

Abstract
In Duchenne muscular dystrophy (DMD), NF‐κB is activated in skeletal muscle from infancy regardless of the underlying dystrophin mutation and drives inflammation and muscle degeneration while inhibiting muscle regeneration. Edasalonexent (CAT‐1004) is a bifunctional orally administered small molecule that covalently links 2 compounds known to inhibit NF‐κB, salicylic acid and docosahexaenoic acid (DHA). Edasalonexent is designed to inhibit activated NF‐κB upon intracellular cleavage to these bioactive components. Preclinical data demonstrate disease‐modifying activity in DMD animal models. Three placebo‐controlled studies in adult subjects assessed the safety, pharmacokinetics, and pharmacodynamics of single or multiple edasalonexent doses up to 6000 mg. Seventy‐nine adult subjects received edasalonexent, and 25 received placebo. Pharmacokinetic results were consistent with the intracellular cleavage of edasalonexent to its active components. Food increased plasma exposures of edasalonexent and salicyluric acid, an intracellularly formed metabolite of salicylic acid. The NF‐κB pathway and proteosome gene expression profiles in peripheral mononuclear cells were significantly decreased (P = .02 and P = .002, respectively) after 2 weeks of edasalonexent treatment. NF‐κB activity was inhibited following a single dose of edasalonexent but not by equimolar doses of salicylic acid and DHA. Edasalonexent was well tolerated, and the most common adverse events were mild diarrhea and headache. In first‐in‐human studies, edasalonexent was safe, well tolerated, and inhibited activated NF‐κB pathways, suggesting potential therapeutic utility in DMD regardless of the causative dystrophin mutation, as well as other NF‐κB–mediated diseases.
Keywords: edasalonexent, CAT‐1004, Duchenne muscular dystrophy, NF‐κB, pharmacokinetics, inflammation
Duchenne muscular dystrophy (DMD) is characterized by progressive loss of skeletal muscle function and is the most common neuromuscular disorder of childhood, with a birth prevalence of approximately 1 in 3500 live male births.1 The X‐linked genetic disorder results from 1 of many possible dystrophin gene mutations. The lack of functional dystrophin is necessary, but not in and of itself sufficient, to cause the progressive and severe loss of muscle mass and function characteristic of DMD.2 Skeletal muscle inflammation and activation of the transcription factor NF‐κB are present from early stages of the disease and precede loss of muscle function.3 Activated NF‐κB is known to drive muscle degeneration and suppress muscle regeneration.4 Although no treatment halts disease progression, current standards of care include glucocorticoids that reduce muscle inflammation and prolong ambulation, reduce cardiomyopathy, and increase life span.5, 6 However, long‐term glucocorticoid use is limited by clinically significant side effects including growth inhibition, puberty delay, weight gain, behavioral changes, hypertension, hyperglycemia, osteoporosis, Cushingoid habitus/facies, and cataracts.5, 7 In addition, glucocorticoids are known to induce muscle atrophy by activating expression of genes, such as Murf1, involved in the proteasomal degradation of skeletal muscle proteins, and by inhibiting expression of genes, such as MyoD, required for skeletal muscle regeneration and development.8 This leads to chronic myopathy that is difficult to distinguish from the underlying DMD pathology.9 Thus, there is an unmet need for disease‐modifying agents for DMD that can be used alone or in combination with drugs that directly target the dystrophin gene to inhibit muscle loss and allow muscle regeneration.
NF‐κB is a multifunctional transcription factor that controls gene expression and is involved in cellular responses to stress, modulating immune and inflammatory responses as well as skeletal muscle development and protein homeostasis.10 NF‐κB is activated in skeletal muscle in DMD as a result of the loss of sarcolemmal dystrophin. Activated NF‐κB results in translocation of the active transcription factor p50/p65 to the nucleus, which regulates expression of genes involved in inflammation such as TNFα and IL‐1β, extracellular matrix such as MMP9, muscle protein homeostasis such as Murf1, and muscle regeneration such as MyoD.8 NF‐κB activation is an early event in DMD pathology and occurs in the muscles of infants with DMD prior to the onset of fibrosis or clinical manifestations.3, 11 Chronic activation of NF‐κB is a key driver of muscle degeneration in DMD.12, 13, 14, 15 NF‐κB signaling pathways are persistently elevated in immune cells and muscle fibers of patients with DMD, and inhibition of NF‐κB pathways13, 16, 17 significantly improves histology, function, and muscle regeneration in mdx mice, even with only a 50 % reduction in NF‐κB activity.13 NF‐κB is specifically activated in skeletal muscles undergoing mechanical stress,18 which may explain in part the disproportionate inflammation, muscle degeneration, and fibrosis in certain muscles (ie, posterior leg muscles)2, 19 of patients with DMD even though every muscle is deficient in dystrophin. In contrast, muscles such as the gracilis and sartorius in the thigh and the extraocular muscles, which are exposed to less mechanical stress and presumably less activation of NF‐κB, are relatively preserved.2, 19 Edasalonexent, [N‐(2‐[(4Z,7Z,10Z,13Z,16Z,19Z)‐docosa‐4,7,10,13,16,19‐hexaenamido] ethyl)‐2‐hydroxybenzamide], is an orally administered novel small molecule in which salicylic acid and docosahexaenoic acid (DHA) are covalently conjugated through an ethylenediamine linker and that is designed to synergistically leverage the ability of both of these compounds to inhibit NF‐κB.20 In preclinical studies salicylic acid has been shown to prevent NF‐κB–mediated muscle atrophy and to decrease protein catabolism in muscle.21, 22, 23 However, toxicity at the doses needed to achieve the high concentrations required to inhibit NF‐κB precludes their clinical utility for this target.24 DHA has been shown to upregulate anti‐inflammatory pathways and suppress proinflammatory pathways via modulation of NF‐κB activity.25, 26 In addition, DHA is metabolized intracellularly to anti‐inflammatory eicosanoids,27, 28, 29, 30 which also play a role in muscle fiber regeneration. Following uptake into cells by endocytosis, edasalonexent is hydrolyzed by the fatty acid amide hydrolase (FAAH) to release salicylic acid and DHA intracellularly and to suppress activated NF‐κB.20 Edasalonexent demonstrates better potency than its individual components separately, presumably because of the simultaneous delivery of the salicylate and DHA.20
Edasalonexent significantly inhibits NF‐κB p65‐dependent inflammatory responses as well as downstream proinflammatory genes modulated by p65 in the golden retriever DMD model.31 Long‐term administration of edasalonexent, or the related analogue CAT‐1041 in which DHA is replaced by eicosapentaenoic acid to maintain equivalent pharmacology, demonstrated several disease‐modifying characteristics. Reduction of fatigue in skeletal muscle following repeated eccentric contractions and increases in skeletal muscle mass were observed.31 In addition, reduction of inflammation and fibrosis resulted in increased exercise endurance in mdx mice and improved diaphragm function in both the mouse and dog DMD models.31
This paper presents the results of the phase 1 clinical program for edasalonexent that includes 3 placebo‐controlled studies in adults (Figure 1): a randomized, double‐blind single‐ascending‐dose study (Study 101; edasalonexent‐101; NCT01440166) to evaluate the safety and tolerability of edasalonexent relative to placebo and pharmacokinetics (PK) under fed and fasted conditions; a randomized, double‐blind multiple‐ascending‐dose study (Study 102; edasalonexent‐102; NCT01511900) to evaluate the safety and tolerability of edasalonexent over 2 weeks, PK, and the effects of edasalonexent treatment on NF‐κB activity; and a crossover biomarker study (Study 103; edasalonexent‐103; NCT01670773) to compare the effects of edasalonexent and equimolar doses of DHA and salicylic acid (in the form of salsalate, which is comprised of 2 molecules of salicylic acid released extracellularly by esterase activity) on NF‐κB activity. In this way the effect of edasalonexent on NF‐κB could be compared with the effect of DHA and salicylic acid delivered extracellularly.
Figure 1.
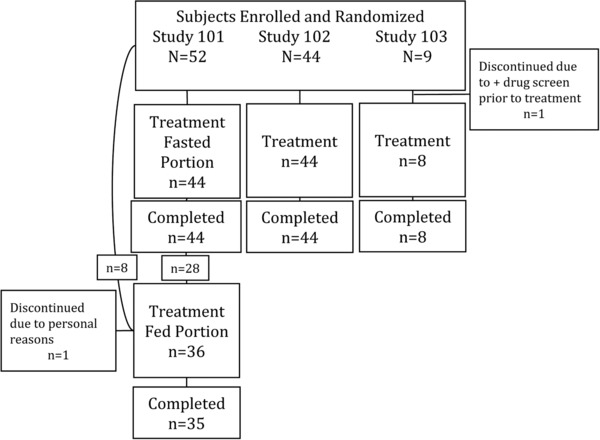
Disposition of subjects.
Subjects and Methods
Studies were approved by independent review boards (IRBs) at all sites (Celerion IRB, Lincoln Nebraska; Independent IRB, Plantation, Florida; New England IRB, Newton, Massachusetts) and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines as set forth by the International Conference on Harmonisation (ICH) and the US Code of Federal Regulations. All subjects gave written informed consent prior to study participation.
Eligible subjects in Studies 101 and 103 were healthy adults 18 (19 for Study 101) to 55 years of age with a body mass index (BMI) of 18 to 30 kg/m2. In Study 102, eligible subjects were male and female adults 18 to 65 years of age with a diagnosis of type 2 diabetes (who are known to have elevated levels of proinflammatory mediators and subclinical inflammation32), a BMI of 25 to 40 kg/m2, and screening glycosylated hemoglobin (HbA1c) ≥7.0% and ≤10.0% if treated with diet and exercise alone or ≥6.5% and ≤9.0% if receiving metformin. Subjects discontinued metformin during the study following a 7‐ to 21‐day washout period prior to dosing.
For all studies, subjects with a history of clinically significant allergies, alcohol/substance abuse, or eating disorders were not eligible. Women not pregnant or lactating were eligible if they were either using acceptable contraception or not of child‐bearing potential. Use of products containing salicylates or ω‐3 fatty acids was not permitted in the 2 weeks prior to study drug dosing, except for low‐dose aspirin permitted for subjects in Study 102. No use of prescription or nonprescription drugs or herbal or dietary supplements was permitted within 7 days of randomization for subjects in Studies 101 and 103. Permitted medications in Study 102 included stable (at least 2 months) use of antihypertensive, lipid‐lowering, and/or thyroid replacement therapies.
Subjects in Study 101 part 1 received a single dose of oral placebo or edasalonexent (300, 1000, 2000, 4000, or 6000 mg) after an overnight fast of at least 8 hours. In parts 2 and 3 of the study, continuing or new subjects received placebo or edasalonexent 1000, 2000, 4000, and 6000 mg immediately following a standardized high‐fat meal (50 g). Continuing subjects had a washout period of at least 10 days. Subjects were monitored for 72 hours in the clinic and had 1 follow‐up visit 7 to 10 days after dosing.
Subjects in Study 102 received oral placebo or edasalonexent at total daily doses of 300 mg, 1000 mg, 2000 mg (1000 mg BID), or 4000 mg (2000 mg BID) administered with a standard meal (up to 22 g of fat) for 14 days. Subjects were monitored as inpatients through day 7, returned for an outpatient visit on day 10, and were admitted to the clinic on day 13 until 24 hours after the last dose. A follow‐up visit occurred 7 to 10 days after the final dosing.
Subjects in Study 103 received a series of 3 single‐dose treatments administered with a high‐fat breakfast on an outpatient basis in the following order and with at least 7‐day washout periods:
edasalonexent 2000 mg →
salsalate 500 mg + DHA 1400 mg → placebo
Amounts of salsalate (comprised of 2 molecules of salicylic acid that are released extracellularly by esterase activity) and DHA corresponded to the approximate contents of salicylic acid and DHA in a 2000‐mg dose of edasalonexent, which corresponded to the highest individual dose in Study 102.
Assessments
Pharmacokinetics
Blood samples for PK assessments in Study 101 were collected predose and through 72 hours postdose on day 1. In Study 102, blood samples were collected prior to the morning doses on days 1 and 14 and for 24 hours postdose. Plasma edasalonexent, salicylic acid, salicyluric acid, and DHA in blood were determined using high‐performance liquid chromatography‐tandem mass spectrometry (HPLC‐MS/MS) methods validated with respect to accuracy, precision, linearity, sensitivity, and specificity at Celerion (Lincoln, Nebraska). The lower limits of quantitation (LLOQ) values were 1.00 ng/mL for edasalonexent, 100 ng/mL for DHA, 100 ng/mL for salicylic acid, and 2.50 ng/mL for salicyluric acid.
Pharmacodynamics
In Study 102, NF‐κB activity was assessed by measuring LPS‐stimulated p65 activation in blood samples obtained from 8 subjects in the 2000 mg BID edasalonexent cohort and 3 subjects in the placebo group predose on day 1 and after 14 days of treatment. Whole blood was stimulated with LPS for 2 hours prior to isolation of CD14‐positive cells using an anti‐CD14 affinity resin. Whole‐cell lysates were prepared, and total protein concentrations were measured using a BCA protein assay (Thermo Scientific Pierce, Rockford, Illinois). An enzyme‐linked immunosorbent assay (ELISA) was used to determine the amount of active p65 (NF‐κB p65 Transcription Factor Kit from Thermo Scientific Pierce, Rockford, Illinois). Gene expression analysis of a predefined NF‐κB gene set, as well as others for comparison (Biocarta and KEGG proteasome gene set), was performed via qPCR following isolation and amplification of mRNA (PAXgene Blood miRNA Kit, PreAnalytix, Hombrechtikon, Switzerland). In Study 103, NF‐κB activity was also assessed by measuring nuclear p65 DNA binding following ex vivo LPS stimulation in blood drawn before and 2 hours after administering edasalonexent, DHA, and salsalate given in combination, or placebo. Whole blood was stimulated with 200 ng/mL LPS for 2 hours before harvesting peripheral blood mononuclear cells (PBMCs). Nuclear lysates from isolated PBMCs were prepared using a commercially available nuclear extract kit (Active Motif, Carlsbad, California). Nuclear protein was quantified using ProStain fluorescent protein quantification (Active Motif). NF‐κB DNA binding activity was determined using a TransAM p65 DNA binding ELISA (Active Motif) in which equal volumes of nuclear lysates were loaded, and data were quantified against purified NF‐κB protein standard and normalized to the amount of protein loaded.
Mean changes or percentage changes from baseline plasma concentrations of adiponectin, high‐sensitivity C‐reactive protein, triglycerides, insulin, total cholesterol, high‐ and low‐density lipoprotein‐cholesterol, and apolipoprotein B for patients in Study 102 were assessed to determine any treatment effects.
Safety
Safety assessments included laboratory evaluations (hematology, chemistry, coagulation, and urinalysis), physical examinations, adverse events, 12‐lead ECGs, and vital signs (blood pressure, pulse, temperature, and respiration). Vital signs, physical exams, clinical labs, and ECGs were assessed/conducted at baseline (predose), throughout the treatment period, and at the end of dosing. Adverse events were monitored throughout the treatment and follow‐up periods.
Analyses and Statistics
Plasma concentrations were summarized by treatment group and day using descriptive statistics. Estimation of PK parameters was performed using WinNonlin v5.2 (Pharsight, Mountain View, California). The primary single‐dose or steady‐state (ss) PK parameters included area under the plasma concentration‐time curve (AUC) (including AUClast and AUCτ, the AUC from time 0 to time of last quantifiable concentration under single‐ and multiple‐dose conditions, respectively, and AUC∞, the AUC time curve from time 0 extrapolated to infinity by dividing the last measurable time point in the AUClast calculation by the apparent terminal elimination rate constant and summing it with AUClast), the observed maximum plasma concentration (Cmax), half‐life (t½), and the time of maximum plasma concentration (Tmax). Apparent oral clearance, CL/F, and apparent volume of distribution, Vz/F, were calculated.
Assessment of dose proportionality after a single‐dose of edasalonexent was made by examining plots of individual dose‐normalized edasalonexent, salicylic acid, salicyluric acid, and DHA AUC∞ and Cmax values vs the edasalonexent doses using the power model (SAS® PROC MIXED, v9.1.3) by linear regression of the natural log (ln)‐transformed PK parameters on the ln‐transformed dose.
The effect of food on PK parameters after a single dose of edasalonexent was evaluated by comparing ln‐transformed edasalonexent, salicylic acid, salicyluric acid, and DHA PK parameters Cmax, AUClast, and AUC∞ using an analysis of variance (ANOVA) model using PROC MIXED of SAS® (Version 9.1.3), with treatment as a fixed effect and subject as a random effect.
Paired 2‐tailed t‐testing compared p65 activity between edasalonexent and placebo groups. Raw qPCR data were used for t‐test analysis of individual genes. A preidentified NF‐κB gene set consisting of 14 genes (Biocarta NF‐κB Pathway) was analyzed along with other gene sets (including Proteasome, MAPK, ROS, Interleukins/Cytokines, and housekeeping genes, total = 75). For statistical analysis, changes from predose to day 14 for the preidentified NF‐κB gene set were assessed using the Student t‐test (2‐sided, equal variance) to test the null hypothesis that there was no change in expression of the preidentified NF‐κB gene set compared to baseline after 14 days of dosing with edasalonexent. Transcripts in the housekeeping and other gene sets were also measured for comparison, and P‐values for these gene sets were not adjusted for multiple comparisons. Descriptive statistics were reported for all safety assessments. ECG and clinical laboratory test results were classified as normal, abnormal not clinically significant, or abnormal clinically significant.
Results
Figure 1 shows the progression of subjects in the studies from screening to study end. A group of 105 subjects were randomized between September 2011 and October 2012, and all but 2 subjects completed the studies. Of these, 79 subjects received edasalonexent, and 25 received placebo. In Study 101, 44 subjects received single doses of placebo (n = 10) or 300 mg (n = 6), 1000 mg (n = 8), 2000 mg (n = 8), 4000 mg (n = 6), or 6000 mg (n = 6) edasalonexent during the fasted portion of the study. For the fed portion of the study, 28 subjects from the fasted portion of the study continued, and 8 additional new subjects were enrolled and randomized to placebo (n = 8) or 1000 mg (n = 8), 2000 mg (n = 7), 4000 mg (n = 6), or 6000 mg (n = 6) edasalonexent. One subject withdrew for personal reasons in the fed portion. In Study 102, 44 subjects were randomized to placebo (n = 12) or 300 mg (n = 6), 1000 mg (n = 8), 2000 mg (n = 9) as 1000 mg BID, or 4000 mg (n = 9) as 2000 mg BID edasalonexent; all subjects completed the study. Nine subjects were enrolled in Study 103. One subject was discontinued due to a positive drug screen prior to receiving edasalonexent.
Subject demographics are shown in Table 1. The majority of subjects in all 3 studies were male. In Studies 101 and 102, the majority of subjects were white, whereas in Study 103 most subjects were African American/black. Mean age across studies ranged from 33 to 53 years.
Table 1.
Subject Demographics
| Study 101 | ||||
|---|---|---|---|---|
| Fasted | Feda | Study 102 | Study 103 | |
| N = 44 | N = 36 | N = 44 | N = 9 | |
| Sex | ||||
| Female | 4 (9%) | 3 (9%) | 15 (34%) | 2 (22%) |
| Male | 40 (91%) | 32 (91%) | 29 (66%) | 7 (78%) |
| Race | ||||
| White | 33 (75%) | 28 (80%) | 34 (77%) | 1 (11%) |
| Black or African American | 10 (23%) | 5 (14%) | 6 (14%) | 6 (67%) |
| Other | 1 (2%) | 3 (9%) | 4 (9%) | 2 (22%) |
| Ethnicity: not Hispanic or Latino | 44 (100%) | 35 (100%) | 11 (25%) | 8 (89%) |
| Age, y, mean (SD) | 33.1 (11.3) | 33.0 (11.5) | 55.3 (7.1) | 40.5 (9.1) |
| Weight, kg, mean (SD) | 75.95 (10.42) | 76.83 (10.99) | 88.11 (15.42) | 83.4 (11.6) |
| Height, cm, mean (SD) | 175.8 (7.3) | 176.3 (7.1) | 167.2 (10.7) | 177.2 (10.5) |
| BMI, kg/mg2, mean (SD) | 24.56 (2.87) | 24.64 (2.57) | 31.48 (4.05) | 26.9 (2.8) |
28 of the 44 subjects in the fasted portion of the study plus 8 new subjects participated in the fed portion.
Pharmacokinetics
Single‐Dose Pharmacokinetics
Pharmacokinetic parameters following a single edasalonexent dose are shown in Table 2 for Study 101 and Table 4 for Study 102. Edasalonexent was rapidly absorbed, with median plasma Tmax values ranging from 0.8 hours to 3 hours across dose levels, and absorption was similar under fasting and fed conditions. Mean edasalonexent plasma exposure (Cmax, AUClast, and AUC∞) generally increased with increasing edasalonexent dose up to 4000 mg under fasting conditions and 6000 mg following a high‐fat meal. Mean t½ values increased with increasing dose and under fed conditions. Plots for edasalonexent AUC vs dose under fed and fasted conditions following a single dose on day 1 for subjects in Studies 101 and 102 are shown in Figure 2A. Exposure levels of edasalonexent taken with the 22‐g fat meal used in Study 102 were lower than those obtained in Study 101 following the standard high‐fat (∼50 g) meal but greater than the exposures when edasalonexent was administered under fasting conditions. Dose proportionality could not be concluded for plasma edasalonexent PK parameters under fed or fasted conditions (data not shown). Mean CL/F and Vz/F values of edasalonexent tended to increase with increasing edasalonexent dose, likely reflecting lack of dose proportionality.
Table 2.
Edasalonexent Plasma Pharmacokinetic Parameters Following Single Edasalonexent Doses (Study 101)
| Edasalonexent Dose (mg) in Fasted Portion of Study | |||||
| 300 | 1000 | 2000 | 4000 | 6000 | |
| Parameter | N = 6 | N = 8a | N = 8 | N = 6 | N = 6 |
| Edasalonexent | Mean ± SD | ||||
| Cmax (ng/mL) | 87 ± 35 | 69 ± 43 | 162 ± 108 | 206 ± 66 | 193 ± 55 |
| AUClast (ng·h/mL) | 130 ± 58 | 155 ± 94 | 359 ± 245 | 596 ± 121 | 546 ± 50 |
| t½ (h) | 2.3 ± 0.4 | 1.9 ± 0.3 | 4.0 ± 3.1 | 10.3 ± 7.6 | 8.7 ± 4.1 |
| CL/F (L/h) | 2832 ± 1872 | 6613 ± 3104 | 9685 ± 9945 | 6730 ± 1636 | 10,726 ± 966 |
| Vz/F (L) | 8867 ± 4454 | 17,360 ± 6967 | 36,109 ± 16,976 | 91,791 ± 59,035 | 136,106 ± 70,386 |
| Tmax (h)b | 0.8 (0.7, 1.5) | 1.5 (1, 6) | 1.5 (0.7, 2) | 1.5 (1.5, 3) | 1.3 (1, 4) |
| Salicyluric acid | N = 5 | N = 7 | N = 8 | N = 5 | N = 6 |
| Cmax (ng/mL) | 116 ± 29 | 128 ± 66 | 202 ± 60 | 185 ± 68 | 257 ± 89 |
| AUClast (ng·h/mL) | 416 ± 181 | 677 ± 279 | 876 ± 493 | 849 ± 367 | 1364 ± 393 |
| t½ (h) | NR | NR | 20.8 ± 10.1ǂ | 12.3 ± 5.7C | 29.4 ± 1.6ǂ |
| Tmax (h)b | 1 (1, 1.5) | 2 (1.5, 6) | 2 (1.5, 2) | 2 (1.5, 3) | 1.75 (1, 4) |
| DHA | N = 6 | N = 8 | N = 8 | N = 6 | N = 6 |
| Cmax (ng/mL) | 693 ± 127 | 738 ± 157 | 744 ± 192 | 746 ± 252 | 733 ± 186 |
| AUClast (ng·h/mL) | 21,375 ± 5208 | 25,245 ± 6165 | 25,325 ± 7589 | 26,606 ± 8774 | 26,956 ± 5262 |
| Tmax (h)** | 5 (3, 18) | 5 (0.7, 6) | 4 (0.3, 18) | 5 (0.3, 24) | 3 (2, 6) |
| Edasalonexent Dose (mg) in Fed Portion of Study | ||||
|---|---|---|---|---|
| 1000 | 2000 | 4000 | 6000 | |
| N = 8 | N = 7 | N = 6 | N = 6 | |
| Edasalonexent | ||||
| Cmax (ng/mL) | 383 ± 305 | 996 ± 174 | 1108 ± 463 | 946 ± 563 |
| AUClast (ng·h/mL) | 801 ± 410 | 2331 ± 638 | 2852 ± 1076 | 3573 ± 1558 |
| AUC∞ (ng·h/mL) | 823 ± 421 | 2366 ± 640 | 2903 ± 1098 | 3643 ± 1576 |
| t½ (h) | 10.4 ± 8.0 | 19.0 ± 2.7 | 21.4 ± 7.8 | 20.3 ± 9.3 |
| CL/F (L/h) | 1747 ± 1450 | 906 ± 280 | 1549 ± 563 | 2764 ± 3247 |
| Vz/F (L) | 17,956 ± 7978 | 24,795 ± 8501 | 43,879 ± 16,125 | 51,552 ± 17,424 |
| Tmax (h)a | 1.8 (1.5, 4) | 1.5 (1.5, 3) | 1.5 (1, 1.5) | 1.7 (1.5, 3) |
| Salicyluric acid | N = 5 | N = 7 | N = 6 | N = 6 |
| Cmax (ng/mL) | 193 ± 129 | 439 ± 130 | 500 ± 212 | 404 ± 220 |
| AUClast (ng·h/mL) | 849 ± 458 | 2004 ± 596 | 2512 ± 1036 | 2686 ± 915 |
| t½ (h) ǂǂ | 16.4 ± 14.3 | 24.7 ± 7.9 | 11.4 ± 12.6 | 19.6 ± 2.8 |
| Tmax (h)b | 2 (1.5, 3) | 3 (2, 4) | 3 (1.5, 3) | 3 (2, 9) |
| DHA | N = 8 | N = 7 | N = 6 | N = 6 |
| Cmax (ng/mL) | 503 ± 138 | 610 ± 181 | 557 ± 138 | 559 ± 115 |
| AUClast (ng·h/mL) | 23,988 ± 5664 | 21,801 ± 4798 | 17,103 ± 5909 | 22,782 ± 4295 |
| Tmax (h)** | 6 (6, 72) | 6 (0.3, 48) | 6 (3, 6) | 7.5 (4, 48) |
DHA, docosahexaenoic acid; NR, not reported; SD, standard deviation; AUClast, area under the serum concentration‐time curve from time 0 to time of last quantifiable concentration; CL/F, apparent oral clearance; CI, confidence interval; Cmax, maximum observed plasma concentration; Tmax, time to Cmax; t½,terminal half‐life; Vz/F apparent volume of distribution.
N = 7 for t½, CL/F, and Vz/F.
Tmax expressed as the median (min, max).
ǂN = 3 for t½ .
ǂǂN = 3, 4, 4, and 5 for the 1000‐, 2000‐, 4000‐, and 6000‐mg dose groups, respectively.
Table 4.
Edasalonexent Plasma Pharmacokinetic Parameters Following Multiple Edasalonexent Doses (Days 1 and 14, Study 102)
| Edasalonexent Dose Day 1 | ||||
|---|---|---|---|---|
| Parameters | 300 mg QD | 1000 mg QD | 1000 mg BID | 2000 mg BID |
| N = 6 | N = 8 | N = 9 | N = 9 | |
| Edasalonexent | ||||
| Cmax (ng/mL) | 20 ± 8.2 | 155 ± 143 | 235 ± 109 | 307 ± 182 |
| AUClast (ng·h/mL)a | 54 ± 15 | 391 ± 244 | 380 ± 122 | 794 ± 349 |
| t½ (h)b | 1.8 ± 0.9 | 8.1 ± 6.3 | U | U |
| CL /F (L/h) | 5034 ± 1435 | 2792 ± 1343 | U | U |
| Vz/F (L) | 13,094 ± 5859 | 26,004 ± 18,287 | U | U |
| Tmax (h) | 1.5 (1.0, 4.0) | 3.0 (2.0, 6.0) | 2.0 (1.0, 3.0) | 2.0 (1.5, 4.0) |
| Salicyluric Acid | N = 5 | N = 2 | N = 4 | N = 3 |
| Cmax (ng/mL) | 21 ± 11 | (132, 154) | 197 ± 79 | 170 ± 80 |
| AUClast (ng·h/mL)a | 169 ± 43 | (473, 529) | 697 ± 406 | 612 ± 174 |
| t½ (h)ǂ | 9.7 ± 4.9 | (6.9, 20.9) | U | U |
| Tmax (h) | 4.0 (3.0, 6.0) | (2.0, 3.0) | 2.0 (2.0, 2.0) | 3.0 (1.5, 4.0) |
| DHA | N = 6 | N = 8 | N = 9 | N = 9 |
| Cmax (ng/mL) | 657 ± 275 | 895 ± 217 | 565 ± 96 | 596 ± 169 |
| AUClast (ng·h/mL)a | 10,219 ± 5193 | 13,093 ± 3349 | 2935 ± 413 | 3654 ± 1238 |
| Tmax (h) | 0 (0, 24) | 16 (0.5, 23) | 0 ± (0, 11) | 0 (0, 11) |
| Edasalonexent Dose Day 14 | ||||
| 300 mg QD | 1000 mg QD | 1000 mg BID | 2000 mg BID | |
| N = 6 | N = 8 | N = 9 | N = 9 | |
| Edasalonexent | ||||
| Cmax,ss (ng/mL) | 34 ± 14 | 212 ± 88 | 237 ± 123 | 354 ± 149 |
| AUCτ(ng·h/mL)a | 103 ± 48 | 582 ± 185 | 512 ± 175 | 1065 ± 414 |
| t½ (h)b | 18.9 ± 22.6 | 14.5 ± 10.0 | U | U |
| CLss/F (L/h) | 3382 ± 1389 | 1854± 509 | 2243 ± 1034 | 2199 ± 999 |
| Vzss /F (L) | 80,164 ± 99,614 | 28,757 ± 16,316 | U | U |
| Tmax,ss (h) | 1.75 (1.5, 6.0) | 3.0 (1.5, 9.0) | 3.0 (1.5, 6.0) | 2.0 (1.0, 6.0) |
| Salicyluric Acid | N = 5 | N = 2 | N = 4 | N = 4 |
| Cmax,ss (ng/mL) | 32 ± 22 | (32, 125) | 226 ± 76 | 234 ± 93 |
| AUCτ(ng·h/mL)a | 227 ± 84 | (448, 478) | 808 ± 336 | 1047 ± 407 |
| t½ (h) | 15.3 ± 4.2 | (53.4 53.4) | 2.1, 3.8 | 3.7 ± 1.3 |
| Tmax,ss (h) | 2.0 (2.0, 6.0) | (3.0, 12.0) | 2.5 (2.0, 4.0) | 2.5 (2.0, 4.0) |
| DHA | N = 6 | N = 8 | N = 9 | N = 9 |
| Cmax,ss (ng/mL) | 694 ± 314 | 749 ± 171 | 695 ± 208 | 806 ± 164 |
| AUCτ(ng·h/mL)a | 10130 ± 3972 | 11127 ± 3177 | 3918 ± 835 | 4654 ± 966 |
| Tmax,ss (h) | 0 (0, 24) | 24 (0, 24) | 0 (0, 10.5) | 0 (0, 10.5) |
Values expressed as mean ± SD except for Tmax presented as median (range) or as individual values where n < 2. BID, twice daily; DHA, docosahexaenoic acid; QD, once daily; U, measurement was unreliable; AUCτ, area under the serum concentration‐time curve for dosing period; CLss/F, steady‐state apparent clearance (as a function of bioavailability); Cmax,ss, maximum observed steady‐state serum concentration; SD, standard deviation; Tmax,ss, steady‐state time to Cmax; t½, terminal half‐life; Vzss/F, steady‐state apparent volume of distribution (as a function of bioavailability).
Dosing interval was 0‐24 hours postdose for QD cohorts and 0‐10.5 hours postdose for BID cohorts.
N = 4 and 7 on day 1, and N = 5 and 3 on day 14 for the 300 and 1000 mg QD groups, respectively.
ǂN = 3 and 2 on day 1 and N = 3 and 1 on day 14 for the 300 and 1000 mg QD groups, respectively.
Figure 2.
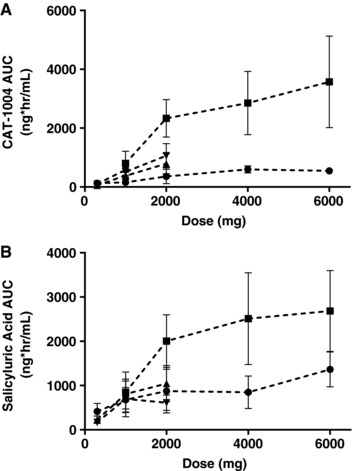
Effect of meals on edasalonexent and salicylic acid exposures. Edasalonexent (A) and salicyluric acid (B) AUClast following dosing under fed (squares, high‐fat meal, 50 g in Study 101; triangles, moderate‐fat meal, 22 g in Study 102, day 1 or 14) or fasted (circles, Study 101) conditions. Bars indicate SD.
Administration of a high‐fat meal resulted in a lack of bioequivalence for mean plasma edasalonexent maximum and overall exposure by approximately 3‐ to 8‐fold (Table 3 and Figure 2A). Mean ratios of fed/fasted ranged from 2.92 to 7.78 for Cmax, 3.69 to 8.13 for AUClast, and 3.64 to 7.97 for AUC∞. Statistical comparisons of plasma edasalonexent PK parameters under fasting and fed conditions resulted in 90% CIs outside the 80% to 125% interval, with the entire 90% CIs above 100% (Table 3).
Table 3.
Comparison of Pharmacokinetic Parameters Under Fed and Fasted Conditions
| Geometric LS Means | |||||
|---|---|---|---|---|---|
| Edasalonexent Dose (mg) | Parameters | Fed | Fasted | Mean Ratio (Fed/Fasted × 100) | 90%CI |
| 1000 | Cmax (ng/mL) | 284 | 48 | 587 | 249–1384 |
| AUClast (ng·hr/mL) | 689 | 119 | 578 | 322–1035 | |
| 2000 | Cmax (ng/mL) | 983 | 126 | 778 | 421–1435 |
| AUClast (ng·hr/mL) | 2265 | 279 | 813 | 504–1309 | |
| 4000 | Cmax (ng/mL) | 1034 | 212 | 489 | 320–746 |
| AUClast (ng·hr/mL) | 2508 | 679 | 369 | 266–513 | |
| 6000 | Cmax (ng/mL) | 770 | 264 | 292 | 112–763 |
| AUClast (ng·hr/mL) | 3096 | 662 | 468 | 184–1188 | |
Parameters were ln‐transformed prior to analysis, and the geometric least‐squares (LS) means were calculated by exponentiating the LS means from the ANOVA.
Plasma salicylic acid concentrations were below quantifiable limits (<100 ng/mL) for all subjects under fed or fasted conditions except 2 subjects with detectable levels of salicylic acid prior to any edasalonexent, (data not shown). In contrast, plasma concentrations of the salicylic acid metabolite salicyluric acid were detectable, with the median Tmax values for salicyluric acid ranging from approximately 1 hour to 2 hours under fasting conditions. Pharmacokinetic parameters for salicyluric acid following a single edasalonexent dose are shown in Table 2 and Table 4. Under fed conditions, median Tmax was slightly delayed, occurring between 2 and 3 hours and consistent with the edasalonexent parent analyte. Significantly higher exposure of salicyluric acid was observed under fed conditions. Higher salicyluric acid levels were generally associated with a slightly later median Tmax than was observed for edasalonexent. Plasma concentrations of edasalonexent and salicyluric acid over time are shown in Figure 3A and 3B, respectively. Mean plasma salicyluric acid exposure (Cmax and AUClast) generally increased with increasing edasalonexent dose, although not in a dose‐proportional manner. Mean t½ values were variable across treatments. Administration of a high‐fat meal increased mean plasma salicyluric acid maximum and overall exposure (Figure 2B), but to a lesser extent than observed for plasma edasalonexent, with mean ratios ranging from approximately 123% to 242%.
Figure 3.
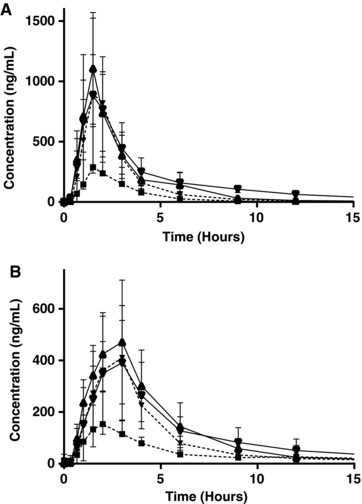
Edasalonexent and salicylic acid exposures over time. Mean edasalonexent (A) and salicyluric acid (B) plasma concentrations over time following a single edasalonexent dose of 1000, 2000, 4000, or 6000 mg under fed conditions. Bars indicate SD.
Mean plasma DHA exposure was not influenced by edasalonexent dosing under either fed or fasting conditions, but instead by timing of meals, as reflected in the lack of effect of edasalonexent dose level on mean plasma DHA Cmax and AUClast values and the timing of Tmax with respect to meals (Table 2); median Tmax was generally prior to or shortly after first postdose meal, ie, 6 hours postdose.
Multiple‐Dose Pharmacokinetics
Pharmacokinetic parameters following daily dosing with edasalonexent for 14 days are shown in Table 4. Although parameters are presented following multiple dosing, the results for the first dose administration were generally consistent with those observed in Study 101. Mean maximum and overall plasma exposure generally increased with increasing doses of edasalonexent up to 4000 mg daily. Mean plasma edasalonexent exposure demonstrated slight accumulation after multiple doses, with accumulation ratio values ranging from 1.35 to 1.67 across dose levels. The median plasma edasalonexent Tmax,ss values following the morning dose on day 14 ranged from 1.75 hours to 3 hours.
Mean, maximum, and overall plasma salicyluric acid exposure generally increased with increasing multiple (day 14) doses of edasalonexent, although not in proportion to dose (Table 4). Plasma salicylic acid concentrations were below quantifiable limits (<100 ng/mL) for all subjects except subjects taking low‐dose aspirin (acetylsalicylic acid, data not shown). Plasma DHA exposure did not appear to increase in response to multiple doses of edasalonexent, as shown by the lack of effect of edasalonexent dose on mean plasma DHA Cmax or AUC values (Table 4).
Pharmacodynamics: NF‐κB Activity
In Study 102, LPS‐stimulated p65 activity was reduced for 5 of 8 subjects receiving 14 days of edasalonexent treatment (4000 mg daily) but not in 3 subjects receiving placebo. Mean (SEM) p65 activity level for subjects in the edasalonexent group was 8600 (3276) LU/μg protein prior to dosing on day –1 and decreased by 76% to 2038 (3184) LU/μg protein at day 14. In contrast, mean p65 activity for 3 subjects receiving placebo was not changed from pretreatment to day 14 (6135 and 7363 LU/μg protein, respectively).
Also, in Study 102 edasalonexent treatment decreased expression of genes in NF‐κB and proteosome gene sets. The expression of a predefined NF‐κB pathway gene set (14 genes: CHUK, FADD, IRF3, MMP9, MYP88, NFKB1, NFKBIA, RIPK1, TICAM2, TLR2, TLR4, TNF, TNFAIP3, and TNFRSF1B) was significantly decreased (P = .02) after 2 weeks of edasalonexent treatment in comparison to day 1. Similarly, a predefined proteosome gene set (8 genes: PSMA5 [alpha5], PSMA7 [alpha4], PSMB1 [beta6], PSMB2 [beta4], PSMB3 [beta3], PSMB5 [beta5], PSMB6 [beta1], and PSMC3 [Rpt5]) was also significantly downregulated (P = .002) on edasalonexent treatment. Expression of these gene sets was not significantly reduced on treatment with placebo.
For subjects in Study 103, there was a statistically significant inhibition of NF‐κB p65 DNA binding activity (P < .005) in PBMC nuclear extracts isolated from LPS‐stimulated whole blood following a single 2000‐mg dose of edasalonexent (Figure 4A). In contrast, inhibition was not observed following a single administration of equimolar doses of salsalate and DHA or placebo. The majority of subjects (7/8) had reductions in p65 activity following edasalonexent administration (Figure 4B) compared to salsalate and DHA (2/8) or placebo (3/8)
Figure 4.
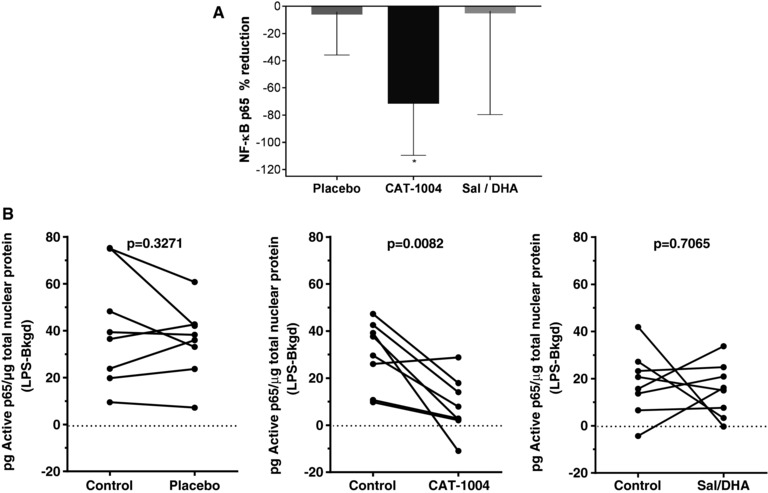
NF‐κB target engagement on treatment with edasalonexent (Study 103). A, Mean (SEM) percentage reduction relative to pretreatment values in nuclear NF‐κB p65 DNA binding following administration of placebo, edasalonexent (2000 mg), or salsalate (500 mg) + DHA (1400 mg). *Statistically significant reduction vs placebo (P < .005). B, Individual p65 responses for the 3 treatment groups.
For subjects with type 2 diabetes in Study 102, dosing of edasalonexent at 300 mg QD to 2000 mg BID for up to 14 days did not appear to alter PD markers of glucose, lipid homeostasis, or hsCRP (data not shown).
Safety and Tolerability
There were no serious adverse events, dosing interruptions, or discontinuations due to adverse events during any of the studies. Vital signs and ECG parameters remained within normal limits postdose, with minimal changes from baseline, and there were no clinically significant changes in laboratory values for any subject. The list of adverse events for Studies 101 and 102 occurring in more than 1 subject are shown in Table 5. There were no adverse events reported in Study 103.
Table 5.
Treatment‐Emergent Adverse Events Occurring in >1 Subject in Studies 101 and 102 a
| Study 101 Daily Dose (mg) | Study 102 Daily Dose (mg) | ||||||||||||
| Placebo | 300 | 1000 | 2000 | 4000 | 6000 | Total | Placebo | 300 | 1000 | 2000 | 4000 | Total | |
| N = 13 | N = 6 | N = 8 | N = 8 | N = 9 | N = 8 | N = 52b | N = 12 | N = 6 | N = 8 | N = 9 | N = 9 | N = 44 | |
| No. of adverse events | 6 | 1 | 2 | 7 | 23 | 15 | 54 | 4 | 7 | 4 | 2 | 10 | 27 |
| Adverse events | Subjects (%) | ||||||||||||
| Diarrhea | 0 | 1 (17) | 0 | 1 (14) | 5 (56) | 2 (25) | 9 (20) | 0 | 0 | 0 | 0 | 2 (22) | 2 (5) |
| Headache | 2 (25) | 0 | 0 | 1 (14) | 1 (11) | 4 (50) | 8 (17) | 0 | 0 | 0 | 1 | 0 | 1 (2) |
| Abdominal pain, upper | 0 | 0 | 0 | 0 | 3 (33) | 1 (13) | 4 (9) | 0 | 0 | 0 | 0 | 0 | 0 |
| Dyspepsia | 0 | 0 | 0 | 0 | 2 (22) | 1 (13) | 3 (7) | 0 | 0 | 1 (13) | 0 | 0 | 1 (2) |
| Nausea | 0 | 0 | 1 (13) | 0 | 0 | 2 (25) | 3 (7) | 0 | 0 | 0 | 0 | 1 (11) | 1 (2) |
| Dizziness | 0 | 0 | 0 | 1 (14) | 1 (11) | 0 | 2 (4) | 0 | 1 (17) | 0 | 0 | 0 | 1 (2) |
| Gastroenteritis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8) | 0 | 1 (13) | 0 | 0 | 2 (5) |
| Upper RT infection | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (22) | 2 (5) |
| Wheezing | 1 (13) | 0 | 1 (13) | 0 | 0 | 0 | 2 (4) | 0 | 0 | 0 | 0 | 0 | 0 |
RT, respiratory tract.
No adverse events were reported in Study 103.
All subjects participating in the fed, fasted, or both fed and fasted parts of the study; 44 subjects participated only in the fasted portion of the study, 23 subjects continued, and 8 new subjects participated in the fed portion. Subjects with adverse events in both portions of the study are counted once. Descriptions of subjects with events during the fasted vs the fed portion of the study are provided in the text.
In Study 101, 54 adverse events were reported overall. In the fasted portion of the study, 11/44 subjects (1 subject in the placebo group and 1, 1, 2, 4, and 3 subjects in the 300, 1000, 2000, 4000, and 6000 mg/day edasalonexent groups, respectively) reported 17 adverse events following the single dose. The majority (15/17) were mild in severity. Two events (1 of headache in a subject in the 4000‐mg group and 1 of herpes zoster in a subject in the 4000‐mg group) were moderate in severity. Headache was the most common event in the fasted portion of the study. In the fed portion of the study, 18/35 subjects (4 subjects in the placebo group, and 1, 2, 16, and 13 subjects in the 1000‐, 2000‐, 4000‐, and 6000‐mg edasalonexent groups, respectively) reported 37 adverse events following the single dose, and all were mild. Headache and diarrhea were the most common events. Gastrointestinal adverse events occurred primarily with single doses of 4000 and 6000 mg and were infrequent at lower doses.
For the 44 subjects in Study 102 receiving edasalonexent or placebo for 14 days, 16 (4 subjects in the placebo group, and 7, 4, 2, and 10 subjects in the 300, 1000, 2000, and 4000 mg/day edasalonexent groups, respectively) reported 27 treatment‐emergent adverse events, the majority of which (25/27) were mild in severity. There was no dose‐dependent trend in adverse event incidence. One subject in the 2000‐mg group had a headache considered severe and another headache event of moderate intensity. A subject in the 300‐mg group had a urinary tract infection of moderate intensity. Diarrhea and upper respiratory tract infection were the most common adverse events in subjects receiving edasalonexent.
Discussion
The 3 phase 1 studies described in this paper support the safety and tolerability of edasalonexent in healthy adults and in adults with chronic inflammation (ie, type 2 diabetes32, 33). No safety concerns were identified in the 79 subjects treated with single doses up to 6000‐mg under fed or fasted conditions or multiple doses up to 4000 mg daily for 14 days under fed conditions. No subject withdrew due to an adverse event, and there were no clinically significant abnormalities in laboratory, ECG, or vital sign assessments. Neither quantifiable salicylic acid levels nor dose‐dependent increases in DHA were detected in plasma, indicating that cleavage of edasalonexent to release its active components does not occur outside of the cellular compartment.
Proof of concept for the mechanism of action of edasalonexent was demonstrated by the statistically significant 70% reduction in LPS‐stimulated NF‐κB activity as well as decreased NF‐κB p65 DNA binding activity. After 14 days of dosing with 4000 mg daily, edasalonexent inhibited unstimulated NF‐κB target genes and proinflammatory genes in subjects with type 2 diabetes mellitus. There is substantial evidence that patients with type 2 diabetes have low‐grade inflammation.34 In fact, inhibition of NF‐κB with salicylates has been shown to improve parameters of glucose homeostasis.35 We believe that demonstration of NF‐κB inhibition in subjects not affected by DMD is a relevant first step supporting studies in pediatric patients with DMD.
NF‐κB is a key signaling molecule in skeletal muscle disease.14 In in vitro systems, DHA and salicylic acid suppress translocation of activated NF‐κB from the cytoplasm to the nucleus and decrease nuclear p65 expression.15, 25, 36 Specific FAAH‐mediated hydrolysis of the covalent link that conjugates salicylic acid to DHA in edasalonexent allows the simultaneous intracellular delivery of both of these inhibitors of activated NF‐κB without significant systemic exposure, thereby synergistically increasing the activity of these molecules to inhibit NF‐κB while potentially eliminating extracellular activities that may underlie nonspecific toxicity and side effects of the component molecules, something that cannot be achieved by coadministration of the individual molecules. This approach was validated for this novel small‐molecule drug candidate as demonstrated by significant reduction in NF‐κB p65 DNA binding activity following edasalonexent administration, in contrast to oral administration of equimolar concentrations of unconjugated salicylic acid and DHA, which had no effect on NF‐κB p65 DNA binding activity.
Although background sources from diet and the environment37, 38 can complicate interpretation of salicylic acid results, edasalonexent did not produce quantifiable plasma salicylic acid. These data support the hypothesis that edasalonexent is stable in plasma and absorbed by cells intact. Edasalonexent is then cleaved intracellularly, where salicylic acid is released and converted to salicyluric acid by glycine conjugation.39 Edasalonexent and plasma salicyluric acid levels increased with edasalonexent doses and with a high‐fat meal. DHA levels did not increase with increasing edasalonexent dose, presumably due to prolonged residence time of DHA in cellular membranes, where the half‐life for DHA following oral administration is greater than 2 days.40
A substantial difference in edasalonexent exposure was observed in Study 101 between the fasted and high‐fat meal‐fed groups. The AUC for edasalonexent in subjects fed a moderate‐fat meal in Study 102 was substantially higher than that observed with fasting administration in Study 101, supporting the administration of edasalonexent with meals containing some fat in future clinical studies. Observations of plateaus in Cmax and AUC above the 2000‐mg dose may be due to limitations in absorption given the hydrophobic nature of the molecule. Initial PK and safety studies in pediatric patients with DMD include divided daily doses of 33, 67, and 100 mg/kg, which correspond to 2000‐, 4000‐, and 6000‐mg doses for a 60‐kg adult.41
The edasalonexent doses used in the phase 1 studies included those that will be tested on a weight‐adjusted basis in the DMD pediatric patient population. Of note, the exposures following these single and multiple doses were greater than those found to be pharmacologically active in mouse and dog models of DMD, in which edasalonexent or analogues demonstrated disease‐modifying activity.31 The level of NF‐κB inhibition necessary for clinical benefit in DMD patients is not known. However, ablation of 1 allele of the p65 subunit of NF‐κB (ie, a 50% reduction in amount of activated NF‐κB) was sufficient to significantly improve pathology in mdx mice, implying that suppression rather than complete inhibition of activated NF‐κB may be sufficient for improvement in muscle pathology and function.13 Because muscle progenitor cells are exhausted very early in muscles of DMD patients due to futile rounds of degeneration and regeneration cycles, the ability of edasalonexent to block NF‐κB activity and maintain or allow replenishing of the progenitor population even partially could be disease modifying.
Targeting NF‐κB with edasalonexent has the potential to be effective in all DMD patients, regardless of the underlying dystrophin mutation, either as monotherapy or when used in combination with other therapies, including dystrophin‐ or myostatin‐targeted therapies. Several drug candidates in clinical development for DMD (eg, ataluren42 and eteplirsen43) target specific gene mutations in order to produce a partially functional dystrophin protein. A recent study in patients with Becker muscular dystrophy investigated the molecular basis for variable dystrophin levels in muscle and proposed that chronic inflammation in muscle microenvironments produces pathological microRNAs that inhibit dystrophin expression.44 Patients with DMD receiving dystrophin‐targeted therapies may benefit from a cotreatment that reduces muscle degeneration and inflammation while it enhances muscle regeneration.
A limitation of the current studies is that they were conducted in adults, and effects in pediatric populations need to be understood. A 2‐part phase 1/2 clinical trial in children with DMD is under way (NCT02439216) to assess PK, safety, and effects on MRI and functional endpoints.
In conclusion, in 3 first‐in‐human studies in adults, edasalonexent was observed to be safe and well tolerated and also to inhibit activated NF‐κB pathways, suggesting potential utility as a therapy in DMD regardless of the causative dystrophin mutation, as well as in other NF‐κB–mediated diseases.
Disclosures and Contributions
J.M.D., M.J., and M.Z. are or were employees of, and hold stock in, Catabasis Pharmaceuticals, Inc. E.O. and T.G. are employees of the clinical contract research organization contracted to perform bioanalytical and pharmacokinetic analysis for the studies.
All authors participated in the design, conduct, and/or analysis of the studies and approved the final manuscript. Catabasis Pharmaceuticals, Inc provided financial support for the conduct of the research and data analyses and for the preparation of the manuscript. Patrice C. Ferriola, PhD, of KZE PharmAssociates, LLC provided assistance in preparation of the manuscript.
Declaration of Conflicting Interests
JD, MZ, and MJ are/were employees of Catabasis.
Acknowledgments
We thank the subjects for their participation and the staff of the clinical research organizations (Celerion, Lincoln, Nebraska; Profil Institute for Clinical Research, Inc, Chula Vista, California; and ProMedica Clinical Research Center, Inc, Brighton, Massachusetts) for conduct of the studies. The authors gratefully acknowledge colleagues at Catabasis Pharmaceuticals, Inc for their contributions to the development of edasalonexent.
References
- 1. Emery AE. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromusc Disord. 1991;1(1):19–29. [DOI] [PubMed] [Google Scholar]
- 2. Porter JD, Merriam AP, Leahy P, Gong B, Khanna S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin‐deficient (mdx) mice. Hum Mol Genet. 2003;12(15):1813–1821. [DOI] [PubMed] [Google Scholar]
- 3. Chen YW, Nagaraju K, Bakay M, et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65(6):826–834. [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg AS, Puig M, Nagaraju K, et al. Immune‐mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. 2015;7(299):299rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008(1):CD003725. [DOI] [PubMed] [Google Scholar]
- 6. Schram G, Fournier A, Leduc H, et al. All‐cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013;61(9):948–954. [DOI] [PubMed] [Google Scholar]
- 7. Ricotti V, Ridout DA, Scott E, et al. Long‐term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. 2013;84(6):698–705. [DOI] [PubMed] [Google Scholar]
- 8. Hanaoka BY, Peterson CA, Horbinski C, Crofford LJ. Implications of glucocorticoid therapy in idiopathic inflammatory myopathies. Nat Rev Rheumatol. 2012;8(8):448–457. [DOI] [PubMed] [Google Scholar]
- 9. Schakman O, Gilson H, Kalista S, Thissen J. Mechanisms of muscle atrophy induced by glucocorticoids. Horm Res. 2009;72(suppl 1):36–41. [DOI] [PubMed] [Google Scholar]
- 10. Mourkioti F, Rosenthal N. NF‐kappaB signaling in skeletal muscle: prospects for intervention in muscle diseases. J Mol Med (Berl). 2008;86(7):747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. Activation of nuclear factor‐kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60(6):993–997. [DOI] [PubMed] [Google Scholar]
- 12. Shin J, Tajrishi MM, Ogura Y, Kumar A. Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol. 2013;45(10):2266–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Acharyya S, Villalta SA, Bakkar N, et al. Interplay of IKK/NF‐kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117(4):889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H, Malhotra S, Kumar A. Nuclear factor‐kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl). 2008;86(10):1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peterson JM, Bakkar N, Guttridge DC. NF‐kappaB signaling in skeletal muscle health and disease. Curr Top Dev Biol. 2011;96:85–119. [DOI] [PubMed] [Google Scholar]
- 16. Lu A, Proto JD, Guo L, et al. NF‐kappaB negatively impacts the myogenic potential of muscle‐derived stem cells. Mol Ther. 2012;20(3):661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mourkioti F, Kratsios P, Luedde T, et al. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest. 2006;116(11):2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar A, Boriek AM. Mechanical stress activates the nuclear factor‐kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17(3):386–396. [DOI] [PubMed] [Google Scholar]
- 19. Hu X, Blemker S. Musculoskeletal simulation can help explain selective muscle degeneration in Duchenne muscular dystrophy. Muscle Nerve. 2015;52:174–182. [DOI] [PubMed] [Google Scholar]
- 20. Vu CB, Bemis JE, Benson E, et al. Synthesis and characterization of fatty acid conjugates of niacin and salicylic acid. J Med Chem. 2016;59(3):1217–1231. [DOI] [PubMed] [Google Scholar]
- 21. Yin MJ, Yamamoto Y, Gaynor RB. The anti‐inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase‐beta. Nature. 1998;396(6706):77–80. [DOI] [PubMed] [Google Scholar]
- 22. Kopp E, Ghosh S. Inhibition of NF‐kappa B by sodium salicylate and aspirin. Science. 1994;265(5174):956–959. [DOI] [PubMed] [Google Scholar]
- 23. Cai D, Frantz JD, NE Tawa Jr, et al. IKKbeta/NF‐kappaB activation causes severe muscle wasting in mice. Cell. 2004;119(2):285–298. [DOI] [PubMed] [Google Scholar]
- 24. Rainsford K. Side effects and toxicology of the salicylates In:Rainsford K, Ed. Aspirin and related drugs. Boca Raton, FL: CRC Press; 2004:367–554. [Google Scholar]
- 25. Williams‐Bey Y, Boularan C, Vural A, et al. Omega‐3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF‐kappaB activation and enhancing autophagy. PLoS One. 2014;9(6):e97957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zwart SR, Pierson D, Mehta S, Gonda S, Smith SM. Capacity of omega‐3 fatty acids or eicosapentaenoic acid to counteract weightlessness‐induced bone loss by inhibiting NF‐kappaB activation: from cells to bed rest to astronauts. J Bone Mineral Res. 2010;25(5):1049–1057. [DOI] [PubMed] [Google Scholar]
- 27. Vedin I, Cederholm T, Freund‐Levi Y, et al. Effects of DHA‐rich n‐3 fatty acid supplementation on gene expression in blood mononuclear leukocytes: the OmegAD study. PLoS One. 2012;7(4):e35425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simopoulos AP. Omega‐3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 2002;21(6):495–505. [DOI] [PubMed] [Google Scholar]
- 29. Bouwens M, van de Rest O, Dellschaft N, et al. Fish‐oil supplementation induces antiinflammatory gene expression profiles in human blood mononuclear cells. Am J Clin Nutr. 2009;90(2):415–424. [DOI] [PubMed] [Google Scholar]
- 30. Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega‐3 fatty acid receptor mediating potent anti‐inflammatory and insulin‐sensitizing effects. Cell. 2010;142(5):687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hammers DW, Sleeper MM, Forbes SC, et al. Disease modifying effects of orally bioavailable NF‐κB inhibitors in dystrophin‐deficient muscle. JCI Insight. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pradhan A. Obesity, metabolic syndrome, and type 2 diabetes: inflammatory basis of glucose metabolic disorders. Nutr Rev. 2007;65(12 Pt 2):S152–156. [DOI] [PubMed] [Google Scholar]
- 33. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13(6):465–476. [DOI] [PubMed] [Google Scholar]
- 34. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goldfine AB, Fonseca V, Jablonski KA, et al. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2010;152(6):346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weldon SM, Mullen AC, Loscher CE, Hurley LA, Roche HM. Docosahexaenoic acid induces an anti‐inflammatory profile in lipopolysaccharide‐stimulated human THP‐1 macrophages more effectively than eicosapentaenoic acid. J Nutr Biochem. 2007;18(4):250–258. [DOI] [PubMed] [Google Scholar]
- 37. Duthie GG, Wood AD. Natural salicylates: foods, functions and disease prevention. Food Funct. 2011;2(9):515–520. [DOI] [PubMed] [Google Scholar]
- 38. Wood A, Baxter G, Thies F, Kyle J, Duthie G. A systematic review of salicylates in foods: estimated daily intake of a Scottish population. Mol Nutr Food Res. 2011;55 Suppl 1:S7–S14. [DOI] [PubMed] [Google Scholar]
- 39. Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet. 1985;10(2):164–177. [DOI] [PubMed] [Google Scholar]
- 40. Zuijdgeest‐van Leeuwen SD, Dagnelie PC, Rietveld T, van den Berg JW, Wilson JH. Incorporation and washout of orally administered n‐3 fatty acid ethyl esters in different plasma lipid fractions. Br J Nutr. 1999;82(6):481–488. [DOI] [PubMed] [Google Scholar]
- 41. Donovan J, Sweeney H, Vandenborne K, et al. CAT‐1004, an oral agent targeting NF‐κB in development for treatment of Duchenne muscular dystrophy: Phase 1/2 study design. Neuromusc Disord. 2015;25:S262. [Google Scholar]
- 42. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mendell JR, Rodino‐Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–647. [DOI] [PubMed] [Google Scholar]
- 44. Fiorillo AA, Heier CR, Novak JS, et al. TNF‐alpha‐induced microRNAs control dystrophin expression in Becker muscular dystrophy. Cell Rep. 2015;12(10):1678–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]