

Abstract
Current treatment regimens for gastric cancer are not adequate. Cancer stem cells (CSCs) may be a key driving factor for growth and metastasis of this tumor type. In contrast to the conventional clonal evolution hypothesis, CSCs can initiate tumor formation, self‐renew, and differentiate into tumor‐propagating cells. Because gastric cancer can originate from CSCs, it is necessary to review current targets of signaling pathways for CSCs in gastric cancer that are being studied in clinical trials. These pathways are known to regulate the self‐renewal and differentiation process in gastric CSCs. A better understanding of the clinical results of trials that target gastric CSCs will lead to better outcomes for patients with gastric cancer. Cancer 2017;123:1303–1312. © 2017 The Authors. Cancer published by Wiley Periodicals, Inc. on behalf of American Cancer Society. This is an open access article under the terms of the Creative Commons Attribution NonCommercial License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Keywords: cancer stem cells, clinical trials, gastric cancer, napabucasin, targeted therapy, vismodeqib
Short abstract
Cancer stem cells may be a key driving factor in the growth and metastasis of gastric cancer. Because gastric cancer can originate from cancer stem cells, it is necessary to review current targets of signaling pathways for cancer stem cells in gastric cancer that are being studied in clinical trials.
EPIDEMIOLOGY OF GASTRIC CANCER
In the United States, it was estimated that 24,590 people were diagnosed with gastric cancer and 16,980 were diagnosed with esophageal cancer in 2015 and that 26,310 men and women died from upper gastrointestinal tumors.1 Globally, gastric and gastroesophageal junction (GEJ) cancers are the second leading cause of death. The highest incidence of gastric/GEJ cancers is observed in Eastern Asia, Central and Eastern Europe, and South America.2, 3
RISK FACTORS FOR GASTRIC/GEJ CANCERS
Several risk factors have been identified for gastric cancer, including Epstein‐Barr virus, Helicobacter pylori infection, obesity, and dietary factors, such as red meat and pickled food consumption. There is a distinct familial risk for gastric cancer, and studies have demonstrated that the risk is higher for siblings than for parents and offspring.4 A high salt intake has been identified as a risk factor for gastric cancer in case‐control studies.5 Red and processed meat intake was associated with a 43% increase in the risk for gastric cancer.6 Epstein‐Barr virus‐encoded small RNA has been detected in up to 17.9% of patients with gastric cancer.7 H. pylori infection is associated with increased incidence of gastric cancer and mortality, and the risk of mortality from gastric cancer was 6‐fold higher in a population infected with H. pylori than in those with no infection.8 A body mass index >30 kg/m2 was associated with a greater than 2‐fold increased risk of esophageal and gastric adenocarcinoma.9 In the United States, there are currently no screening recommendations for gastric/GEJ cancers; however, surveillance should be conducted by endoscopy 1 year after the removal of adenomatous polyps and then no more frequently than at 3‐year to 5‐year intervals.10
CURRENT CLINICAL PRACTICE GUIDELINES AND TREATMENT FOR GASTRIC CANCER
Treatment choices for gastric cancer are based on clinical stage and overall health.11 Patients with no distant metastases (M0 disease) usually require multimodality treatment, which may include surgery, radiation, and chemotherapy, and should preferably be evaluated by a multidisciplinary team.11 When distant metastases are present (M1 disease), treatment options include systemic chemotherapy, clinical trial, or palliative/best supportive care.11
For patients with locally advanced, unresectable, or metastatic disease, preferred first‐line chemotherapy regimens are based on a platinum compound and a fluoropyrimidine doublet or triplet combination.11 Patients with human epidermal growth factor receptor 2 (HER2‐neu) overexpression should receive trastuzumab in addition to chemotherapy. Preferred second‐line regimens include: ramucirumab and paclitaxel, docetaxel, paclitaxel, irinotecan, or ramucirumab.11
Cytotoxic chemotherapy can reduce tumor burden, but its effect is usually transient, because resistance to chemotherapy lies mainly in the cancer stem cell (CSC) population.12 Even when a complete radiologic response is observed, CSCs are still present and eventually lead to tumor regrowth.12 Therefore, targeting CSCs is a rational approach to preventing tumor regrowth and the development of resistance.12 The combination of standard chemotherapy and anti‐CSC therapy may more efficiently eliminate both CSCs and non‐CSC tumor cells. In addition, the use of both standard chemotherapy and anti‐CSC therapies may improve the efficacy of standard chemotherapy and reduce the likelihood of acquired chemotherapy resistance.13
CANCER STEM CELLS
CSCs are characterized by their ability to generate tumor cells with different phenotypes.14 CSC populations account for a small proportion of the tumor bulk and usually remain quiescent for extended periods of time.14 CSCs are resistant to conventional therapies like chemotherapy and radiation because of activation of prosurvival and antiapoptotic pathways, overexpression of drug efflux pumps, and increased DNA repair capacity. Furthermore, chemotherapy and radiation can induce stemness genes in cancer cells, converting non‐CSCs to CSC‐like cells. These cells may remain after chemotherapy or radiation therapy and may be responsible for relapse after treatment.14
There are 3 primary models of tumor heterogeneity (Fig. 1). The clonal evolution model was the first to be described.15 In this model, cancer cell populations evolve progressively during tumorigenesis because of inherited genetic and epigenetic changes. The acquisition of accumulating mutations leads to the growth of novel cell populations. The second model, the classical CSC model, proposes that cancer cells within a given tumor exist in different states of stemness or differentiation.15 In this model, CSC to non‐CSC differentiation is a unidirectional process. The most tumorigenic cells reside at the top of the cellular hierarchy. These cells divide to generate identical CSCs (self‐renewal) and can also undergo asymmetric division to create non‐CSCs with limited tumorigenic and metastatic potential. Finally, the plastic CSC model describes a model in which bidirectional conversion exists between non‐CSCs and CSCs.15
Figure 1.
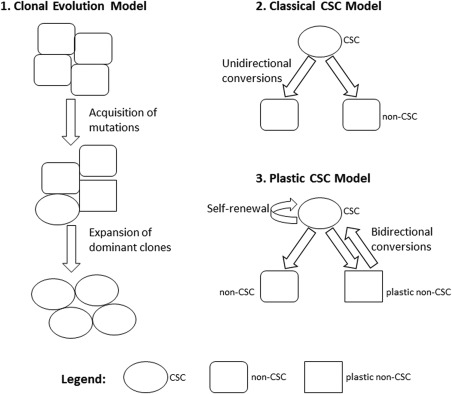
Primary models of cancer stem cells (CSCs) and tumor heterogeneity are illustrated.
A unified model of clonal evolution and CSC theory attempts to account for solid tumor heterogeneity and tumorigenesis. In this unified approach, the originating CSC that sustained the first oncogenic mutation is proposed to give rise to subclones with self‐renewal capabilities. These subclones then accumulate epigenetic and genetic changes over time. Each different CSC subclone gives rise to intermediate progenitors that lack self‐renewal capabilities. A subset of these progenitors follows a model of tumor cell plasticity and bidirectional conversion between non‐CSC and CSC states. This conversion between nontumorigenic and tumorigenic cell states is likely modulated by microenvironmental stimuli and endogenous transcription factors.16 This model suggests that the tumor cell is a dynamic state with highly adaptable CSCs and non‐CSCs that are capable of transient evolution and plasticity.16
CSCs in Gastric Cancer
Gastric CSCs were first described in 2007 by Yang et al.17 The telomerase expression in the gastric stem cells studied was up‐regulated by exposure to a chemical carcinogen (1 methyl‐3 nitro‐1 nitrosoguanidine [MNNG]) or H. pylori culture products. Unlike normal human cells or tissue, most human tumors demonstrate telomerase activity. Telomerase activation is significantly different between tumor tissue and noncancerous gastric tissue.
There are several proposed mechanisms by which H. pylori may interact with CSCs. It may induce CSC‐like properties to promote the development of gastric cancer by the up‐regulation of NANOG and octamer‐binding transcription factor 4 (Oct4) through Wnt/β‐catenin signaling, cytotoxin‐associated gene A (CagA)‐induced shatterproof 2 (SHP2) dysfunction, activation of bone morphogenetic protein/transforming growth factor‐β signaling, down‐regulation of sonic Hedgehog signaling, and recruitment of mesenchymal stem cells through interleukin 6 (IL‐6) and IL‐8 or bone marrow‐derived stem cells by means of chronic inflammation and C‐X‐C chemokine receptor type 4 (CXCR‐4) expression.18, 19 It has also been demonstrated that H. pylori infection up‐regulates aquaporin‐3 (AQP3) in gastric cancer and that ACP3 increases the expression of cluster of differentiation 44 (CD44 [a cell‐surface glycoprotein]) through the Wnt/glycogen synthase kinase 3β/β‐catenin signaling pathways and promotes CSC‐like properties in gastric cancer cells.20
It has been demonstrated that, because of chronic inflammation caused by H. pylori infection, bone marrow‐derived cells (BMDCs) repopulate the gastric mucosa and may contribute to carcinogenesis.21 It is believed that BMDCs differentiate in the gastric mucosa by cell‐cell fusion with local gastric epithelial cells and induce CSCs. However, the majority of dysplastic lesions do not arise from BMDCs.21 More studies are needed to fully understand the pathogenesis of gastric CSCs.
Gastric CSC Markers
Specific cellular markers have been used to identify, isolate and therapeutically target CSCs.21, 22, 23 Several potential gastric CSC markers have been identified. These include cluster of differentiation 44 (CD44, and its variants); CD24/CD44; CD54/CD44; CXCR4; epithelial cell adhesion molecule (EpCAM)/CD44; aldehyde dehydrogenase 1 (ALDH1); CD90; CD71‐negative; CD133; CD166; leucine‐rich, repeat‐containing, G‐protein–coupled receptor 5 (LGR5), Oct4; and sex‐determining region Y‐box 2 (Sox2). Table 1 lists examples of the CSC markers that have been studied in gastric cancer.24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 When CSCs are positive for several of these markers, the cells display a phenotype of tumorigenicity and chemoresistance. For instance, Nguyen and colleagues observed that CD44 and ALDH are the most specific biomarkers for detecting and isolating tumorigenic and chemoresistant gastric CSCs independent of the histologic classification of the tumor.25
Table 1.
Examples of Potential Gastric Cancer Stem Cell Biomarkers
| Cell‐Surface Marker | Phenotype of Marker‐Positive CSCs | Reference(s) |
|---|---|---|
| CD44 | Tumorigenicity, spheroid formation, chemoresistance | Takaishi 200924 |
| CD24/CD44 | Tumorigenicity | Nguyen 201625 |
| CD54/CD44 | Tumorigenicity, hierarchical organization | Chen 201226 |
| CD44/CD166/ALDH | Tumorigenicity, chemoresistance | Nguyen 201625 |
| CXCR4 | Tumorigenicity, chemoresistance | Fujita 201527 |
| EpCAM/CD44 | Tumorigenicity, phenotypical heterogeneity, chemoresistance | Han 201128 |
| ALDH1 | Tumorigenicity, phenotypical heterogeneity | Katsuno 201229 |
| CD90 | Tumorigenicity, trastuzumab‐reduced CD90‐positive population | Jiang 201230 |
| CD71‐negative | Tumorigenicity, chemoresistance, tumor cell invasion | Ohkuma 201231 |
| CD133 | Poorly differentiated gastric cancer, independent prognostic factor | Jiang 2012,32 Hashimoto 2014,33 Chen 201634 |
| LGR5 | Tumorigenicity | Gong 201635 |
| Oct4 | Tumorigenicity, tumor progression | Chen 201634 |
| Sox2 | Well or moderately differentiated gastric cancer | Chen 201634 |
Abbreviations: ALDH, aldehyde dehydrogenase; ALDH1, aldehyde dehydrogenase 1; CD133, cluster of differentiation 133 (a pentaspan membrane glycoprotein); CD166, activated leukocyte cell adhesion molecule (ALCAM); CD24, heat‐stable antigen CD24; CD44, cellular protein; CD54, intercellular adhesion molecule 1; CD71, transferrin receptor protein; CD90, Thy‐1 cell surface antigen; CSC, cancer stem cells; CXCR4, C‐X‐C chemokine receptor type 4; EpCAM, epithelial cell adhesion molecule; LGR5, leucine‐rich repeat‐containing G‐protein coupled receptor 5; Oct4, octamer‐binding transcription factor 4; Sox2, sex‐determining region Y‐box 2.
Other phenotypes conferred by CSC markers include hierarchical organization, with the presence of a small population of tumorigenic cells that give rise to a larger population of phenotypically diverse, nontumorigenic cells, and tumor cell invasion. CD133‐positive CSCs have demonstrated resistance to traditional chemotherapies,36, 37 and quantitative polymerase chain reaction analyses indicate that high CD133 expression is a marker for a poor prognosis.34 In addition, CD44 is expressed in up to 80% of primary gastric cancer resection specimens and is associated with more advanced clinicopathologic features and a poorer prognosis.21 CD44 expression also denotes a subpopulation of gastric cancer cells in which Hedgehog signaling pathway proteins are up‐regulated and also promote chemotherapy resistance and thus a poorer prognosis.21, 38 A variant of CD44 (CD44v8‐10) was identified as the predominant CD44 variant expressed in gastric cancer cells and contributes to tumor initiation, possibly through enhancing oxidative stress defense.39 It has also been demonstrated that CD44 and CD133 expression in gastric cancers is associated with statistically significant histology for intestinal‐type tumors and that CD44v5 is significantly associated with Signet ring histology.21 However, markers of CSCs have limitations, in that some CSC populations do not express these markers, whereas non‐CSC cancer cells may express them. For this reason, the markers can be used to identify CSC‐rich subpopulations (stemness‐high) but might not be able to isolate all CSCs.23
Several preclinical studies have indicated that targeting markers of CSCs may be an effective approach to treating gastric cancer. For instance, 2 anti‐LGR5 antibody‐drug conjugates effectively induced cytotoxicity in LGR5‐high gastrointestinal cancer cells, but not in LGR5‐negative or LGR4‐knockdown cancer cell lines.35 In another preclinical study, all‐trans retinoic acid down‐regulated the expression of CSC markers CD44 and ALDH and of stemness genes, including Kruppel‐like factor 4 (Klf4) and Sox2.40 In mouse xenograft models, 2 weeks of daily all‐trans retinoic acid treatment were sufficient to inhibit gastric tumor progression in vivo.40
A clinical study has demonstrated the potential for targeting CSC markers in gastric cancer.41 A variant form of CD44 (CD44v) reportedly interacts with xCT, a cysteine‐glutamate transporter subunit that maintains high levels of intracellular‐reduced glutathione (GSH), which helps to defend the cell against oxidative stress. Sulfasalazine (SSZ) is an inhibitor of xCT and can suppress the survival of CD44v‐positive stem‐like cancer cells both in vitro and in vivo. Eleven patients were enrolled in a dose‐ranging study and received daily SSZ doses from 8 to 12 g daily given 4 times daily with 2 weeks as 1 cycle. Among the 8 patients in that study who had CD44v‐positive cells in their pretreatment biopsies, the CD44v‐positive cancer cell population was reduced in the post‐treatment biopsies of 4 patients. Intratumoral GSH levels were also decreased in 2 patients. Thus, SSZ may be a promising treatment for targeting CSCs in gastric cancer.42
CSC SIGNALING PATHWAYS
Because CSCs are resistant to traditional chemotherapy treatments,13 the cellular pathways that drive stemness represent another rational target for therapy. There are several molecular signaling pathways known to be involved in the induction and maintenance of stemness in both normal and cancer cells.
Hedgehog Pathway
The Hedgehog signaling cascade plays a major role in many processes, such as cell differentiation and organ formation during normal vertebrate embryonic development.42 The Hedgehog pathway becomes inactive in most adult tissues, but it regulates adult stem cells and is involved in tissue maintenance and repair. Activation of the Hedgehog signaling pathway plays an important role in the pathogenesis of various types of cancers, including cancers of the skin, mammary gland, brain, lung, and prostate. Basal cell carcinoma, a form of skin cancer, has been associated with disruptions in Hedgehog signaling. Mutations in the genes PTCH and SMO, which code for Patched and Smoothened proteins, respectively, were observed in patients with this disease.43
NANOG Pathway
The NANOG transcription factor cooperates with signal transducer and activator of transcription 3 (STAT3) to transcribe stemness genes required for pluripotency. In embryonic stem cells, STAT3 forms a complex with NANOG, which translocates to the nucleus to transcribe genes required for maintaining pluripotency. The NANOG gene is expressed in a variety of cancers, and its expression correlates with poor survival in patients with cancer. Many studies suggest that NANOG enhances the defined characteristics of CSCs and may function as an oncogene to promote carcinogenesis.44
STAT3 Pathway
STAT proteins are located in the cytoplasm in resting (quiescent) cells as inactive proteins. Phosphorylation of a specific tyrosine residue is essential for STAT activation. The STAT family includes proteins STAT1, STAT3, STAT4, STAT5a, STAT5b, and STAT6. STAT3 is excessively active in many cancers and plays a major role in tumor growth and metastasis.45 Evidence supports the finding that STAT3 activation plays a critical role in each step of metastasis, including cellular proliferation, invasion, migration, and angiogenesis. Malignant transformation of cells by various protein tyrosine kinases, oncogenes, and viruses is mediated through STAT3 activation. In addition, it has been demonstrated that activation of the downstream phosphorylated STAT3 transcription factor pathway is facilitated by IL‐17 and also that IL‐17 is positively correlated with the transformation of quiescent gastric CSCs into invasive gastric CSCs.46 STAT3 signaling is associated with the up‐regulation of cyclin D1 and cMyc expression, contributing to accelerated cell‐cycle progression. In addition, STAT3 signaling provides survival signals and suppresses apoptosis in cancerous cells. STAT3 also has a crucial role in cellular migration, which is required for cell invasion and cancer metastases. STAT3 signaling is required for cell motility. Depletion of STAT3 reduces the rate of cellular migration. STAT3 activation protects tumor cells from immune surveillance and increases the number of surviving tumor cells that invade distant organs. Targeting STAT3 activation inhibits tumor growth and metastasis, both in vitro and in vivo, without affecting normal cells, thus suggesting that STAT3 could be a valid molecular target for cancer therapy.47
Wnt/β‐Catenin Pathway
The Wnt/β‐catenin pathway has been identified as 1 of the pathways for CSC renewal.48 Wnt ligands are produced from cells in the stem cell microenvironment, serving as a self‐renewal signal for the stem cells. Wnt signaling is reportedly involved in a process called epithelial‐to‐mesenchymal transition (EMT). Because the CSC population represents the only cells that propagate tumors, it can be extrapolated that CSCs are responsible for tumor metastases. EMT is 1 of the crucial, early steps in the invasion‐metastases cascade and has been associated with a poor clinical outcome. Epithelial cells that undergo EMT acquire CSC‐like phenotypes.49
CSC AGENTS IN CLINICAL DEVELOPMENT FOR GASTRIC CANCER
Residual CSCs that survive standard chemotherapy and radiation are sufficient for cancer recurrence. Because CSCs are considered a source of metastasis, combination therapy with agents that target CSCs along with standard chemotherapy may have a profound impact on the management of gastric cancer.
The rationale for targeting CSCs in gastric/GEJ cancers was first described in a study evaluating CTCs that expressed CD44, which is thought to be a gastric CSC marker.50 In that study, CTCs were identified in 27 of 45 patients. The presence of CTCs was significantly associated with lymph node metastases, distant metastases, and disease recurrence. Of the 27 patients who had CTCs, 19 had CD44‐positive CTCs, indicative of CSCs. Those who had CD44‐positive CTCs were more likely to develop metastases and recurrence. In addition, patients with recurrent disease and those with CD44‐positive cells had higher CTC counts. In 13 of 19 patients who had CD44‐positive CTCs, recurrent disease developed, and the time to recurrence was shorter than in those who had CD44‐negative CTCs.50 Thus, identifying and targeting a subset of CTCs could be clinically useful in patients with gastric/GEJ cancers. Two agents targeting CSC pathways have been studied in gastric/GEJ cancer: vismodegib and napabucasin (also known as BBI608).
Vismodegib is an oral, small‐molecule antagonist of the Hedgehog pathway (Fig. 2), which is activated in gastric/GEJ tumors. Preclinically, Hedgehog inhibitors have demonstrated a reduction in gastroesophageal tumor growth, cell motility, and invasiveness. Vismodegib is currently approved for basal cell carcinoma51 and has been combined with leucovorin, fluorouracil (5‐FU), and oxaliplatin (FOLFOX) in the treatment of gastric/GEJ cancers, as described below.52
Figure 2.
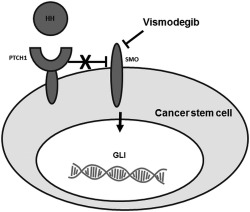
Inhibiting the hedgehog (HH) signaling pathway with vismodegib is illustrated. Binding of an HH protein to the transmembrane receptor patched 1 (PTCH1) prevents PTCH1‐mediated inhibition of signaling by the transmembrane protein smoothened (SMO), leading to activation of the GLI family of transcription factors and the regulation of target genes. Vismodegib inhibits the HH pathway by binding to SMO.
Napabucasin is an orally administered STAT3 inhibitor (Fig. 3). It inhibits CSC self‐renewal and induces cell death in CSCs. Napabucasin targets the STAT3, β‐catenin, and NANOG signaling pathways and inhibits the critical genes necessary for maintaining stemness. Targeting STAT3 activation with napabucasin has produced antitumor and antimetastatic activity in both in vitro and in vivo models of cancer without affecting normal cells, suggesting that STAT3 could be a valid molecular target for cancer therapy.53, 54
Figure 3.
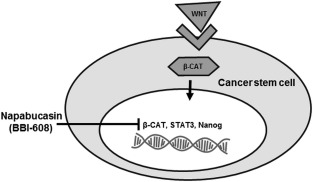
Inhibiting the signal transducer and activator of transcription 3 (STAT3) signaling pathway with napabucasin is illustrated. STAT proteins are located in the cytoplasm in resting cells as inactive proteins. Phosphorylation of a specific tyrosine residue is essential for STAT activation. Once activated, STAT dimerizes, leading to its translocation into the nucleus, which then leads to the initiation of transcription. Napabucasin inhibits the STAT3, β‐catenin (β‐CAT), and NANOG signaling pathways and inhibits the critical genes necessary for maintaining stemness.
Vismodegib in Clinical Trials of Gastric/GEJ Cancers
Vismodegib combined with FOLFOX has been studied in a randomized, double‐blind, phase 2 clinical trial in patients with advanced gastric and GEJ carcinomas (National Clinical Trials identifier NCT00982592; www.clinicaltrials.gov).52 In total, 124 patients with untreated, metastatic, or locally advanced gastric or GEJ adenocarcinoma were randomized 1:1 to receive either FOLFOX (oxaliplatin 85 mg/m2, leucovorin 200 mg/m2, and 5‐FU bolus 400 mg/m2 plus 5‐FU infusion 2400 mg/m2 over 48 hours) every 14 days plus vismodegib 150 mg daily; or FOLFOX plus placebo daily. Responses were assessed every 8 weeks (according to the Response Evaluation Criteria in Clinical Trials [RECIST] 1.1). The primary endpoint was progression‐free survival, and the secondary objectives were overall survival, the response rate, and toxicity. The median progression‐free survival in an intent‐to‐treat population for the FOLFOX plus vismodegib and FOLFOX plus placebo cohorts was 11.5 (95% confidence interval [CI], 8.5‐14.4 months) and 9.3 months (95% CI, 6.7‐11.9 months), respectively (P = .34), and the median overall survival was 12.2 months (95% CI, 10.2‐14.3 months) and 13.9 months (95% CI, 11.5‐16.3 months), respectively (P = .48). The most common grade 3 or higher toxicities with the combination FOLFOX plus vismodegib versus FOLFOX plus placebo were neutropenia (50% vs 31.7%, respectively), neuropathy (23.1% vs 14.3%, respectively), fatigue (15.4% vs 9.5%, respectively), thrombosis (13.5% vs 11.1%, respectively), anemia (9.6% vs 9.5%, respectively), hypokalemia (9.6%vs 4.8%, respectively), and nausea (7.7% vs 9.5%, respectively). In general, the addition of vismodegib to FOLFOX did not improve antitumor activity over FOLFOX alone in this biomarker‐unselected population with advanced gastric/GEJ carcinoma.52 A biomarker analysis of cell lines from that trial was conducted to determine whether a subset of patients could potentially derive benefit with combination vismodegib and FOLFOX treatment.38
Yoon et al examined 97 available tumor samples from patients in the phase 2 trial described above for CD44‐expressing cells.38 Those authors observed that CD44‐positive cells in gastric cancer cell lines had up‐regulation of Hedgehog pathway proteins. Two patients in the FOLFOX plus vismodegib group had a complete response, and these 2 patients had median CD44 expression rates that were significantly higher compared with those who had a partial response, stable disease, and progressive disease (P = .001). In the FOLFOX‐alone group, high CD44 expression was associated with decreased survival; whereas, in the FOLFOX plus vismodegib group, high CD44 expression was associated with improved survival. Thus, these in vitro data indicate that a subset of patients who have tumor cells with high CD44 expression may have improved survival with a combination of chemotherapy plus vismodegib over chemotherapy alone.38 In these patients with high CD44‐expressing tumor cells, Hedgehog pathway inhibition with vismodegib (or another Hedgehog inhibitor) potentially may reverse chemotherapy resistance in a select patient population.
Napabucasin in Clinical Trials of Gastric/GEJ Cancers
Napabucasin combined with paclitaxel was studied in a phase 1, open‐label study in Japan (JapicCTI‐142420) in 6 patients with gastric cancer.55 Napabucasin 480 mg twice daily was administered in combination with paclitaxel 80 mg/m2 weekly on days 1, 8, and 15 of each 28‐day study cycle until patients developed disease progression or unacceptable toxicity. Of the 6 patients enrolled, 3 had previously received taxane therapy. Two patients achieved a partial response, including 1 who maintained a response for more than 7.5 months. Two additional patients achieved stable disease that lasted 2.8 months or nonprogressive disease that lasted 7.5 months. Diarrhea was the most common adverse reaction, but it was only grade 1 (mild) in all 6 patients. Grade 1 anorexia also was observed in 2 patients. No other severe side effects were observed. In that study, napabucasin plus weekly paclitaxel was safely administered at full dose with promising efficacy signals.55
In a phase 1b dose‐escalation study (National Clinical Trials identifier NCT01325441; www.clinicaltrials.gov), patients with advanced solid tumors received napabucasin plus weekly paclitaxel.56 In total, 24 patients received continuous oral administration of escalating doses of napabucasin in combination with paclitaxel 80 mg/m2 weekly for 3 of every 4 weeks of a 28‐day cycle. Napabucasin was received by 3 patient dose cohorts of 200 mg twice daily, 400 mg twice daily, and 500 mg twice daily. Treatment continued until disease progression, unacceptable toxicity, or other discontinuation criteria were met. Of the 24 patients enrolled, 5 had gastric or gastric/GEJ tumors. Of those 5 patients, 3 had tumor regression (reductions of 45%, 48%, and 24%), 2 had prolonged stable disease for >5 months, and the median progression‐free survival was 23 weeks for the cohort with gastric/GEJ cancers. The most common adverse events included grade 1 or 2 diarrhea, abdominal cramps, nausea, and vomiting. Four patients experienced grade 3 events related to therapy, including diarrhea, dehydration, and weakness. In this study, napabucasin plus weekly paclitaxel was safely combined and produced antitumor activity across several tumor types, particularly in patients who had gastric and GEJ adenocarcinoma.56
A phase 1b/2 extension study (National Clinical Trials identifier NCT01325441; www.clinicaltrials.gov) of napabucasin combined with paclitaxel was conducted in patients with advanced gastric and GEJ adenocarcinoma.57 All patients received daily, continuous napabucasin 480 mg twice daily or 500 mg twice daily plus weekly paclitaxel 80 mg/m2 for 3 of every 4 weeks in a 28‐day cycle. Objective tumor response assessments occurred every 8 weeks. In total, 46 patients were treated on the study, and 35 were evaluated for response according to the protocol, including 19 who had received taxanes in the metastatic setting and 16 who had received no prior taxanes for metastatic disease. In addition, 10 patients (22%) had received 1 line of prior therapy, 16 (35%) had received 2 prior lines of therapy, and 20 (43%) had received ≥3 prior lines of therapy. On average, patients had received 2.4 lines of prior therapy, with an overall response rate of 15% for all 46 patients. Of the 35 patients who could be evaluated per protocol, the overall response rate was 20%, the disease control rate was 71%, the median progression‐free survival was 14.6 weeks, and the median overall survival was 34 weeks. In 6 evaluable patients who had not previously received a taxane in the metastatic setting and who had received 1 prior line of therapy, an objective response rate of 50% was observed. In heavily pretreated patients, including those who had failed an average of more than 2 lines of prior therapies and who had not previously received a taxane in the metastatic setting (n = 16), the objective response rate was 31% in the per‐protocol population. The disease control rate was 75%, the median progression‐free survival was 20.6 weeks, and the median overall survival was 39.3 weeks. The most common adverse events were grade 1 to 2 diarrhea, nausea, vomiting, and abdominal pain. Grade 3 adverse events included vomiting, diarrhea lasting ≥5 days, fatigue, abdominal and gastrointestinal pain, nausea, dehydration, anorexia, white blood cell decrease, and acute kidney injury. Results from the study indicate that napabucasin plus weekly paclitaxel is well tolerated in patients with advanced gastric/GEJ cancer, and activity was observed even in heavily pretreated patients.57 Continued evaluation of the combination of napabucasin and paclitaxel in patients with gastric/GEJ cancer who received only 1 prior line of therapy is currently underway in the phase 3 BBI608 Plus Weekly Paclitaxel to Treat Gastric and Gastroesophageal Junction Cancer (BRIGHTER) study, as described below.
On the basis of encouraging anticancer activity using the combination of napabucasin with paclitaxel in patients with gastric/GEJ adenocarcinoma in early phase trials, a phase 3 trial is being conducted. This trial—the BRIGHTER trial—is a randomized, double‐blind, placebo‐controlled trial of napabucasin plus weekly paclitaxel versus placebo plus weekly paclitaxel in patients with advanced, previously treated gastric/GEJ adenocarcinoma (National Clinical Trials identifier NCT02178956; www.clinicaltrials.gov).58 Enrolled patients must have failed 1 previous line of therapy that contained a fluoropyrimidine/platinum doublet for unresectable gastric/GEJ carcinoma. Patients are to be randomized to receive napabucasin 480 mg or placebo twice daily continuously plus weekly paclitaxel 80 mg/m2 for 3 of every 4 weeks. Treatment will continue until disease progression, death, intolerable toxicity, or patient or investigator decision to stop. The primary endpoint is overall survival in the general study population. Secondary endpoints include progression‐free survival, the objective response rate, the disease control rate, overall survival progression‐free survival in a predefined biomarker (β‐catenin)‐positive subpopulation, and safety. Twenty‐eight patients had been randomized as of January 2015, and recruitment is ongoing at multiple sites in North America, Europe, Australia, and Japan.58 The goal of the BRIGHTER trial is to determine whether napabucasin plus paclitaxel as second‐line therapy will extend survival compared with paclitaxel alone and whether a biomarker‐selected patient population exhibits improved efficacy with the combined treatment versus chemotherapy alone.
DISCUSSION AND CONCLUSIONS
Worldwide, gastric cancer is the most frequent malignancy; and, because of the high recurrence rate, it is the second leading cause of cancer mortality.2 Although many improvements have been made in the early diagnosis and surgical treatment of gastric cancer, patient prognosis remains poor. The major cause of death from gastric cancer is the inability to detect and prevent metastasis. Because of the resistance of CSCs to chemotherapy and radiation, the few that persist after standard chemotherapy and radiation are sufficient for cancer recurrence. Targeting CSCs by blocking or modifying the signaling pathways characteristic of these cells holds the promise of preventing disease metastasis and relapses. However, the development of CSC inhibitors will require a better understanding of key connections in the stem cell signaling network.
To date, there are very limited clinical data on the use of CSC pathway inhibitors in gastric/GEJ cancer. Two agents, vismodegib and napabucasin, have been studied. Data from a trial comparing FOLFOX plus vismodegib versus FOLFOX alone did not suggest an added benefit with the combination; however, post hoc analyses using CD44 as a biomarker for CSCs demonstrated more activity in tumor samples that had increased CD44 expression. Conversely, data from phase 1 and 2 studies of napabucasin suggest that targeting CSC signaling pathways by STAT3 inhibition may hold promise. A phase 3 study—the BRIGHTER trial in refractory gastric cancer—is currently underway.58 This study will provide definitive data on whether napabucasin will add a clinical benefit to paclitaxel in the studied patient population.
It is hypothesized that patients who would benefit most from agents that target CSCs have tumors that exhibit up‐regulation of the CSC signaling pathways. Preclinical studies suggest that gastric tumors expressing the CSC marker CD44 are associated with up‐regulation of the Hedgehog pathway and with a worse outcome.50 A biomarker study of tumor samples from a phase 2 study with vismodegib plus FOLFOX in gastric cancer confirmed these findings.38 Further study of phase 2 tumor samples revealed that, when tumors were selected for positive CD44 expression, the combination of vismodegib and FOLFOX produced increased activity over FOLFOX alone.38 Thus, a study of combined chemotherapy plus a CSC pathway inhibitor in patients with gastric cancer who are biomarker‐selected to have high expression of CD44 tumor cells might be warranted. On a larger scale, biomarker analyses are being conducted in the BRIGHTER study and hopefully will help to clarify whether a subset of patients with gastric cancer whose tumors overexpress β‐catenin will further benefit from the addition of napabucasin to chemotherapy.
The BRIGHTER study and other ongoing studies in various malignancies may demonstrate a benefit from targeting CSCs in combination with chemotherapy. In addition, the identification and further validation of biomarkers that better identify patients who have up‐regulated CSC pathways should be prioritized during clinical development. Future incorporation of these specific biomarkers into clinical trials may help identify patient subpopulations that are most likely to respond to the combination of chemotherapy and the CSC inhibitor class of drugs.
Funding Support
Medical writing and editorial support was provided by Keith Lantz (Link Health Group, LLC, Bridgewater, NJ) and was funded by Boston Biomedical, Inc.
CONFLICT OF INTEREST DISCLOSURES
Tanios Bekaii‐Saab reports personal fees from Genentech outside the submitted work. Bassel El‐Rayes reports grants from Boston Biomedical outside the submitted work.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. [DOI] [PubMed] [Google Scholar]
- 2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 3. Park JY, von Karsa L, Herrero R. Prevention strategies for gastric cancer: a global perspective. Clin Endosc. 2014;47:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hemminki K, Sundquist J, Ji J. Familial risk for gastric carcinoma: an updated study from Sweden. Br J Cancer. 2007;96:1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kono S, Hirohata T. Nutrition and stomach cancer. Cancer Causes Control. 1996;7:41–55. [DOI] [PubMed] [Google Scholar]
- 6. Zhu H, Yang X, Zhang C, et al. Red and processed meat intake is associated with higher gastric cancer risk: a meta‐analysis of epidemiological observational studies [serial online]. PLoS One. 2013;8:e70955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen XZ, Chen H, Castro FA, Hu JK, Brenner H. Epstein‐Barr virus infection and gastric cancer: a systematic review [serial online]. Medicine (Baltimore). 2015;94:e792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet. 1993;341:1359–1362. [PubMed] [Google Scholar]
- 9. Turati F, Tramacere I, La Vecchia C, Negri E. A meta‐analysis of body mass index and esophageal and gastric cardia adenocarcinoma. Ann Oncol. 2013;24:609–617. [DOI] [PubMed] [Google Scholar]
- 10. Hirota WK, Zuckerman MJ, Adler DG, et al. Standards of Practice Committee, American Society for Gastrointestinal Endoscopy ASGE guideline: the role of endoscopy in the surveillance of premalignant conditions of the upper GI tract. Gastrointest Endosc. 2006;63:570–580. [DOI] [PubMed] [Google Scholar]
- 11. National Comprehensive Cancer Network (NCCN) . NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Gastric Cancer. Fort Washington, PA: NCCN, Inc; 2015. [Google Scholar]
- 12. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. [DOI] [PubMed] [Google Scholar]
- 13. Vidal SJ, Rodriguez‐Bravo V, Galsky M, Cordon‐Cardo C, Domingo‐Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2014;33:4451–4463. [DOI] [PubMed] [Google Scholar]
- 14. Adams JM, Strasser A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008;68:4018–4021. [DOI] [PubMed] [Google Scholar]
- 15. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cabrera MC, Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. 2015;7:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang YC, Wang SW, Hung HY, et al. Isolation and characterization of human gastric cell lines with stem cell phenotypes. J Gastroenterol Hepatol. 2007;22:1460–1468. [DOI] [PubMed] [Google Scholar]
- 18. Pilpilidis I, Kountouras J, Zavos C, Katsinelos P. Upper gastrointestinal carcinogenesis: H. pylori and stem cell cross‐talk. J Surg Res. 2011;166:255–264. [DOI] [PubMed] [Google Scholar]
- 19. Yong X, Tang B, Xiao YF, et al. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/beta‐catenin signaling pathway to promote cancer stem cell‐like properties in human gastric cancer. Cancer Lett. 2016;374:292–303. [DOI] [PubMed] [Google Scholar]
- 20. Zhou Y, Wang Y, Wen J, et al. Aquaporin 3 promotes the stem‐like properties of gastric cancer cells via Wnt/GSK‐3beta/beta‐catenin pathway. Oncotarget. 2016;7:16529–16541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M. Gastric cancer stem cells: evidence, potential markers, and clinical implications. J Gastroenterol. 2016;51:313–326. [DOI] [PubMed] [Google Scholar]
- 22. Singh SR. Gastric cancer stem cells: a novel therapeutic target. Cancer Lett. 2013;338:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol 42(suppl. 1):S3–S17, 2015. [DOI] [PubMed] [Google Scholar]
- 24. Takaishi S, Okumura T, Tu S, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27:1006–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen PH, Giraud J, Chambonnier L, et al. Characterization of biomarkers of tumorigenic and chemoresistant cancer stem cells in human gastric carcinoma [published online ahead of print September 12, 2016]. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-15-2157. [DOI] [PubMed] [Google Scholar]
- 26. Chen T, Yang K, Yu J, et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 2012;22:248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujita T, Chiwaki F, Takahashi RU, et al. Identification and characterization of CXCR4‐positive gastric cancer stem cells [serial online]. PLoS One. 2015;10:e0130808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han ME, Jeon TY, Hwang SH, et al. Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell Mol Life Sci. 2011;68:3589–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katsuno Y, Ehata S, Yashiro M, Yanagihara K, Hirakawa K, Miyazono K. Coordinated expression of REG4 and aldehyde dehydrogenase 1 regulating tumourigenic capacity of diffuse‐type gastric carcinoma‐initiating cells is inhibited by TGF‐beta. J Pathol. 2012;228:391–404. [DOI] [PubMed] [Google Scholar]
- 30. Jiang J, Zhang Y, Chuai S, et al. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene. 2012;31:671–682. [DOI] [PubMed] [Google Scholar]
- 31. Ohkuma M, Haraguchi N, Ishii H, et al. Absence of CD71 transferrin receptor characterizes human gastric adenosquamous carcinoma stem cells. Ann Surg Oncol. 2012;19:1357–1364. [DOI] [PubMed] [Google Scholar]
- 32. Jiang Y, He Y, Li H, et al. Expressions of putative cancer stem cell markers ABCB1, ABCG2, and CD133 are correlated with the degree of differentiation of gastric cancer. Gastric Cancer. 2012;15:440–450. [DOI] [PubMed] [Google Scholar]
- 33. Hashimoto K, Aoyagi K, Isobe T, Kouhuji K, Shirouzu K. Expression of CD133 in the cytoplasm is associated with cancer progression and poor prognosis in gastric cancer. Gastric Cancer. 2014;17:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen XL, Chen XZ, Wang YG, et al. Clinical significance of putative markers of cancer stem cells in gastric cancer: a retrospective cohort study [published online ahead of print August 19, 2016]. Oncotarget. doi: 10.18632/oncotarget.11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gong X, Azhdarinia A, Ghosh SC, et al. LGR5‐targeted antibody‐drug conjugate eradicates gastrointestinal tumors and prevents recurrence. Mol Cancer Ther. 2016;15:1580–1590. [DOI] [PubMed] [Google Scholar]
- 36. Zhang C, Li C, He F, Cai Y, Yang H. Identification of CD44+CD24+ gastric cancer stem cells. J Cancer Res Clin Oncol. 2011;137:1679–1686. [DOI] [PubMed] [Google Scholar]
- 37. Bertolini G, Roz L, Perego P, et al. Highly tumorigenic lung cancer CD133+ cells display stem‐like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106:16281–16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoon C, Park DJ, Schmidt B, et al. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res. 2014;20:3974–3988. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39. Lau WM, Teng E, Chong HS, et al. CD44v8‐10 is a cancer‐specific marker for gastric cancer stem cells. Cancer Res. 2014;74:2630–2641. [DOI] [PubMed] [Google Scholar]
- 40. Nguyen PH, Giraud J, Staedel C, et al. All‐trans retinoic acid targets gastric cancer stem cells and inhibits patient‐derived gastric carcinoma tumor growth [published online ahead of print October 27, 2016]. Oncogene. doi: 10.1038/onc.2016.87. [DOI] [PubMed] [Google Scholar]
- 41. Shitara K, Doi T, Nagano O, et al. Dose‐escalation study for the targeting of CD44v+ cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205) [published online ahead of print April 7, 2016]. Gastric Cancer. doi: 10.1007/s10120-016-0610-8. [DOI] [PubMed] [Google Scholar]
- 42. Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303–312. [DOI] [PubMed] [Google Scholar]
- 43. Xie J, Murone M, Luoh SM, et al. Activating Smoothened mutations in sporadic basal‐cell carcinoma. Nature. 1998;391:90–92. [DOI] [PubMed] [Google Scholar]
- 44. Gong S, Li Q, Jeter CR, Fan Q, Tang DG, Liu B. Regulation of NANOG in cancer cells. Mol Carcinog. 2015;54:679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aggarwal BB, Kunnumakkara AB, Harikumar KB, et al. Signal transducer and activator of transcription‐3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang YX, Yang SW, Li PA, et al. The promotion of the transformation of quiescent gastric cancer stem cells by IL‐17 and the underlying mechanisms [published online ahead of print August 15, 2016]. Oncogene. doi: 10.1038/onc.2016.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances [serial online]. Biomed Res Int 2013:421821, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mohammed MK, Shao C, Wang J, et al. Wnt/beta‐catenin signaling plays an ever‐expanding role in stem cell self‐renewal, tumorigenesis and cancer chemoresistance. Genes Dis. 2016;3:11–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vaiopoulos AG, Kostakis ID, Athanasoula K, Papavassiliou AG. Targeting transcription factor corepressors in tumor cells. Cell Mol Life Sci. 2012;69:1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li M, Zhang B, Zhang Z, et al. Stem cell‐like circulating tumor cells indicate poor prognosis in gastric cancer [serial online]. Biomed Res Int 2014:981261, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Genentech USA, Inc. Erivedge (vismodegib) [package insert]. San Francisco, CA: Genentech USA, Inc; 2015. [Google Scholar]
- 52. Cohen DJ, Christos PJ, Sparano JA, et al. A randomized phase II study of vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX in patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: a New York Cancer Consortium led study [abstract]. J Clin Oncol. 2013;31(suppl 4). Abstract 67. [Google Scholar]
- 53. Li Y, Rogoff HA, Keates S, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A. 2015;112:1839–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y, Jin Z, Zhou H, et al. Suppression of prostate cancer progression by cancer cell stemness inhibitor napabucasin. Cancer Med. 2016;5:1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shitara K, Kuboki Y, Nakamura Y, et al. A phase I study of BBI608, a cancer stemness inhibitor, administered with paclitaxel (PTX) as combination therapy (Rx) for pretreated unresectable or recurrent gastric cancer [abstract]. J Clin Oncol. 2015;33(suppl). Abstract e15089. [Google Scholar]
- 56. Hitron M, Stephenson J, Chi KN, et al. A phase 1b study of the cancer stem cell inhibitor BBI608 administered with paclitaxel in patients with advanced malignancies [abstract]. J Clin Oncol. 2014;32(suppl). Abstract 2530. [Google Scholar]
- 57. Becerra C, Stephenson J, Jonker DJ, et al. Phase Ib/II study of cancer stem cell (CSC) inhibitor BBI608 combined with paclitaxel in advanced gastric and gastroesophageal junction (GEJ) adenocarcinoma [abstract]. J Clin Oncol. 2015;33(suppl). Abstract 4069. [Google Scholar]
- 58. Shah MA, Muro K, Shitara K, et al. The BRIGHTER trial: a phase III randomized double‐blind study of BBI608 + weekly paclitaxel versus placebo (PBO) + weekly paclitaxel in patients (pts) with pretreated advanced gastric and gastro‐esophageal junction (GEJ) adenocarcinoma [abstract]. J Clin Oncol. 2015;33(suppl). Abstract TPS4139. [Google Scholar]