with DNase (Thermo), reverse transcribed using Superscipt II, and PCR was performed to detect miR22hg (GCATCAGGAACCTGTGCCTCCCACAC, CCGGATGATAGGCAAAGCTGCTGC) or Gapdh (ACCACAGTCCATGCCATCAC, CATGCCAGTGAGCTTCCCGT). Appropriate minus RT controls were performed to confirm amplification of RNA by the PCR. The RNA immunoprecipitation experiment was repeated twice with similar outcome.
Statistical analysis and RNA secondary structure prediction
RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (Lorenz et al, 2011) was used to predict secondary structures of pri‐miR‐22 molecules using default parameters. No statistical method was used to predetermine sample size. Animals were assigned to experimental groups according to genotype. No blinding of investigators was done for animal studies. GraphPad Prism 6 was used for statistical analysis. Normal distribution of data was assumed, and data are represented as mean ± SEM unless otherwise indicated. F‐test was used to compare variances, and ROUT method was used to identify outliers. Two‐tailed Student's t‐tests were performed unless otherwise indicated. Log‐rank (Mantel–Cox method) test and the Gehan–Breslow–Wilcoxon test were used for survival analysis. P‐values < 0.05 were regarded to be significant.
Accession codes
Microarray data are available via ArrayExpress (http://www.ebi.ac.uk/arrayexpress/; Accession# E‐MTAB‐3518 and E‐MTAB‐5409).
Author contributions
JS and TBo conceived the experimental design; JS, CS, SW, AW, BG, PC, SD, and TBo performed the experiments and analyzed the data. TBo, TBr, and KSK wrote the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We are grateful to Sylvia Thomas, Marion Wiesnet, Ursula Hofmann, Ulrike Neckmann, Sonja Krüger, and Susanne Kreuzer for technical assistance. We would like to thank Antonia Sassmann (MPI Bad Nauheim) for help with the metabolic cages, Sandra Schoors (KU Leuven, Belgium) for advice regarding the FAO protocol, Antje M. Richter (Universität Giessen, Germany) for cell lines, and Vincent Cavailles (Université Montpellier, France) for the vector expressing FLAG‐tagged ERα. Research support to KSK came from the NIEHS Division of Intramural Research. The work was supported by the Max‐Plank‐Society, the DFG (Excellence Cluster Cardio‐Pulmonary System; ECCPS), and LOEWE research cluster “Medical RNomics”.
The EMBO Journal (2017) 36: 1199–1214
Contributor Information
Thomas Braun, Email: thomas.braun@mpi-bn.mpg.de.
Thomas Boettger, Email: thomas.boettger@mpi-bn.mpg.de.
References
- Aguer C, Fiehn O, Seifert EL, Bezaire V, Meissen JK, Daniels A, Scott K, Renaud JM, Padilla M, Bickel DR, Dysart M, Adams SH, Harper ME (2013) Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete beta‐oxidation. FASEB J 27: 4213–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badia E, Escande A, Balaguer P, Metivier R, Cavailles V (2009) New stably transfected bioluminescent cells expressing FLAG epitope‐tagged estrogen receptors to study their chromatin recruitment. BMC Biotechnol 9: 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal NC, Maurya SK, Sopariwala DH, Sahoo SK, Gupta SC, Shaikh SA, Pant M, Rowland LA, Bombardier E, Goonasekera SA, Tupling AR, Molkentin JD, Periasamy M (2012) Sarcolipin is a newly identified regulator of muscle‐based thermogenesis in mammals. Nat Med 18: 1575–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros RP, Gustafsson JA (2011) Estrogen receptors and the metabolic network. Cell Metab 14: 289–299 [DOI] [PubMed] [Google Scholar]
- Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin KK, Winders BR, Olson EN (2015) Muscle as a “mediator” of systemic metabolism. Cell Metab 21: 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366 [DOI] [PubMed] [Google Scholar]
- Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119: 2634–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettger T, Braun T (2012) A new level of complexity: the role of microRNAs in cardiovascular development. Circ Res 110: 1000–1013 [DOI] [PubMed] [Google Scholar]
- Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR (1998) A muscle‐specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2: 559–569 [DOI] [PubMed] [Google Scholar]
- Capozza F, Combs TP, Cohen AW, Cho YR, Park SY, Schubert W, Williams TM, Brasaemle DL, Jelicks LA, Scherer PE, Kim JK, Lisanti MP (2005) Caveolin‐3 knockout mice show increased adiposity and whole body insulin resistance, with ligand‐induced insulin receptor instability in skeletal muscle. Am J Physiol Cell Physiol 288: C1317–C1331 [DOI] [PubMed] [Google Scholar]
- Cavalcanti‐de‐Albuquerque JP, Salvador IC, Martins EL, Jardim‐Messeder D, Werneck‐de‐Castro JP, Galina A, Carvalho DP (2014) Role of estrogen on skeletal muscle mitochondrial function in ovariectomized rats: a time course study in different fiber types. J Appl Physiol 116: 779–789 [DOI] [PubMed] [Google Scholar]
- Cortes VA, Curtis DE, Sukumaran S, Shao X, Parameswara V, Rashid S, Smith AR, Ren J, Esser V, Hammer RE, Agarwal AK, Horton JD, Garg A (2009) Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2‐deficient mouse model of congenital generalized lipodystrophy. Cell Metab 9: 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, Hata A (2008) SMAD proteins control DROSHA‐mediated microRNA maturation. Nature 454: 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M et al (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Volonte D, Chu JB, Li M, Fine SW, Fu M, Bermudez J, Pedemonte M, Weidenheim KM, Pestell RG, Minetti C, Lisanti MP (2000) Transgenic overexpression of caveolin‐3 in skeletal muscle fibers induces a Duchenne‐like muscular dystrophy phenotype. Proc Natl Acad Sci USA 97: 9689–9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Sun Z, Li P (2009) CIDE proteins and metabolic disorders. Curr Opin Lipidol 20: 121–126 [DOI] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R (2004) The Microprocessor complex mediates the genesis of microRNAs. Nature 432: 235–240 [DOI] [PubMed] [Google Scholar]
- Guil S, Caceres JF (2007) The multifunctional RNA‐binding protein hnRNP A1 is required for processing of miR‐18a. Nat Struct Mol Biol 14: 591–596 [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurha P, Wang T, Larimore AH, Sassi Y, Abreu‐Goodger C, Ramirez MO, Reddy AK, Engelhardt S, Taffet GE, Wehrens XH, Entman ML, Rodriguez A (2013) microRNA‐22 Promotes Heart Failure through Coordinate Suppression of PPAR/ERR‐Nuclear Hormone Receptor Transcription. PLoS One 8: e75882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524 [DOI] [PubMed] [Google Scholar]
- Hamadeh MJ, Devries MC, Tarnopolsky MA (2005) Estrogen supplementation reduces whole body leucine and carbohydrate oxidation and increases lipid oxidation in men during endurance exercise. J Clin Endocrinol Metab 90: 3592–3599 [DOI] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS (2000) Increased adipose tissue in male and female estrogen receptor‐alpha knockout mice. Proc Natl Acad Sci USA 97: 12729–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS (2010) Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J 24: 4660–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZP, Chen J, Seok HY, Zhang Z, Kataoka M, Hu X, Wang DZ (2013) MicroRNA‐22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res 112: 1234–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nature Rev Genet 12: 99–110 [DOI] [PubMed] [Google Scholar]
- Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER (2000) Aromatase‐deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA 97: 12735–12740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchy A, Edvardsson K, Aydogdu E, Williams C (2012) Estradiol‐activated estrogen receptor alpha does not regulate mature microRNAs in T47D breast cancer cells. J Steroid Biochem Mol Biol 128: 145–153 [DOI] [PubMed] [Google Scholar]
- Klinge CM (2012) miRNAs and estrogen action. Trends Endocrinol Metab 23: 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: 4051–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA package 2.0. Algorithms Mol Biol 6: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundsgaard AM, Kiens B (2014) Gender differences in skeletal muscle substrate metabolism – molecular mechanisms and insulin sensitivity. Front Endocrinol 5: 195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL (2011) Pyruvate kinase M2 is a PHD3‐stimulated coactivator for hypoxia‐inducible factor 1. Cell 145: 732–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Zhang H, Yuan L, Jing H, Thacker P, Li D (2011) CREBL2, interacting with CREB, induces adipogenesis in 3T3‐L1 adipocytes. Biochem J 439: 27–38 [DOI] [PubMed] [Google Scholar]
- Mauvais‐Jarvis F, Clegg DJ, Hevener AL (2013) The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34: 309–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon A, Rhead WJ (1987) Complementation analysis of fatty‐acid oxidation disorders. J Clin Invest 79: 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran VA, Niland CN, Khalil AM (2012) Co‐Immunoprecipitation of long noncoding RNAs In Methods in molecular biology, Engel N. (ed.), pp 219–228. New York City, NY: Humana Press; [DOI] [PubMed] [Google Scholar]
- Pandey DP, Picard D (2009) miR‐22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol Cell Biol 29: 3783–3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CJ, Zhao Z, Glidewell‐Kenney C, Lazic M, Chambon P, Krust A, Weiss J, Clegg DJ, Dunaif A, Jameson JL, Levine JE (2011) Genetic rescue of nonclassical ERalpha signaling normalizes energy balance in obese Eralpha‐null mutant mice. J Clin Invest 121: 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, Hevener AL (2010) Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha‐deficient mice. Am J Physiol Endocrinol Metab 298: E304–E319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, Mohammad L, Henstridge DC, Febbraio MA, Hewitt SC, Korach KS, Bensinger SJ, Hevener AL (2011) Myeloid‐specific estrogen receptor alpha deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci USA 108: 16457–16462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V, Drew BG, Zhou ZQ, Phun J, Kalajian NY, Soleymani T, Daraei P, Widjaja K, Wanagat J, Vallim TQD, Fluitt AH, Bensinger S, Le T, Radu C, Whitelegge JP, Beaven SW, Tontonoz P, Lusis AJ, Parks BW, Vergnes L et al (2016) Skeletal muscle action of estrogen receptor a is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci Transl Med 8: 334ra54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Becerril S, Ezquerro S, Mendez‐Gimenez L, Fruhbeck G (2016) Crosstalk between adipokines and myokines in fat browning. Acta Physiol 219: 362–381 [DOI] [PubMed] [Google Scholar]
- Rolfe DF, Brown GC (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77: 731–758 [DOI] [PubMed] [Google Scholar]
- Sakakibara I, Fujino T, Ishii M, Tanaka T, Shimosawa T, Miura S, Zhang W, Tokutake Y, Yamamoto J, Awano M, Iwasaki S, Motoike T, Okamura M, Inagaki T, Kita K, Ezaki O, Naito M, Kuwaki T, Chohnan S, Yamamoto TT et al (2009) Fasting‐induced hypothermia and reduced energy production in mice lacking acetyl‐CoA synthetase 2. Cell Metab 9: 191–202 [DOI] [PubMed] [Google Scholar]
- Salehzadeh F, Rune A, Osler M, Al‐Khalili L (2011) Testosterone or 17{beta}‐estradiol exposure reveals sex‐specific effects on glucose and lipid metabolism in human myotubes. J Endocrinol 210: 219–229 [DOI] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B (2004) JASPAR: an open‐access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32: D91–D94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandona D, Desaphy JF, Camerino GM, Bianchini E, Ciciliot S, Danieli‐Betto D, Dobrowolny G, Furlan S, Germinario E, Goto K, Gutsmann M, Kawano F, Nakai N, Ohira T, Ohno Y, Picard A, Salanova M, Schiffl G, Blottner D, Musaro A et al (2012) Adaptation of mouse skeletal muscle to long‐term microgravity in the MDS mission. PLoS One 7: e33232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoors S, Bruning U, Missiaen R, Queiroz KCS, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, Heggermont W, Godde L, Vinckier S, Van Veldhoven PP, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M, De Bock K et al (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520: 192–U113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63 [DOI] [PubMed] [Google Scholar]
- Simpson ER, Misso M, Hewitt KN, Hill RA, Boon WC, Jones ME, Kovacic A, Zhou J, Clyne CD (2005) Estrogen–the good, the bad, and the unexpected. Endocr Rev 26: 322–330 [DOI] [PubMed] [Google Scholar]
- Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469: 336–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3 [DOI] [PubMed] [Google Scholar]
- Su H, Mills AA, Wang X, Bradley A (2002) A targeted X‐linked CMV‐Cre line. Genesis 32: 187–188 [DOI] [PubMed] [Google Scholar]
- Subauste AR, Das AK, Li X, Elliott BG, Evans C, El Azzouny M, Treutelaar M, Oral E, Leff T, Burant CF (2012) Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes 61: 2922–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbotina E, Sierra A, Zhu Z, Gao Z, Koganti SRK, Reyes S, Stepniak E, Walsh SA, Acevedo MR, Perez‐Terzic CM, Hodgson‐Zingman DM, Zingman LV (2015) Musclin is an activity‐stimulated myokine that enhances physical endurance. Proc Natl Acad Sci USA 112: 16042–16047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K (2009) Modulation of microRNA processing by p53. Nature 460: 529–533 [DOI] [PubMed] [Google Scholar]
- Trabucchi M, Briata P, Garcia‐Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG (2009) The RNA‐binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459: 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss AK, Thomas T, Gruss P (1997) Germ line chimeras from female ES cells. Exp Cell Res 230: 45–49 [DOI] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33: e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welboren WJ, van Driel MA, Janssen‐Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG (2009) ChIP‐Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J 28: 1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wystub K, Besser J, Bachmann A, Boettger T, Braun T (2013) miR‐1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet 9: e1003793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, Zhang X, Zou F, Gent LM, Hahner LD, Khan SA, Elias CF, Elmquist JK, Clegg DJ (2011) Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab 14: 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K (2006) Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 13: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File