

Abstract
The ST18 gene has been proposed to act either as a tumor suppressor or as an oncogene in different human cancers, but direct evidence for its role in tumorigenesis has been lacking thus far. Here, we demonstrate that ST18 is critical for tumor progression and maintenance in a mouse model of liver cancer, based on oncogenic transformation and adoptive transfer of primary precursor cells (hepatoblasts). ST18 messenger RNA (mRNA) and protein were detectable neither in normal liver nor in cultured hepatoblasts, but were readily expressed after subcutaneous engraftment and tumor growth. ST18 expression in liver cells was induced by inflammatory cues, including acute or chronic inflammation in vivo, as well as coculture with macrophages in vitro. Knocking down the ST18 mRNA in transplanted hepatoblasts delayed tumor progression. Induction of ST18 knockdown in pre‐established tumors caused rapid tumor involution associated with pervasive morphological changes, proliferative arrest, and apoptosis in tumor cells, as well as depletion of tumor‐associated macrophages, vascular ectasia, and hemorrhage. Reciprocally, systemic depletion of macrophages in recipient animals had very similar phenotypic consequences, impairing either tumor development or maintenance, and suppressing ST18 expression in hepatoblasts. Finally, RNA sequencing of ST18‐depleted tumors before involution revealed down‐regulation of inflammatory response genes, pointing to the suppression of nuclear factor kappa B–dependent transcription. Conclusion: ST18 expression in epithelial cells is induced by tumor‐associated macrophages, contributing to the reciprocal feed‐forward loop between both cell types in liver tumorigenesis. Our findings warrant the exploration of means to interfere with ST18‐dependent epithelium–macrophage interactions in a therapeutic setting. (Hepatology 2017;65:1708‐1719).
Abbreviations
- CSF
colony stimulating factor
- CSF‐R1
colony stimulating factor 1 receptor
- DMEM
Dulbecco's modified Eagle's medium
- E
embryonic day
- ECCD‐1
rat anti‐mouse E‐cadherin
- EGF
epidermal growth factor
- FBS
fetal bovine serum
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- IgG
immunoglobulin G
- IHC
immunohistochemistry
- L1
LINE‐1
- LPS
lipopolysaccharide
- mRNA
messenger RNA
- NF‐kB
nuclear factor kappa B
- PCR
polymerase chain reaction
- PFIC
progressive familial intrahepatic cholestasis
- shRNA
short hairpin RNA
- TAM
tumor‐associated macrophage
ST18 was originally identified as a candidate tumor suppressor in breast cancer, hence its name (suppression of tumorigenicity 18).1 Successive studies revealed activation of the same gene in pediatric acute myeloid leukemia2, 3 and hepatocellular carcinoma (HCC),4 suggesting an oncogenic function of ST18. In particular, the mapping of integration sites of the endogenous retrotransposon LINE‐1 (L1) in HCC led to the identification of 12 tumor‐specific L1 insertions, one of which activated ST18.4
ST18 (also called NZF‐3 or MYT3) is a member of the NZF/MyT1 family of transcription factors, a nonclassical zinc finger family that includes two other members, NZF‐1 and Myt1, with six C2HC‐type fingers arranged in two main clusters, each of which might in principle bind DNA.1, 5, 6 The three genes are expressed in neural tissue and, based on expression patterns and overexpression experiments, are involved in the induction of neuronal differentiation.7 ST18 is expressed at low levels in a number of different rat tissues (including liver) and is required for fatty acid– and cytokine‐induced apoptosis in pancreatic β cells.8 Finally, RNA interference and messenger RNA (mRNA) profiling studies in human fibroblasts have indicated that ST18 regulates proapoptotic and proinflammatory genes in response to tumor necrosis factor α,5 although the relevance of these regulatory events in cancer remains unclear.
Here, we unravel a critical role for ST18 in a mouse model of HCC based on the adoptive transfer of transformed mouse embryonic hepatoblasts.9 ST18 was undetectable in either cultured cells or normal livers but was induced in subcutaneous tumors under the control of inflammatory cues, and in particular tumor‐associated macrophages (TAMs). Systemic depletion of macrophages in recipient animals prevented ST18 expression in transformed hepatoblasts and impaired both tumor development and maintenance. Reciprocally, ST18 knockdown in hepatoblasts delayed tumor progression or, when induced in preformed tumors, led to rapid tumor involution associated with loss of TAMs and down‐regulation of an inflammatory gene expression signature. Therefore, ST18 expression in the epithelial compartment contributes to the tight connection between inflammation and tumorigenesis in the liver.10
Materials and Methods
ISOLATION, CULTURE, RETROVIRAL INFECTION, AND SUBCUTANEOUS TRANSPLANTATION OF LIVER PROGENITOR CELLS
We derived hepatoblasts from the two mouse strains C57/JHsd (Harlan laboratories) and TRP53/C57 (Jackson laboratories), according to a protocol published previously.9 Liver cell suspensions from fetal livers of embryonic day (E) 14.5‐18.5 mice were diced and treated with Dispase (Gibco, 1000 U/mL) for 1 hour at 37°C. The livers were dispersed into single cells by pipetting and filtrated through a nylon mesh filter (pore size 100μm). The cellular pellet was washed with hypotonic lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 100 μM EDTA) for 3 minutes at 4°C, centrifuged, and placed in ice. Purification of E‐cadherin positive hepatoblasts was performed using the MACS magnetic cell sorting system (Miltenyi) through indirect labeling with the rat anti‐mouse E‐cadherin (ECCD‐1) antibody (Calbiochem).11 Before loading onto MACS MS size columns, liver cell suspensions were incubated with the antibody complex for 45 minutes at 4°C. Antibody complexes were prepared by incubating 4 μg of ECCD‐1 antibody with 20 μL of immunomagnetic beads at room temperature for 1 hour. Eluted cells were plated onto laminin‐coated plates (Sigma) in Dulbecco's modified Eagle's medium (DMEM) (Lonza), supplemented with 10% fetal bovine serum (FBS) NA (HyClone), 1% glutamine (Euroclone), 1% penicillin/streptomycin (Life Technologies), hepatocyte growth factor (HGF) (40 ng/mL, Peprotech), epidermal growth factor (EGF) (20 ng/mL, Peprotech) and dexamethasone (×10−6 M, Sigma). After 48 hours, cultured hepatoblasts were transduced with a combination of retrovirus encoding for c‐myc, oncogenic RAS (H‐RasV12), or shp53. Human hepatocellular liver carcinoma cell line HepG2 (DSMZ) cells were grown in Roswell Park Memorial Institute 1640 medium (Lonza) supplemented with 10% FBS NA (HyClone), 1% glutamine (Euroclone), and 1% penicillin/streptomycin (Life Technologies) under 5% CO2 at 37°C.
To generate short hairpin RNA (shRNA) hairpins targeting both human and mouse ST18, we designed 97 bp oligonucleotides including the desired shRNA sequences (in a mir‐30 format) flanked on either side by tails complementary to the following polymerase chain reaction (PCR) primers: XhoI_fw (5′‐ CAG AAG GCT CGA GAA GGT ATA TTG CTG TTG ACA GTG AGC G‐3′) and EcoRI_rv (5′‐ CTA AAG TAG CCC CTT GAA TTC CGA GGC AGT AGG CA‐3′). Amplified PCR products were digested with XhoI/EcoRI and cloned in the mir‐30 based retroviral vector TtRMPVIR.12 The target sequences used in this study were TTCATGCTTAAGTCCAATGtg (ST18_1), TTTAAAGACTGGATACTGCtg (ST18_6) and TTGATTCAGGAAATGGTGGtg (ST18_7). The efficacy of knockdown was initially tested in 293T cells. Retroviruses were produced in Phoenix packaging cells and collected in DMEM (Lonza) supplemented with 10% tetracycline‐free FBS NA (HyClone), 1% glutamine (Euroclone), and 1% penicillin/streptomycin (Life Technologies). Supernatant was passed through a 0.45‐μm filter and supplemented with polybrene (2 μg/mL, Sigma). The infection procedure was repeated three times every 4 hours before adding fresh medium supplemented with HGF (40 ng/mL, Peprotech), EGF (20 ng/mL, Peprotech), and dexamethasone (×10−6 M, Sigma).
For in vivo tumor formation experiments, 3 × 105 genetically modified hepatoblasts were injected subcutaneously into CD1 nude mice (Charles River Laboratories) in a volume of 0.3 mL of phosphate‐buffered saline. Animals were monitored twice a week for signs of distress or disease, and tumor size was measured using a caliper. Ex vivo imaging of Venus fluorescence in dissected tumors was performed on an IVIS Lumina III platform and analyzed with Living Image Software, version 4.2 (Caliper Life Sciences). The average radiant efficiency was calculated based on the epifluorescence signal as indicated in the user manual.
Experiments involving animals were performed in accordance with Italian laws (D.L.vo 116/92 and following additions), which enforces the EU 86/609 directive (Council Directive 86/609/EEC of 24 November 1986 on the approximation of laws, regulations, and administrative provisions of the Member States regarding the protection of animals used for experimental and other scientific purposes).
MACROPHAGE DEPLETION IN VIVO AND CULTURE IN VITRO
To deplete macrophages in vivo, we used the anti–colony stimulating factor 1 (CSF‐1) receptor antibody (ASF98), a rat monoclonal anti‐murine CD115 antibody (immunoglobulin G [IgG] 2a) that inhibits CSF‐1–dependent cell growth by blocking the binding of CSF‐1 to its receptor (CSF‐1R), the selectivity of which has been characterized previously.13 As a control, we injected an isotype‐matched anti‐rat IgG (Sigma‐Aldrich). Mice were injected intraperitoneally at doses of 2 mg/mouse.
Clodronate, encapsulated in liposomes, was also used to deplete macrophages in Mdr2 −/− and CD1‐nude mice. Clodronate liposomes were prepared as described previously.14 Control liposomes contained phosphate‐buffered saline only. Each animal received 0.01 mL/g (5 mg of clodronate per 1 mL of the total suspension volume) of clodronate liposomes or control liposomes via intraperitoneal injection. The clodronate and control liposomes were obtained from the Foundation Clodronate Liposomes (Amsterdam, Netherlands).
The murine macrophage cell line RAW264.7 (ECACC) was grown in DMEM (Lonza) supplemented with 10% FBS NA (HyClone), 1% glutamine (Euroclone) and 1% penicillin/streptomycin (Life Technologies) under 5% CO2 at 37°C. Normal bone marrow macrophages were derived from of C57/BL6 mice (Harlan).15
OTHER METHODS
Other protocols and reagents are described in the Supporting Materials and Methods.
Results
We used a model based on the genetic manipulation of embryonic liver progenitors (hepatoblasts) ex vivo followed by their adoptive transfer into recipient mice.9 Hepatoblasts were isolated from fetal livers at E14.5‐18.5 and transduced with different combinations of retroviruses expressing c‐myc, oncogenic Ras (H‐RasV12), or an shRNA targeting p53 (unless using p53‐null cells). As expected,9 these cells became immortal in culture and acquired a transformed phenotype, as shown by their ability to generate liver‐derived tumors with histological subtypes16 and markers characteristic of human HCC when injected subcutaneously in immunocompromised CD‐1 nude mice (Supporting Fig. S1A,B). RNA analysis and immunostaining revealed that ST18 was expressed in tumors but not in normal liver1, 8 or transformed hepatoblasts in vitro before injection into recipient mice (Fig. 1A), indicating that the gene was induced by tumor‐associated microenvironmental signals.
Figure 1.
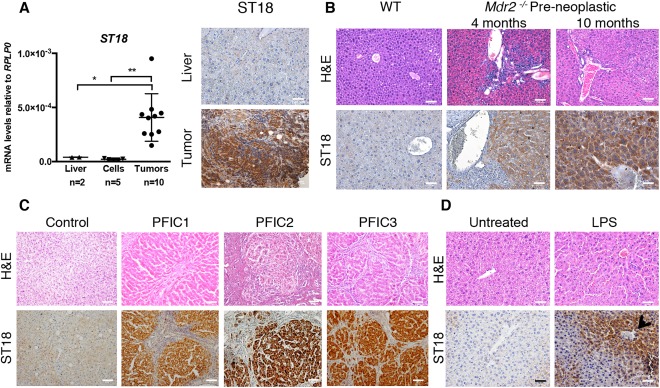
ST18 is induced by inflammatory cues in hepatoblasts. (A) Left: Quantitative reverse‐transcription PCR analysis of ST18 mRNA levels in adult liver in cell lines derived from mouse liver progenitors (including E14.5 shp53‐Myc, E14.5 shp53‐RasV12, E14.5 Myc‐RasV12, E18.5 shp53‐Myc‐RasV12, and E18.5 shp53‐RasV12) and in subcutaneous tumors derived from the same cells. *P = 0.002. **P = 0.0462. Right: ST18 is expressed in subcutaneous hepatoblast‐derived tumors but not normal liver. (B) Hematoxylin and eosin (H&E) staining shows portal inflammatory infiltrates in preneoplastic Mdr2 −/− livers at the indicated ages. Staining of IHC sections with ST18 antibodies (ST18) shows positivity in Mdr2 −/− but not wild‐type livers. (C) Hepatic lesions and ST18 positivity in liver biopsies from PFIC1, PFIC2, and PFIC3 patients. (D) Mouse liver sections 24 hours after LPS treatment showing irregular hepatocyte arrangement, inflammatory infiltrates, and induction of ST18, in particular nearby blood vessels (arrow) in both the periportal and centrilobular zones.
ST18 is expressed in tumors arising in mice knockout for the Abcb4 or Mdr2 gene (henceforth Mdr2 −/− mice),4 a model of inflammation‐driven HCC.17 Preneoplastic livers in 4‐ to 10‐month‐old Mdr2 −/− animals also became positive for ST18 (Fig. 1B), suggesting that expression was triggered by chronic inflammation even before the onset of tumorigenesis. Mdr2 −/− mice lack a P‐glycoprotein of the bile canalicular membrane, causing defective secretion by hepatocytes of lipids required to neutralize bile salts.17 The ensuing high concentration of monomeric bile salts causes persistent damage of the hepatic epithelium forming the initial bile canaliculi, with the consequent inflammatory response preceding tumor development.18, 19, 20 These lesions are analogous to those observed in progressive familial intrahepatic cholestasis (PFIC), a recessive autosomal disorder involving a chronic hepatic inflammation that progresses to fatal liver failure during childhood. PFIC comes in three types—PFIC1, PFIC2, and PFIC3—with mutations in the hepatocyte membrane transporters ATP8B1, ABCB11, and ABCB4, respectively.21 The most severe form, PFIC2, may further progress to HCC or cholangiocarcinoma.22 Liver biopsies from PFIC1, PFIC2, and PFIC3 patients showed expression of ST18 in the three conditions, clearly above the levels detected in control tissue (Fig. 1C). Hence, membrane transporter deficiencies lead to analogous effects in mice and humans, with chronic inflammation and induction of ST18 expression.
To further investigate the link between inflammation and ST18 expression, we induced an acute inflammatory response in wild‐type C57BL/6 mice by intraperitoneal injection of bacterial lipopolysaccharide (LPS).23 ST18 was induced in the liver 24 hours after LPS injection, especially nearby blood vessels (Fig. 1D). Thus, either acute or chronic inflammatory conditions could trigger ST18 expression in liver cells, owing most likely to the exposure to proinflammatory cytokines or cellular contacts.
Based on above results, we hypothesized that expression of ST18 in hepatoblast‐derived tumors (Fig. 1A) might be driven by the inflammatory microenvironment characteristic of many cancers.10, 24 TAMs have been shown to play important cancer‐promoting functions in a variety of models, including HCC.25, 26 Consistent with this notion, we observed significant infiltration of TAMs in mouse hepatoblast–derived tumors, as measured by immunohistochemistry (IHC) for the pan‐macrophage marker Iba127 (Supporting Fig. S1C). To mimic the interplay between macrophages and liver cancer cells in vitro, we cocultured transformed hepatoblasts for 12 hours with either primary bone marrow–derived macrophages or the macrophage cell line RAW 264.7, and purified back liver cells before RNA isolation. Coculture led to the acute induction of the ST18 mRNA in hepatoblasts, which was further reinforced by pretreatment of the macrophages with LPS for 1 hour (Supporting Fig. S1D). Milder activation of the gene was also observed upon treatment of hepatoblasts with RAW 264.7 culture supernatants (Supporting Fig. S1E). Thus, macrophages trigger ST18 induction in liver cells in vitro: this involves at least in part soluble cues, but remains most effective with cell–cell contacts.
To address the role of ST18 in tumorigenesis, c‐myc– and H‐RasV12–transformed p53−/− hepatoblasts were transduced with an shRNA hairpin targeting the ST18 mRNA, expressed from the doxycycline‐inducible vector TtRMPVIR.12 Infected cells were then sorted for expression of the associated Venus marker and injected subcutaneously into CD‐1 nude mice. Induction of the shRNA by exposure to doxycycline from the day of seeding (day 0) significantly suppressed tumor development relative to either untreated mice (shST18 off) or tumors infected with the control vector (shREN.713) (Supporting Fig. S2A,B). Thus, expression of ST18 is required for tumor development in vivo.
We then let tumors develop for 2 weeks before inducing the knockdown: 4 hours after activation, the ST18 shRNA caused hemorrhages that extended progressively from the tumor site to adjacent subcutaneous areas (Fig. 2A). Dissection and ex vivo imaging 24 hours after shRNA activation revealed hemorrhagic and friable tumor masses with loss of Venus fluorescence relative to untreated controls (Fig. 2B). RNA analysis and IHC staining confirmed a decrease in ST18 abundance with residual expression confined to the progressively reduced nonnecrotic areas (Fig. 2C,D). Similar effects were induced by two other ST18 shRNAs, but not the shREN.713 control (Supporting Fig. S3A‐D). Closer pathological analysis (Fig. 2E, insets) revealed that in untreated mice, the subcutaneous tumor mass was consistent with a poorly differentiated tumor, composed of highly cohesive atypical cells. Doxycycline‐treated tumors showed severe intratumoral hemorrhages and necrotic areas that increased over time. Cells became multifocally less cohesive, arranged in bundles, and spindle‐shaped. Staining with the vascular endothelial cell marker VE‐cadherin28 revealed a dilatation of intratumoral blood vessels (ectasia), coincident with a decrease in Ki67 and appearance of cleaved caspase‐3, none occurring with the control shRNA (Supporting Fig. S4A,B). Altogether, silencing of ST18 led to proliferative arrest and induction of apoptosis, concomitant with vascular ectasia and hemorrhage: these combined effects are all likely to contribute to acute tumor regression.
Figure 2.
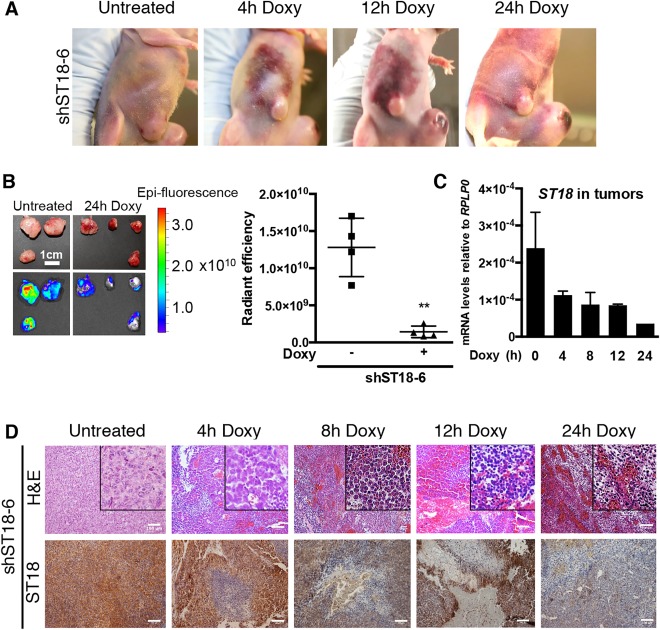
Expression of ST18 is required for tumor maintenance. (A) Hemorrhages initiating from the tumor and extending to the adjacent subcutaneous areas were noticeable 4 hours after ST18 silencing and increased progressively throughout the indicated time course. All experiments were performed using induction of the shST18‐6 hairpin with administration of doxycycline (at time 0) by way of oral gavage. (B) Left: Ex vivo fluorescent imaging of tumors dissected from either untreated recipient mice or 24 hours after doxycycline administration. The fluorescent Venus marker was constitutively expressed from the same TtRMPVIR vector12 expressing doxycycline‐inducible shST18; loss of fluorescence after ST18 knockdown is thus due to loss of the targeted cells. Right: quantification of average radiant efficiency in the same tumors shown at left. **P = 0.0013. (C) Quantitative reverse‐transcription PCR analysis of ST18 mRNA levels in tumors confirmed knockdown 4 hours after shST18 induction. (D) Induction of shST18 induced hemorrhages and necrosis concomitant with a progressive decrease of ST18 expression. Insets denote that in untreated mice, the tumor was composed of highly cohesive atypical cells; in doxycycline‐treated tumors, cells were multifocally less cohesive, arranged in bundles, and spindle‐shaped. Scale bars = 100 μm.
Knockdown of ST18 in liver cancer cells also led to a rapid loss of TAMs in subcutaneous tumors, residual IBA1+ cells showing dramatic changes in morphology suggestive of impaired functionality (Supporting Fig. S4C, D). Importantly, TAMs themselves did not express ST18, and our experimental system ensures hepatocyte‐specific knockdown. Thus, the maintenance of TAMs relied on sustained expression of ST18 in cancer cells.
To further investigate the interplay between ST18 expression in hepatoblasts, TAMs and tumor maintenance, we predepleted macrophages systemically in recipient animals by intraperitoneal injection of a monoclonal antibody that blocks the murine CSF‐1 receptor (or CD115)13 or isotype‐matched IgG as control. Anti–CD‐115 effectively caused selective depletion of circulating CD115+ cells 4 days after injection, with no effects on LY6G+ neutrophils (Fig. 3A). At this stage (day 0, Fig. 3B), transformed hepatoblasts were injected subcutaneously: examination of the animals over time revealed continued depletion of circulating CD115+ monocytes/macrophages (Fig. 3B) associated with a significant delay in tumor development (Fig. 3C). At day 15, CD115+ cell numbers were minimal and tumors virtually undetectable. At the last time point (day 24), partial recovery of circulating CD115+ cells (Fig. 3B) was accompanied by the growth of small tumor masses (Fig. 3C); relative to untreated controls, however, these tumors still lacked macrophages (assessed by way of IBA1 staining), failed to induce ST18, and showed elevated apoptosis (Fig. 3D). These findings suggest that macrophages are required for ST18 induction and tumor development.
Figure 3.
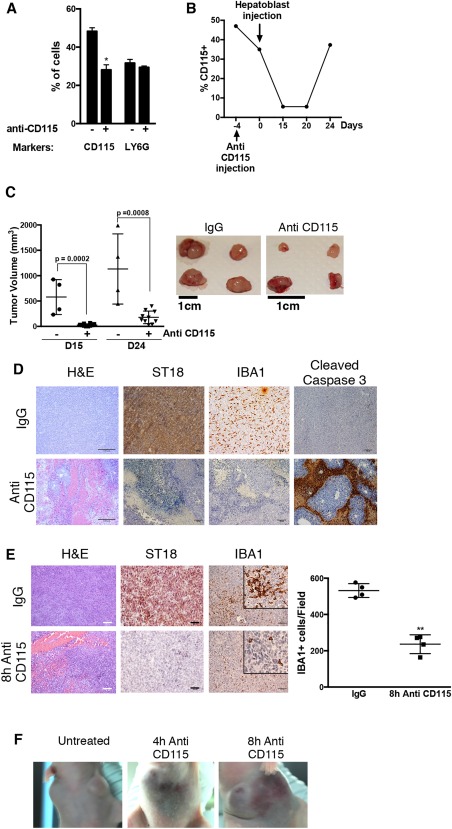
Depletion of macrophages in vivo prevents ST18 expression in hepatoblasts and affects both tumor development and maintenance. (A) Fluorescence‐activated cell sorting of macrophages (CD115+) and neutrophils (LY6G+) in peripheral blood 4 days after injection of mice with anti‐CD115 (+), compared with injection of control IgG (−). The data represent the average and standard deviation for three mice. *P = 0.01. Cell numbers are expressed relative to total white blood cells. (B) Analysis of CD115+ cell numbers over time in one of the above animals (note that day 0 here is the 4th day after anti‐CD115 treatment (i.e., the same time point analyzed in panel A). (C) Left: Tumor volumes in mice pretreated with either control IgG (−) or anti‐CD115 (+) at days 15 and 24 after tumor seeding. Right: Photographs of tumors dissected (at day 24) from carriers pretreated with IgG (control) or anti‐CD115+, as indicated. (D) Hematoxylin and eosin (H&E) and IHC staining for the indicated proteins (IBA1, ST18, cleaved caspase‐3) reveal hemorrhage, macrophage depletion, lack of ST18 expression, and increased apoptosis in tumors arising in anti‐CD115–treated mice compared with IgG‐treated mice. (E) Hematoxylin and eosin (H&E) and IHC staining 8 hours after anti‐CD115 or control IgG injection in established tumors revealed effects analogous to the above, including intratumoral hemorrhage and necrosis (H&E), decreased ST18 expression, and, as expected, loss of IBA1+ macrophages. The mean numbers of IBA1+ cells in four different microscopic fields in anti‐CD115 and IgG‐injected tumors are shown. **P < 0.0001. Scale bars = 100 μm. (F) Subcutaneous hemorrhages were noticeable as early as 4 hours after anti‐CD115 injection in established tumors and became more severe over time.
We then addressed the effect of macrophage depletion in preformed tumors by injecting anti‐CD115 or control IgG in tumor‐bearing animals. Four hours after treatment, loss of IBA1+ cells was accompanied by down‐regulation of ST18 with intratumoral and subcutaneous hemorrhages (Fig. 3E,F). As an independent means to deplete macrophages, we injected liposome‐encapsulated clodronate.14 Again, loss of IBA1+ cells coincided with dramatic changes in tumor morphology, with necrosis, hemorrhage, loss of ST18 expression, and induction of apoptosis (Supporting Fig. S5A). Finally, we injected clodronate in preneoplastic and neoplastic Mdr2 −/− animals, once again causing the parallel loss of IBA1 and ST18 staining (Supporting Fig. S5B,C). Altogether, we conclude that TAMs are required both for ST18 expression in cancer cells and tumor development/maintenance.
To complement our findings, we addressed the function of ST18 in the human HCC cell line HepG2 because, unlike mouse hepatoblasts, these cells expressed ST18 constitutively in the absence of inflammatory stimuli.4 ST18 knockdown in cultured HepG2 cells induced cell death (Supporting Fig. S6A, B), indicating a cell‐autonomous requirement for ST18. As with mouse hepatoblasts, induction of the shRNA in subcutaneous HepG2 tumors caused intratumoral hemorrhages and tumor regression (Supporting Fig. S6C‐E).
We finally profiled ST18‐dependent gene expression in our mouse model. RNA sequencing of control and ST18‐depleted tumors (analyzed 4 hours following induction of shST18‐6) allowed us to identify 677 and 467 up‐regulated and down‐regulated genes, respectively (Supporting Tables S1 and S2). The latter included known inflammation‐related genes, such as members of the nuclear factor kappa B (NF‐kB) family of transcription factors, as well as inflammatory cytokines and chemokines (most notably Ccl2) that are known to control the recruitment and activation of myeloid cells to the tumor sites (Fig. 4A).29, 30, 31, 32, 33 Gene set enrichment analysis (Supporting Tables S3 and S4) showed that ST18 knockdown led to the down‐regulation of inflammation‐associated genes, including in particular a set of genes up‐regulated in hepatic stellar cells after stimulation with LPS34 (Fig. 4B) as well as genes containing the binding motif for the inflammatory transcription factor NF‐kB (Fig. 4C). Use of Ingenuity pathway analyzer software further pointed to a role of the transcription factor NF‐kB in the regulation of ST18‐dependent genes (Fig. 4C). Immunostaining analysis of the NF‐kB subunits P65 and P50 revealed rapid decreases in the levels of both proteins after ST18 knockdown (Fig. 4D,E) a result confirmed for p65 in macrophage‐depleted tumors (Fig. 4E, Anti‐CD115). Although the mechanisms linking ST18 and NF‐kB activities remain to be addressed, and indirect effects due to macrophage loss cannot be excluded at this stage, these data point to a role of ST18 upstream of NF‐kB in controlling the transcriptional response of liver cells to inflammatory cues, thereby sustaining the mutual interaction between tumor cells and TAMs.
Figure 4.
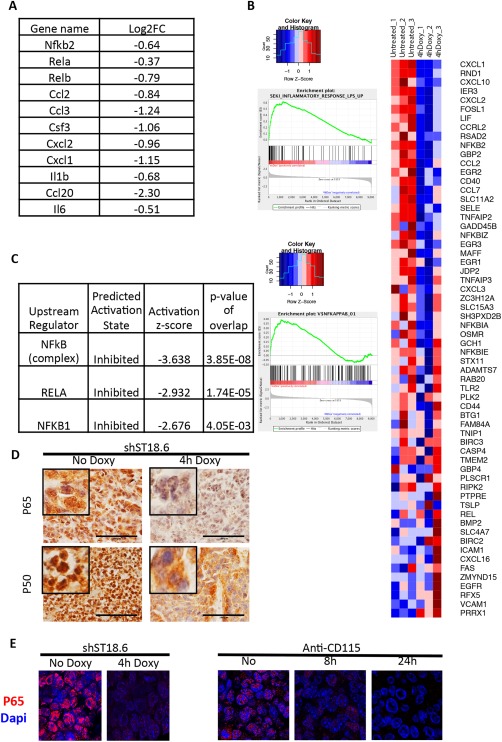
ST18 knockdown in hepatoblast‐derived tumors affects inflammation‐ and NF‐kB–associated genes. RNA sequencing was used to identify genes whose expression was affected 4 hours after induction of ST18 knockdown with the shST18‐6 hairpin in subcutaneous tumors, with three mice per condition. (A) Genes involved in NF‐kB signaling and/or positive regulation of myeloid cell activity that showed ST18‐dependent expression. (B) Gene set enrichment analysis revealed that genes induced by LPS in hepatic stellar cells (gene set SEKI_INFLAMMATORY_RESPONSE_LPS_UP)34 were down‐regulated after ST18 knockdown. The heatmap shows the relative expression of the mRNAs included in this gene set in hepatoblast‐derived tumors, without (untreated) or with (4h Doxy) ST18 knockdown (three independent samples for each condition). (C) Gene set enrichment analysis revealed that genes containing the NF‐kB binding motif (V$NFKAPPAB_01) were down‐regulated after ST18 knockdown. (D) Ingenuity pathway analyzer software pointed to a central role of NF‐kB in the regulation of ST18‐dependent genes. (E) IHC staining of the NF‐kB subunits P65 and P50 revealed rapid decreases in the levels of both proteins after ST18 knockdown. (F) Immunofluorescent detection of the NF‐kB subunit P65 revealed its rapid decrease in hepatoblast‐derived tumors after ST18 knockdown and macrophage depletion.
Discussion
Previous observations, including L1 insertional mutagenesis in human HCC, as well as gene amplification and activation in inflammation‐driven HCC nodules in Mdr2 −/− mice, pointed to ST18 as a candidate oncogene in HCC.4 However, direct evidence for a role of ST18 in cancer was missing altogether. Using a mouse model of HCC based on ex vivo transformation and adoptive transfer of liver progenitor cells (hepatoblasts),9 we demonstrate that ST18 is important for tumor development and maintenance. Detailed analysis revealed an unexpected mode of action for ST18, as a central component in a feed‐forward loop between neoplastic epithelial cells and TAMs.
The ST18 mRNA and protein were undetectable in either normal liver1, 8 or cultured hepatoblasts but were expressed in subcutaneous tumors derived from the same cells. These results led us to hypothesize that ST18 expression in vivo may require tumor‐derived microenvironmental signals and in particular inflammatory infiltrates, a recurrent feature in solid tumors.10, 26 In line with this concept, hepatoblast‐derived tumors contained infiltrating macrophages, the ablation of which led to rapid down‐regulation of ST18 in the tumor cells. Furthermore, ST18 could be induced in liver cells, either in vitro by coculture with macrophages or in vivo by exposure to inflammatory conditions. Taken together, these findings indicate that ST18 expression in tumors is triggered by inflammatory cues emanating from TAMs.
The role of ST18 in tumor cells was addressed through inducible knockdown of the gene in the transduced hepatoblasts; this led to rapid tumor involution associated with a series of adverse events (see below), including a marked loss of TAM infiltrates. This mutual dependency between expression of ST18 in hepatoblasts and maintenance of TAMs pointed to a regulatory loop between both cell types, with a central role in tumor maintenance. Indeed, the alternative modalities of intervention used in our experiments—ST18 knockdown in tumor cells versus ablation of TAMs—had strikingly similar consequences. Both treatments suppressed tumor progression (compare Supporting Fig. S2 and Fig. 3C,D) or, if applied to preformed tumors, caused rapid tumor involution associated with loss of TAMs (Supporting Fig. S4C,D and Fig. 3E), down‐regulation of ST18 (Figs. 2D and 3E), proliferative arrest, and apoptosis in the tumor cells (Supporting Fig. S4A and Fig. 3D), as well as pervasive hemorrhage, spreading from the tumor into the adjacent subcutaneous areas (Figs. 2A and 3F).
The precise origin of the TAMs infiltrating subcutaneous tumor lesions remains to be determined. Like most tissues, both the liver and the skin are home to resident macrophage populations, which are intimately linked to tissue homeostasis and recruit blood monocytes when damage occurs.26, 35 At the steady state, most tissue‐resident macrophage populations (with some notable exception such as the intestine and, to some extent, the liver) are originated during embryonic development by yolk sac–derived precursors, with minimal contribution from adult bone marrow hematopoiesis. The contribution of circulating monocytes to the resident macrophage pool is instead dominant in inflammatory contexts, such as infection and cancer.33, 35, 36, 37, 38
The origin of TAMs notwithstanding, a large body of literature indicates that these cells—in particular in the M2 polarized state—tend to have strong protumoral activities in most cancer types.26, 29 In HCC, in particular, TAM infiltration was shown to correlate with poor prognosis.39 Our macrophage‐depletion experiments with anti‐CD115 or Clodronate directly support this notion and are consistent with previous observations in glioblastoma and breast cancer29, 40, 41; however, it remains to be addressed whether TAMs may also elicit expression of ST18 in those tumors. Reciprocally, whether ST18 controls TAM function elsewhere than in the liver remains unclear. It is noteworthy here that TAMs can either promote or antagonize tumor growth,29 which may conceivably also explain the contrasting roles attributed to ST18 in cancer.1, 2, 3, 4
The signaling pathways through which TAMs elicit ST18 expression in tumor cells, and ST18 in turn feeds back to TAMs, also remain to be unraveled. Our RNA sequencing analysis indicated that ST18 might regulate part of the transcriptional response to inflammatory signals in hepatoblasts, in particular through its requirement for NF‐kB activation. Indeed, among the genes showing ST18‐dependent expression in our tumor model, we found a number of known NF‐kB regulated genes that may have a direct role in TAM homeostasis; for example, CCL2 and Cxcl2 are both involved in the recruitment of inflammatory monocytes (and myeloid cells) to tumors.32, 33, 42 Other proinflammatory cytokines such as interleukin‐1b, interleukin‐6, and CSF‐3 have been shown to control the phenotype of tumor‐elicited myeloid cells.43, 44 Although not investigated experimentally in this study, the reduced expression levels of these cytokines in ST18‐depleted tumors supports a model whereby (1) TAMs derive from circulating monocytes in our tumor model and (2) ST18 controls the expression of critical environmental factors that control the recruitment/activation of TAMs.
An essential question raised by our study pertains to the primary outputs of the aforementioned regulatory loop in tumor maintenance, with two extreme—albeit not exclusive—possibilities. First, ST18 may have an intrinsic (i.e., cell‐autonomous) role in proliferation and survival of tumor cells, as suggested by our in vitro data on the HepG2 cell line. In this setting, the tumorigenic properties of TAMs in the liver would rely at least in part on their ability to elicit ST18 expression in tumor cells. Second, ST18 may act mainly in a nonautonomous manner, allowing the maintenance of TAMs, which in turn signal tumor cell proliferation/survival. This cell‐extrinsic effect of ST18 on TAMs may thus be an important, if not the principal determinant of its tumor‐promoting activity in the liver.4
In either of the above scenarios, our findings lend further support to the concept that macrophage targeting may be an effective therapeutic strategy in liver cancer.26 Among others, macrophage depletion enhanced the antitumoral effects of the kinase inhibitor sorafenib,45 and the chemotherapeutic agent trabectedin acted in part through its toxic effect of TAMs.46 Consequently, elucidation of the molecular mechanisms through which ST18 mediates epithelium–macrophage interactions bears significant potential for future therapeutic development.
Finally, the mutual relationship between macrophages and epithelial cells in liver tumorigenesis, as well as the involvement of ST18—and possibly NF‐kB—in this process may have important counterparts in tissue homeostasis. Indeed, clodronate‐mediated depletion of Kupffer cells impaired liver regeneration upon partial hepatectomy due to loss of NF‐kB activation.47 In the same conditions, direct inhibition of NF‐kB in hepatocytes increased apoptosis and decreased proliferation.48 It will thus be interesting to address whether Kupffer cells mediate the activation of ST18 in the regenerating liver epithelium and whether ST18 is required for activation of NF‐kB after partial hepatectomy. It is tempting to speculate that this role of ST18 may extend to other tissues, representing perhaps a conserved element of macrophage–epithelium interactions.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28942/suppinfo.
Supporting Information Tables.
Supporting Information
Acknowledgment
We thank Andrea Piontini, Alberto Gobbi, and Manuela Capillo for help with the management of mouse colonies; Salvatore Bianchi, Luca Rotta, and Thelma Capra for assistance with the Illumina HiSeq platform; Federica Pisati for the preparation of histological samples; Alessia Curina for the preparation of primary macrophages; and Maria Rescigno, Elisabetta Dejana, Luigia Pace, Antonio Sica, and Elena Riboldi for insightful comments and discussion. We also thank the members of the MODHEP ethical advisory board—Giuseppe Testa, Göran Hermerén, Inez de Beaufort, and Luca Chiapperino—for their guidance.
Potential conflict of interest: Nothing to report.
This study was supported by grants from the European Community's Seventh Framework Programme (MODHEP consortium), the European Research Council, the Italian Health Ministry and the Italian Association for Cancer Research (to B.A.). G.J.F. acknowledges funding from the Australian National Health and Medical Research Council (grant numbers GNT1045237 and GNT1068789).
REFERENCES
- 1. Jandrig B, Seitz S, Hinzmann B, Arnold W, Micheel B, Koelble K, et al. ST18 is a breast cancer tumor suppressor gene at human chromosome 8q11.2. Oncogene 2004;23:9295–9302. [DOI] [PubMed] [Google Scholar]
- 2. Steinbach D, Schramm A, Eggert A, Onda M, Dawczynski K, Rump A, et al. Identification of a set of seven genes for the monitoring of minimal residual disease in pediatric acute myeloid leukemia. Clin Cancer Res 2006;12:2434–2441. [DOI] [PubMed] [Google Scholar]
- 3. Steinbach D, Bader P, Willasch A, Bartholomae S, Debatin KM, Zimmermann M, et al. Prospective validation of a new method of monitoring minimal residual disease in childhood acute myelogenous leukemia. Clin Cancer Res 2015;21:1353–1359. [DOI] [PubMed] [Google Scholar]
- 4. Shukla R, Upton KR, Munoz‐Lopez M, Gerhardt DJ, Fisher ME, Nguyen T, et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 2013;153:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang J, Siqueira MF, Behl Y, Alikhani M, Graves DT. The transcription factor ST18 regulates proapoptotic and proinflammatory gene expression in fibroblasts. FASEB J 2008;22:3956–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yee KS, Yu VC. Isolation and characterization of a novel member of the neural zinc finger factor/myelin transcription factor family with transcriptional repression activity. J Biol Chem 1998;273:5366–5374. [DOI] [PubMed] [Google Scholar]
- 7. Kameyama T, Matsushita F, Kadokawa Y, Marunouchi T. Myt/NZF family transcription factors regulate neuronal differentiation of P19 cells. Neurosci Lett 2011;497:74–79. [DOI] [PubMed] [Google Scholar]
- 8. Henry C, Close AF, Buteau J. A critical role for the neural zinc factor ST18 in pancreatic beta‐cell apoptosis. J Biol Chem 2014;289:8413–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zender L, Xue W, Cordon‐Cardo C, Hannon GJ, Lucito R, Powers S, et al. Generation and analysis of genetically defined liver carcinomas derived from bipotential liver progenitors. Cold Spring Harb Symp Quant Biol 2005;70:251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature 2008;454:436–444. [DOI] [PubMed] [Google Scholar]
- 11. Nitou M, Sugiyama Y, Ishikawa K, Shiojiri N. Purification of fetal mouse hepatoblasts by magnetic beads coated with monoclonal anti‐e‐cadherin antibodies and their in vitro culture. Exp Cell Res 2002;279:330–343. [DOI] [PubMed] [Google Scholar]
- 12. Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline‐regulated RNAi. Nat Biotechnol 2011;29:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sudo T, Nishikawa S, Ogawa M, Kataoka H, Ohno N, Izawa A, et al. Functional hierarchy of c‐kit and c‐fms in intramarrow production of CFU‐M. Oncogene 1995;11:2469–2476. [PubMed] [Google Scholar]
- 14. van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome‐mediated intracellular delivery of clodronate and propamidine. J Immunol Methods 1996;193:93–99. [DOI] [PubMed] [Google Scholar]
- 15. Austenaa LM, Barozzi I, Simonatto M, Masella S, Della Chiara G, Ghisletti S, et al. Transcription of mammalian cis‐regulatory elements Is restrained by actively enforced early termination. Mol Cell 2015;60:460–474. [DOI] [PubMed] [Google Scholar]
- 16. Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T, et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol 2010;38:5S–81S. [DOI] [PubMed] [Google Scholar]
- 17. Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P‐glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993;75:451–462. [DOI] [PubMed] [Google Scholar]
- 18. Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004;127:261–274. [DOI] [PubMed] [Google Scholar]
- 19. Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, et al. Mice with homozygous disruption of the mdr2 P‐glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol 1994;145:1237–1245. [PMC free article] [PubMed] [Google Scholar]
- 20. Katzenellenbogen M, Mizrahi L, Pappo O, Klopstock N, Olam D, Jacob‐Hirsch J, et al. Molecular mechanisms of liver carcinogenesis in the mdr2‐knockout mice. Mol Cancer Res 2007;5:1159–1170. [DOI] [PubMed] [Google Scholar]
- 21. Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012;36(Suppl. 1):S26–S35. [DOI] [PubMed] [Google Scholar]
- 22. Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006;44:478–486. [DOI] [PubMed] [Google Scholar]
- 23. Zhong J, Deaciuc IV, Burikhanov R, de Villiers WJ. Lipopolysaccharide‐induced liver apoptosis is increased in interleukin‐10 knockout mice. Biochim Biophys Acta 2006;1762:468–477. [DOI] [PubMed] [Google Scholar]
- 24. McAllister SS, Weinberg RA. The tumour‐induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 2014;16:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014;59:2034–2042. [DOI] [PubMed] [Google Scholar]
- 27. Rehg JE, Bush D, Ward JM. The utility of immunohistochemistry for the identification of hematopoietic and lymphoid cells in normal tissues and interpretation of proliferative and inflammatory lesions of mice and rats. Toxicol Pathol 2012;40:345–374. [DOI] [PubMed] [Google Scholar]
- 28. Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. J Clin Invest 1996;98:1949–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 2015;36:229–239. [DOI] [PubMed] [Google Scholar]
- 30. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013;121:4930–4937. [DOI] [PubMed] [Google Scholar]
- 31. Hamilton JA. Colony‐stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008;8:533–544. [DOI] [PubMed] [Google Scholar]
- 32. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature 2011;475:222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014;515:130–133. [DOI] [PubMed] [Google Scholar]
- 34. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF‐beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–1332. [DOI] [PubMed] [Google Scholar]
- 35. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 2014;14:392–404. [DOI] [PubMed] [Google Scholar]
- 36. Bain CC, Bravo‐Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 2014;15:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue‐resident macrophages originate from yolk‐sac‐derived erythro‐myeloid progenitors. Nature 2015;518:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C‐Myb(+) erythro‐myeloid progenitor‐derived fetal monocytes give rise to adult tissue‐resident macrophages. Immunity 2015;42:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High expression of macrophage colony‐stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol 2008;26:2707–2716. [DOI] [PubMed] [Google Scholar]
- 40. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF‐1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013;19:1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fend L, Accart N, Kintz J, Cochin S, Reymann C, Le Pogam F, et al. Therapeutic effects of anti‐CD115 monoclonal antibody in mouse cancer models through dual inhibition of tumor‐associated macrophages and osteoclasts. PLoS One 2013;8:e73310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steele CW, Karim SA, Leach JD, Bailey P, Upstill‐Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016;29:832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor‐induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010;32:790–802. [DOI] [PubMed] [Google Scholar]
- 44. Chittezhath M, Dhillon MK, Lim JY, Laoui D, Shalova IN, Teo YL, et al. Molecular profiling reveals a tumor‐promoting phenotype of monocytes and macrophages in human cancer progression. Immunity 2014;41:815–829. [DOI] [PubMed] [Google Scholar]
- 45. Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of tumor‐associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 2010;16:3420–3430. [DOI] [PubMed] [Google Scholar]
- 46. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013;23:249–262. [DOI] [PubMed] [Google Scholar]
- 47. Abshagen K, Eipel C, Kalff JC, Menger MD, Vollmar B. Loss of NF‐kappaB activation in Kupffer cell‐depleted mice impairs liver regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol 2007;292:G1570–G1577. [DOI] [PubMed] [Google Scholar]
- 48. Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, et al. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest 1998;101:802–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28942/suppinfo.
Supporting Information Tables.
Supporting Information