

Abstract
Understanding the pharmacokinetic (PK) and pharmacodynamic (PD) relationship of a therapeutic monoclonal antibody against proprotein convertase subtilisin/kexin type 9 (PCSK9) exhibiting target‐mediated drug disposition (TMDD) is critical for selecting optimal dosing regimens. We describe the PK/PD relationship of evolocumab using a mathematical model that captures evolocumab binding and removal of unbound PCSK9 as well as reduction in circulating low‐density lipoprotein cholesterol (LDL‐C). Data were pooled from 2 clinical studies: a single‐dose escalation study in healthy subjects (7‐420 mg SC; n = 44) and a multiple‐dose escalation study in statin‐treated hypercholesterolemic patients (14 mg weekly to 420 mg monthly [QM] SC; n = 57). A TMDD model described the time course of unbound evolocumab concentrations and removal of unbound PCSK9. The estimated linear clearance and volume of evolocumab were 0.256 L/day and 2.66 L, respectively, consistent with other monoclonal antibodies. The time course of LDL‐C reduction was described by an indirect response model with the elimination rate of LDL‐C being modulated by unbound PCSK9. The concentration of unbound PCSK9 associated with half‐maximal inhibition (IC50) of LDL‐C elimination was 1.46 nM. Based on simulations, 140 mg every 2 weeks (Q2W) and 420 mg QM were predicted to achieve a similar time‐averaged effect of 69% reduction in LDL‐C in patients on statin therapy, suggesting that an approximate 3‐fold dose increase is required for a 2‐fold extension in the dosing interval. Evolocumab dosing regimens of 140 mg Q2W or 420 mg QM were predicted to result in comparable reductions in LDL‐C over a monthly period, consistent with results from recently completed phase 3 studies.
Keywords: monoclonal antibody, pharmacokinetics, pharmacokinetic/pharmacodynamic, target‐mediated drug disposition, PCSK9, LDL‐C
Low‐density lipoprotein cholesterol (LDL‐C) lowering is a crucial component of the management of atherosclerotic cardiovascular disease (ASCVD) and has broadly proven benefits for primary and secondary prevention of ASCVD complications.1 Low‐density lipoprotein receptors (LDLR) on hepatocytes bind apolipoproteins B and E on circulating lipoprotein particles followed by internalization of the LDLR‐lipoprotein complex. In the acidic environment of the endosome, LDL dissociates from the LDLR and is subsequently catabolized in lysosomes while the LDLR recycles back to the cell surface.2 LDLR recycling is critical for the maintenance of cellular and whole‐body cholesterol balance. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an enzymatically inactive serine protease predominantly secreted by hepatocytes. PCSK9 associates with LDLR and targets the internalized receptor for degradation, preventing recycling and thereby regulating serum LDL‐C levels.3
Compelling human genetic data generated from studies of several thousand patients provide strong validation for the role of PCSK9 in modulating LDL‐C levels and coronary heart disease (CHD) risk in humans. PCSK9 “gain‐of‐function” mutations are associated with elevated LDL‐C levels (eg, >300 mg/dL) and CHD risk, whereas “loss‐of‐function” (LOF) mutations are associated with lower LDL‐C levels (eg, ≤ 100 mg/dL) and reduced CHD risk.4, 5, 6, 7, 8, 9, 10 Two adults with LOF mutations in both alleles have been reported.10, 11 Despite having no detectable circulating PCSK910 and extremely low LDL‐C levels (< 20 mg/dL),10, 11 these individuals are otherwise healthy. Patients with heterozygous LOF mutations exhibit modestly lower serum levels of PCSK9 and as much as an 88% reduction in the incidence of CHD over a 15‐year period relative to noncarriers of the mutations.6
Hydroxymethyl glutaryl coenzyme A reductase inhibitors, or statins, are currently the LDL‐C–lowering treatment of choice for patients with increased cardiovascular risk. However, many patients either do not achieve their LDL‐C targets with statins or are unable to tolerate statins at the dose recommended for their level of risk. Failure to achieve the LDL‐C target may be due to high baseline LDL‐C concentrations, diminished responsiveness, or an inability to tolerate adequate statin dosages.12 Patients with inadequately controlled LDL‐C values are at increased risk of future cardiovascular events.
Several studies indicate that statin therapy increases the levels of PCSK9.3 It is hypothesized that the cholesterol‐lowering effect of statins may be attenuated by this increase in PCSK9 levels. Therefore, the use of a PCSK9 antagonizing therapy, alone or as an add‐on to statin therapy, may be a particularly effective strategy to lower LDL‐C, especially in high‐risk patients and in patients who cannot tolerate high statin doses.
Evolocumab is a fully human monoclonal immunoglobulin IgG2 approved for use as an adjunct treatment for hyperlipidemia. Evolocumab binds to human PCSK9 with high affinity in vitro (dissociation constant [KD] = 8.0 pM) and prevents its interaction with LDLR. Clinical studies have demonstrated robust reductions in LDL‐C following administration of evolocumab in patients with primary hyperlipidemia and mixed dyslipidemia as monotherapy,13 in combination with a statin,14 in statin‐intolerant patients,15 in patients with heterozygous familial hypercholesterolemia (HeFH),16 and in patients with homozygous familial hypercholesterolemia.17
This analysis described the pharmacokinetic (PK) and pharmacodynamic (PD) relationship of evolocumab using a mathematical model that captured evolocumab binding and removal of unbound PCSK9, and subsequent reduction in circulating LDL‐C. This modeling exercise helps to understand the implications of target‐mediated elimination on evolocumab dose and regimen selection, particularly given the higher PCSK9 concentrations observed in hypercholesterolemic patients treated with statins.
Methods
Study Participants
The model included results from a phase 1a study in healthy subjects (Study 20080397) and a phase 1b study in hypercholesterolemic patients stably treated with statins (Study 20080398). Details of the study designs have been reported previously.18 The protocols and study procedures were approved by an institutional review board for each site.
Study Design
In the ascending single‐dose phase 1a study, healthy subjects were randomized to a single dose of placebo or evolocumab ranging from 7 mg to 420 mg administered via subcutaneous (SC) injection. In the ascending multiple‐dose phase 1b study, hypercholesterolemic patients receiving low‐ to moderate‐dose statin therapy were randomized in 5 cohorts to SC placebo or evolocumab at doses of 14 mg once weekly (QW) × 6 doses, 35 mg QW × 6 doses, 140 mg once every 2 weeks (Q2W) × 3 doses, 280 mg Q2W × 3 doses, or 420 mg once monthly (QM; every 4 weeks) × 2 doses. Two additional patient cohorts that received either high‐dose statin therapy or had been diagnosed with HeFH received either SC placebo or evolocumab 140 mg Q2W × 3 doses.
PK/PD Sampling and Assays
Venous blood samples were collected for PK and PD measurements after administration of placebo or evolocumab. All PD blood samples were obtained after overnight fasting (> 10 hours). The blood samples were collected into tubes containing no anticoagulant and allowed to clot at room temperature for 30 to 60 minutes. The samples were then centrifuged, and serum aliquots were prepared and stored at −80°C for unbound evolocumab and unbound PCSK9 measurements.
Unbound evolocumab was measured in serum using a validated enzyme‐linked immunosorbent assay (ELISA). The ELISA was designed to measure unbound evolocumab in test samples by using highly specific anti‐idiotype antibodies for capture and detection of evolocumab. By virtue of binding to the antigen‐combining site of evolocumab, the anti‐idiotype antibodies bound to evolocumab that was not bound to PCSK9. The assay limits of quantification ranged from 0.8 μg/mL to 10 μg/mL. The validated procedure required accuracy (percentage difference from nominal concentration) and precision (percentage coefficient of variation) of ±15% and ≤15%, respectively, for the standard, and ±20% and ≤15%, respectively, for the QC samples.
Unbound PCSK9 was measured in serum using a qualified ELISA method.19 Measurements of PCSK9 quantified the unbound serum concentration by using evolocumab for capture and a second biotin‐conjugated anti‐PCKS9 antibody for detection. Thus PCSK9 that was bound to evolocumab was not measured. Standard and QC samples were prepared in 100% fetal bovine serum because of the high and variable levels of endogenous PCSK9 in human serum. The QC samples in each run were 45.0, 140, and 500 ng/mL PCSK9 in fetal bovine serum. The assay lower and upper limits of quantification were 15 ng/mL and 1200 ng/mL, respectively. The validated procedure required accuracy (percentage difference from nominal concentration) and precision (percentage coefficient of variation) of ±15% and ≤20%, respectively, for the standard and ±25% and ≤20%, respectively, for the QC samples. LDL‐C was measured using a homogeneous direct assay after overnight fasting.
Data Analysis
Maximum observed concentration (Cmax) and area under the serum concentration curve from time zero to the time of the last quantifiable concentration (AUClast) were determined using noncompartmental methods. Population PK/PD data were analyzed using the nonlinear mixed‐effects modeling software program NONMEM (version 7.2)20 on the NONMEM High Performance Cluster. A simultaneous approach to the PK/PD analysis was undertaken using the stochastic approximation expectation‐maximization and importance sampling estimation methods sequentially. The M3 method was used for unbound evolocumab and unbound PCSK9 to simultaneously model observations as either continuous or categorical data when observations were above or below the limit of quantification, respectively.21, 22, 23 All statistical analyses were performed using either TIBCO Spotfire S+ for Windows version 8.2 (TIBCO Software Inc, Palo Alto, California) or R software 2.10.1 or higher (The R Foundation for Statistical Computing, Vienna, Austria).
The target‐mediated drug disposition (TMDD) model was used to analyze concentration‐time data using a previously described model framework.24, 25 Unbound evolocumab and unbound PCSK9 were analyzed using the steady‐state approximation of the TMDD model that incorporated binding between evolocumab and PCSK9, turnover of PCSK9, and elimination of drug‐target complex, as shown in Figure 1.26 Equations used for the model are shown in equations (1) to (3):
| (1) |
| (2) |
Figure 1.
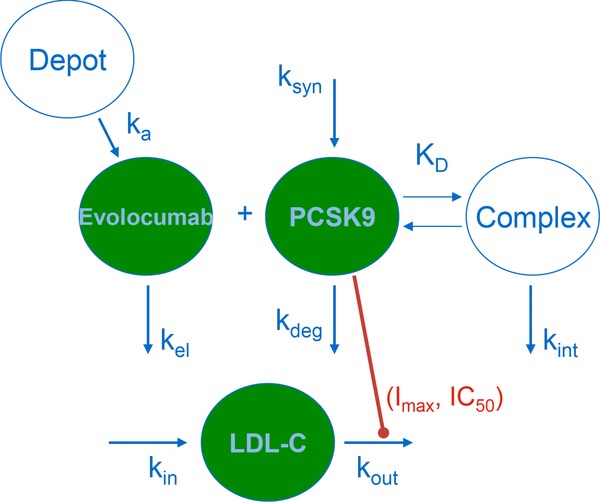
Schematic of the PK/PD model structure. Abbreviations: Imax, maximal inhibition; IC50, PCSK9 concentration associated with half‐maximal inhibition; ka, absorption rate constant; kel, elimination rate constant; ksyn, PCSK9 production rate constant; kdeg, PCSK9 elimination rate constant; KD, dissociation constant; kint, evolocumab‐PCSK9 complex elimination rate constant; kin, LDL‐C production rate constant; kout, LDL‐C elimination rate constant; PCSK9, proprotein convertase subtilisin/kexin type 9.
| (3) |
These equations described the PK of evolocumab in the depot (Adepot) after SC administration, the total drug amount in the central compartment (TDA), and the total ligand concentration (TLC), respectively. A 1‐compartment open model with linear elimination from the central compartment was parameterized by volume of distribution in the central compartment (V) and drug clearance (CL). Absorption after SC administration was described by a first‐order process (ka) from the depot compartment to the central compartment. Bioavailability (F) was fixed to 72% (Amgen internal data). The total drug concentration (TDC; nM) was calculated as shown in equation (4):
| (4) |
A zero‐order production rate constant (ksyn) and a first‐order degradation rate constant (kdeg) described the production and degradation of PCSK9, related to the baseline PCSK9 (BASEPCSK9) level as ksyn/kdeg. The quasi‐steady‐state approximation of the full TMDD model was selected.25, 27 In this model approximation, binding of evolocumab and PCSK9 to form a complex was described by the steady‐state constant (kss), which represented the ratio of the sum of the complex internalization rate (kint, here representing the elimination of complex) and dissociation rate constant (koff) to the drug‐target association rate constant (kon), where the KD was defined as the ratio of koff/kon.
The free drug concentration (FDC; nM) was determined from TLC in equation (5):
| (5) |
The free ligand concentration (FLC; nM) was calculated as shown in equation (6):
| (6) |
Total drug or ligand referred to the sum of bound and free species under the assumption of one‐to‐one stoichiometry of binding between drug and target.
An indirect response model was used to describe the effect of PCSK9 on LDL‐C removal,28, 29 consistent with PCSK9's role in the recycling of LDLR as shown in equation (7):
| (7) |
The model included parameters for the zero‐order production rate constant (kin) and the first‐order degradation rate constant (kout) of LDL‐C. The maximal kout was the rate of LDL‐C degradation in the absence of PCSK9. The ability of PCSK9 to modulate kout was estimated with the potency parameters IC50 (the serum unbound PCSK9 concentration associated with 50% of the maximal kout) and Imax (the theoretical maximal proportional change in kout due to PCSK9).
Between‐subject variability (BSV) was modeled as log‐normally distributed. BSV for kdeg and kint were fixed at 0%. Proportional error models described residual error for unbound evolocumab and unbound PCSK9. A proportional and additive error model described residual error for LDL‐C.
The assessment of model adequacy and decisions about increasing model complexity were driven by the data and guided by goodness‐of‐fit criteria including (1) visual inspection of diagnostic scatter plots (predicted vs observed concentration), histograms of individual random effects, and visual predictive checks30; (2) precision of parameter estimates; and (3) objective function value (OFV) and the stability of the objective function. A decrease in OFV of 10.8 points, corresponding to a χ2 P‐value of 0.001 with 1 degree of freedom, was considered significant. Model evaluation also included prediction‐corrected visual predictive checks (VPC).31 PK, PCSK9, and LDL‐C observations were placed in time bins corresponding to the nominal collection times listed in the study protocol: 4, 8, 12, or 24 hours postdose on day 1 and on days 3, 4, 5, 6, 7, 8, 11, 15, 22, 29, 36, 43, 57, 71, 85. Observations were assigned to bins by determining the closest nominal time postdose corresponding to the actual time postdose.
Simulations
Simulations were performed with the final PK/PD model using Berkeley Madonna Version 8.3.14.32 The time course of LDL‐C response was simulated following repeated SC administration of evolocumab at 140 mg Q2W, 280 mg QM, and 420 mg QM for 12 weeks in a patient population stably treated with statins with the predicted baseline PCSK9 in the population PK/PD model (5.27 nM [379 ng/mL]).
Results
Baseline characteristics are shown in Table S1. A total of 73 patients were treated with evolocumab, and 28 patients received placebo, in the phase 1a and 1b studies. Most were male (73.3%) and white (83.2%), with a mean age and body weight of 45.5 years and 81.4 kg, respectively. The baseline PCSK9 was 1.58‐fold higher in the phase 1b study relative to the phase 1a study due to the inclusion of hypercholesterolemic patients on stable statin therapy (> 97%) in the phase 1b study. Baseline LDL‐C was similar between the evolocumab and placebo groups in both studies.
Figure 2 shows the observed mean ± SD serum unbound evolocumab concentration‐time profiles after a single SC administration of 21, 70, 210, or 420 mg evolocumab in healthy subjects. Unbound evolocumab concentrations were below the detection limit for all healthy subjects after a single 7 mg SC dose of evolocumab and were not included in the figure. Treatment with a single SC administration of evolocumab resulted in nonlinear elimination of unbound evolocumab over the dose range studied in healthy subjects. The terminal half‐life of unbound evolocumab was dose dependent with the longest half‐life observed after 420 mg SC evolocumab.
Figure 2.
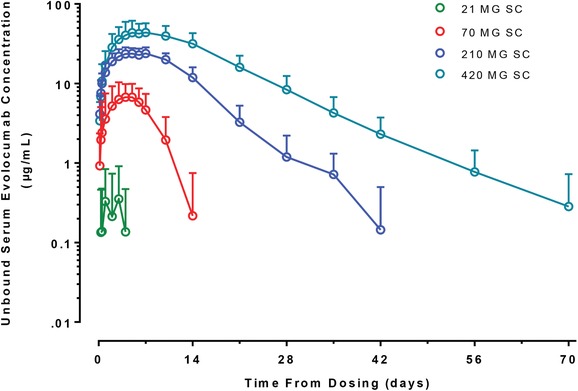
Observed unbound evolocumab concentration‐time profiles after single administration to non‐statin‐treated healthy subjects. The open circles represent the arithmetic mean of the observed data, and the error bars represent the standard deviation.
Unbound evolocumab noncompartmental PK parameters following 140 mg Q2W in hypercholesterolemic patients treated with low‐ to moderate‐intensity statins or high‐intensity statins are shown in Table 1. A 26% increase in baseline PCSK9 and a 20% decrease in unbound evolocumab Cmax and AUClast were observed in patients on high‐intensity statins compared with patients on low‐ to moderate‐intensity statins, consistent with target‐mediated elimination of evolocumab.
Table 1.
Summary of Unbound Evolocumab Pharmacokinetic Parameters After 140 mg SC Evolocumab Every 2 Weeks in Hypercholesterolemic Patients Treated With Low‐ to Moderate‐Intensity or High‐Intensity Statins
| Parameter (Mean ± SD) | Low‐Moderate (n = 6) | High (n = 9) | Ratio (High/Low‐Moderate) |
|---|---|---|---|
| Baseline PCSK9 (ng/mL) | 385 ± 88 | 487 ± 214 | 1.26 |
| Cmax (μg/mL) | 20 ± 13 | 16 ± 11 | 0.80 |
| AUClast (day·μg/mL) | 226 ± 249 | 181 ± 157 | 0.80 |
Low‐moderate: atorvastatin <40 mg, rosuvastatin <20 mg, simvastatin <40 mg, or simvastatin 80 mg. High: atorvastatin 80 mg or rosuvastatin 40 mg. AUClast , area under the concentration‐time curve from time zero to the last quantifiable concentration; Cmax, maximum observed concentration; PCSK9, proprotein convertase subtilisin/kexin type 9.
Following a single SC administration of evolocumab, rapid decreases in unbound PCSK9 were observed within 4 hours and resulted in > 80% suppression of unbound PCSK9 after doses ≥ 21 mg SC evolocumab. Increasing the dose of evolocumab increased the duration of full suppression of unbound PCSK9 up to a maximum of 14 days with the highest dose evaluated of 420 mg SC evolocumab. On elimination of unbound evolocumab from serum, unbound PCSK9 concentrations gradually returned toward the baseline without evidence of rebound above the initial baseline values.
Based on the nonlinear elimination profile of evolocumab and consequent reductions in unbound PCSK9, a TMDD model was selected to describe evolocumab PK and PD (Figure 1). The data set used in the analysis included 101 individuals and 4910 observations including unbound evolocumab, unbound PCSK9, and LDL‐C (Figure 3). Overall, there was agreement between observed and predicted values for the 3 PK and PD endpoints of unbound evolocumab, unbound PCSK9, and LDL‐C for both population and individual predictions. The diagnostic plots indicated that the TMDD model provided an adequate description of the data.
Figure 3.
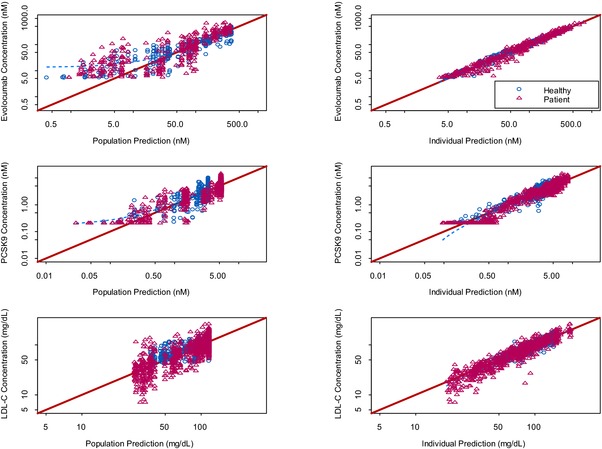
Diagnostic plots of model‐predicted and observed unbound evolocumab concentrations, unbound PCSK9 concentrations, and LDL‐C.
Population PK parameter estimates for the TMDD model of evolocumab and unbound PCSK9 are presented in Table 2. The final TMDD model provided reasonably precise estimates of the structural parameters (≤23% relative standard error [RSE]) of all the fixed effect parameters. The residual variabilities for unbound evolocumab and unbound PCSK9 were 24% and 31%, respectively. The intersubject variabilities (%CV) for ka, V, CL, BASEPCSK9, and kss were 90%, 40%, 96%, 19%, and 108%, respectively. A covariate indicated that the baseline PCSK9 was 3.36 nM (or 242 ng/mL) in healthy subjects and 5.27 nM (or 379 ng/mL) in hypercholesterolemic patients stably treated with statins. The prediction‐corrected VPC for all doses and the VPC for 140 mg Q2W indicated good agreement between the simulations and the observed data for unbound evolocumab and unbound PCSK9 (Figure 4).
Table 2.
Population Pharmacokinetic Parameter Estimates for Evolocumab Using a Target‐Mediated Drug Disposition Model
| Fixed Effects: Population Mean Parameter | Random Effects: Intersubject/Residual Variance | ||||
|---|---|---|---|---|---|
| Parameter | Units | Estimate | SE (%RSE) | Estimate (∼CV%) | SE (%RSE) |
| ka | 1/day | 0.245 | 0.0290 (12) | 0.807 (90) | 0.157 (19) |
| V | L | 2.66 | 0.156 (5.9) | 0.158 (40) | 0.0362 (23) |
| CL | L/day | 0.256 | 0.0439 (17) | 0.924 (96) | 0.260 (28) |
| kdeg | 1/day | 2.12 | 0.156 (7.3) | 0.00a | ___ |
| BASEPCSK9 | nM | 5.27 | 0.141 (2.7) | 0.0348 (19) | 0.00583 (17) |
| kss | nM | 0.253 | 0.0579 (23) | 1.17 (108) | 0.233 (20) |
| kint | 1/day | 0.0529 | 0.00184 (3.5) | 0.00a | ___ |
| θ1 | ___ | 0.637 | 0.0259 (4.1) | 0.00a | ___ |
| Covariance (ka − V) | 0.253 (70) | 0.0650 (26) | |||
| Covariance (ka − CL) | −0.671 (–78) | 0.182 (27) | |||
| Covariance (V − CL) | −0.183 (–48) | 0.0775 (42) | |||
| σ2 (evolocumab) | ___ | ___ | ___ | 0.0576 (24) | 0.00994 (4.1) |
| σ2 (PCSK9) | ___ | ___ | ___ | 0.0942 (31) | 0.00625 (2.0) |
CV%, coefficient of variation calculated as ; %RSE, relative standard error of the estimate calculated as (SE/Estimate)·100; SE, standard error of the estimate, ka, absorption rate constant; V, volume of distribution; CL, clearance; kdeg, PCSK9 degradation rate constant; BASEPCSK9, baseline PCSK9; kss, steady‐state constant; kint, complex elimination rate constant; θ1, fold change in baseline PCSK9 for healthy subjects vs statin‐treated patients.
Intersubject random variance was fixed at 0 in the pharmacokinetic model.
Figure 4.
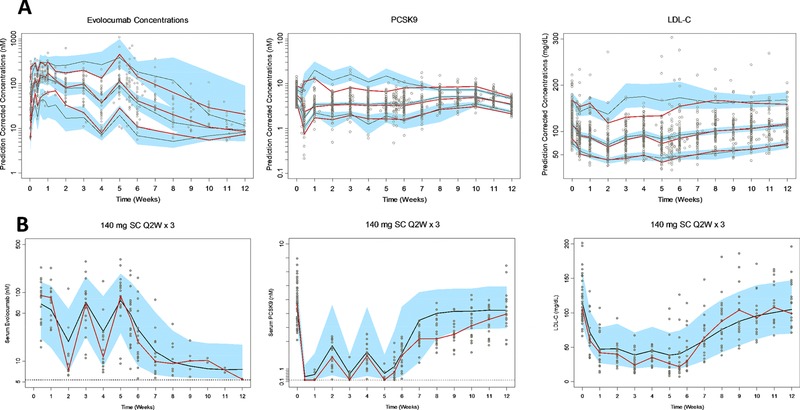
(A) Prediction‐corrected visual predictive check. The red and black lines represent the median and the 5th and 95th percentiles of observed and predicted data, respectively. The blue‐shaded region represents the 95% confidence interval of the respective median and 5th and 95th percentiles of predicted data. (B) Visual predictive check of 140 mg SC evolocumab Q2W × 3, median ± 90% prediction interval of simulated pharmacokinetics and pharmacodynamics (solid black line and blue‐shaded region, respectively, for n = 100 simulations) and observed data (open circles). Red line represents the observed median.
An indirect response model described the turnover of LDL‐C in which PCSK9 inhibited the elimination of LDL‐C, such that decreasing PCSK9 levels due to evolocumab administration led to increased elimination of LDL‐C, thereby decreasing serum LDL‐C levels (Figure 1). The final PK/PD model described the time course of LDL‐C. The final model provided reasonably precise estimates of the structural parameters (≤10% RSE) of all the fixed effect parameters, with proportional and additive residual variability of 11% and 7.8 mg/dL, respectively (Table 3). The intersubject variabilities for BASELDL‐C and IC50 were 22% and 69%, respectively. Initial attempts to estimate Imax suggested that it was near the boundary of 1.00, so its value was fixed to 1.00. The mean (± standard error of the estimate) value of IC50 for inhibition of the elimination of LDL‐C was 1.46 ± 0.152 nM. The prediction‐corrected VPC for all doses and the VPC for 140 mg Q2W indicated good agreement between the simulations and the observed data for LDL‐C (Figure 4).
Table 3.
Population Pharmacokinetic/Pharmacodynamic Parameter Estimates for Evolocumab Using an Indirect Response Model for LDL‐C
| Fixed Effects: Population Mean Parameter | Random Effects: Intersubject/Residual Variance | ||||
|---|---|---|---|---|---|
| Parameter | Units | Estimate | SE (%RSE) | Estimate (∼CV%) | SE (%RSE) |
| kout | 1/day | 0.305 | 0.0121 (4.0) | 0.00a | ___ |
| BASELDL‐C | mg/dL | 116 | 3.36 (2.9) | 0.0465 (22) | 0.00698 (15) |
| Imax | ___ | 1.00 (fix) | ___ | 0.00a | ___ |
| IC50 | nM | 1.46 | 0.152 (10) | 0.481 (69) | 0.106 (22) |
| σ2 (proportional error) | ___ | ___ | ___ | 0.0130 (11) | 0.00505 (4.4) |
| σ2 (additive error) | ___ | ___ | ___ | 60.1 (7.8)b | 0.519 (6.7) |
CV%, coefficient of variation calculated as ; %RSE, relative standard error of the estimate calculated as (SE/Estimate)·100; SE, standard error of the estimate; kout, elimination rate constant for LDL‐C; BASELDL‐C, baseline LDL‐C; Imax, maximal inhibition; IC50, concentration associated with half‐maximal inhibition.
Intersubject random variance was fixed at 0 in the pharmacokinetic/pharmacodynamic model.
Represents the standard deviation and calculated as .
Based on the final PK/PD model, simulations were performed to investigate the time course of LDL‐C response after 140 mg SC Q2W, 280 mg SC QM, and 420 mg SC QM evolocumab in patients treated with stable statins (Figure5). The simulations indicated that doubling the evolocumab dose from 140 mg SC Q2W to 280 mg SC QM to extend the dosing interval did not adequately maintain the reductions in LDL‐C over the entire monthly dosing interval from weeks 8 to 12 after LDL‐C reductions reached steady state. The time‐averaged effects in the area under the LDL‐C effect curve based on the simulations for evolocumab doses of 140 mg Q2W, 280 mg QM, and 420 mg QM were 68.9%, 63.5%, and 68.9%, respectively. Therefore, based on simulations from the PK/PD model, an approximate 3‐fold increase in the dose to 420 mg SC QM evolocumab appeared to be required to maintain stable LDL‐C reductions observed after 140 mg SC Q2W and to limit fluctuations in LDL‐C over the dosing interval.
Figure 5.
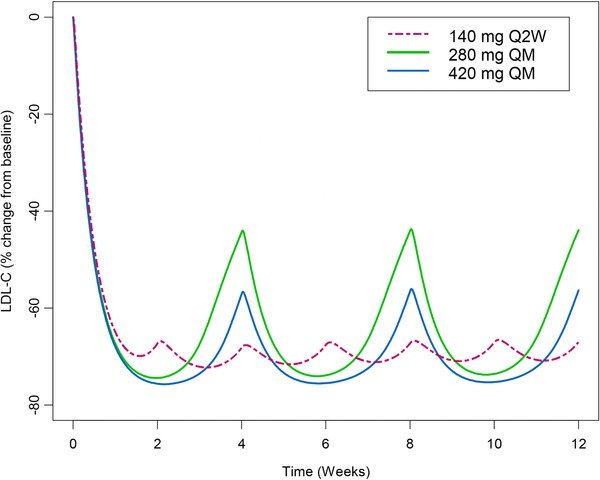
Model‐predicted time course of LDL‐C after multiple SC evolocumab doses.
Discussion
Knowledge of the PK/PD relationship including the onset and offset of response is critical to defining optimal doses and regimens for novel therapeutics in different patient populations. Simulations based on the PK/PD relationship among unbound evolocumab, unbound PCSK9, and LDL‐C following evolocumab administration were used to help support dose and regimen selection for clinical studies. The model was based on intensive, longitudinal data collected in 101 individuals (44 healthy subjects and 57 hypercholesterolemic patients treated with statins), including data from single administration or repeated dosing of evolocumab for 2‐months. This PK/PD analysis leveraged the target‐mediated interaction between evolocumab and PCSK9, and the impact on LDL‐C, to evaluate the dose increment required to maintain maximal reduction in LDL‐C while extending the dosing interval from Q2W to QM.
Empirical approaches to posology would assume that doubling the dose would be sufficient to extend the drug effect from 2 weeks to 4 weeks. However, given the nonlinear PK of evolocumab due to TMDD and the nonlinear PK/PD relationship between PCSK9 and LDL‐C, this simplification was inappropriate for a monoclonal antibody directed against PCSK9. A 3‐fold increase in the dose of evolocumab from 140 mg to 420 mg was required to obtain similar time‐averaged reductions in LDL‐C when the dosing interval was extended from Q2W to QM. Both doses were associated with more than 5% greater time‐averaged reduction of LDL‐C compared with the 280‐mg QM dose of evolocumab. For statins, a similar difference (approximately 4% to 6%) in LDL‐C reduction between lower‐intensity and higher‐intensity therapy has been used to support high‐dose statin therapy in clinical practice.33
The TMDD model and its approximations have been applied to describe nonlinear PK and reflect the influence of the target on drug disposition.24, 25, 27 Concentration‐dependent binding between drug and target and subsequent elimination of the drug‐target complex augment the elimination of the drug over the endogenous elimination pathways such as catabolism by the reticuloendothelial system. Here, availability of serum unbound PCSK9 concentrations facilitated model development and provided additional confirmation of the mechanism of action. The TMDD model parameters gave an indication of the extent and duration of PCSK9 suppression required to maintain consistent reduction in LDL‐C. The linear PK parameters (ie, CL and V) for evolocumab were similar to those for other IgG antibodies.34 Similarly, the kss value was within the reported range for TMDD models of other drugs and their targets. In comparing the in vivo kss to the in vitro KD, kss is always greater than KD, but it may be within the same range or up to 30 times greater.26
PCSK9 may facilitate LDLR degradation via 2 routes: (1) intracellular endosomal association with LDLR and (2) internalization of secreted PCSK9 following cell surface association with LDLR.35, 36, 37, 38 Intracellular cholesterol regulates the synthesis of PCSK9 and LDLR through the sterol regulatory element binding proteins known as SREBPs.3 In addition, clearance of PCSK9 is primarily mediated through its association with LDLR.3 The estimated kdeg of PCSK9 in this study was 2.12 day−1, suggesting an elimination half‐life of approximately 8 hours, consistent with the short half‐life of injected recombinant PCSK9 observed after administration to mice.36, 39 The higher baseline PCSK9 observed in patients treated with statins compared to healthy subjects with a similar baseline LDL‐C was consistent with other reports.40, 41 In addition, in hypercholesterolemic patients with a higher baseline PCSK9 due to high‐intensity statin therapy, there was a 20% reduction in unbound evolocumab exposure. Thus, it is plausible that the high‐turnover soluble target PCSK9 represents a sink for exogenous therapeutic proteins directed against PCSK9.
The indirect response model has been applied to capture the PD of drugs acting on LDL‐C including treatment with statins in hypercholesterolemic patients42, 43 and with corticosteroids in rats.44 Dietary cholesterol consumption and de novo hepatic cholesterol synthesis regulate production of very low density lipoprotein cholesterol and ultimately LDL‐C, and internalization by hepatic LDLR is the predominant elimination mechanism for circulating LDL‐C.45 Here, the production and elimination mechanisms were represented as system parameters (kin and kout, respectively) in the indirect response model, with PCSK9 regulating kout according to its known role in regulating LDLR expression. The maximal elimination rate (kout) gave an estimate of the time to achieve a new equilibrium in the absence of PCSK9, taking into account the half‐life of LDL‐C. The estimated maximal kout for LDL‐C was 0.305 day−1, suggesting an approximate half‐life of approximately 2.3 days for the elimination of LDL‐C in humans in the absence of PCSK9. Previously reported values of kout in healthy subjects and patients treated with statins ranged from 0.105 to 0.350 day−1.42, 45, 46 It is necessary to account for these delays in the onset and offset of LDL‐C response to accurately estimate LDL‐C reduction after administration of a monoclonal antibody such as evolocumab.
The model described in this report represented a simplified version of the biology of cholesterol metabolism as well as interactions between the drug and PCSK9 and between PCSK9 and rates of LDL‐C elimination. This model did not include other known endogenous binding partners for PCSK9 such as LDL‐C and LDLR.36, 47 As mentioned, the TMDD model assumed there was no competition for the drug binding to its target. Exploration of a more complex physiologic model may help to better understand these interactions. In addition, we performed simulations using the typical values of PK/PD parameters using Berkley Madonna. Thus, the impact of dose regimen on interindividual variability in the response was not evaluated.
Conclusions
Based on simulations that included PCSK9‐mediated nonlinear evolocumab elimination, 140 mg Q2W and 420 mg QM were predicted to achieve similar LDL‐C responses, suggesting that an approximate 3‐fold dose increase was required for a 2‐fold extension in the dosing interval. Evolocumab 140 mg Q2W and 420 mg QM resulted in comparable reductions in LDL‐C over a monthly period. Conclusions from modeling and simulation exercises using phase 1 study data were consistent with results of phase 3 studies,13, 14 demonstrating the value of early characterization of PK/PD relationships to help inform dose and regimen selection.
Funding
This study was sponsored by Amgen, Inc. Meera Kodukulla of Amgen and Jonathan Latham of PharmaScribe, LLC (on behalf of Amgen) assisted the authors with preparation of the manuscript.
Declaration of Conflicting Interests
J.P.G., S.D., M.K., A.G., M.G.E., M.D., M.A.G., R.S., and S.M.W. were employees and stockholders of Amgen, Inc at the time this work was conducted. D.B. received honoraria from Amgen Inc, Sanofi‐Aventis, Merck Sharp & Dohme, Pharma Dynamics, AstraZeneca, Pfizer, and Aegerion; and was a consultant to board: Merck Sharp & Dohme, Sanofi‐Aventis, Gemphire, and Aegerion.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplemental Table S1 Baseline Characteristics in Integrated Phase 1a and Phase 1b Studies
Acknowledgments
We thank the volunteer participants and clinical teams that conducted the studies. Additionally, we would like to thank Alex Colbert for his bioanalytical support. Meera Kodukulla (Amgen Inc) and Jonathan Latham (on behalf of Amgen Inc) assisted the authors with the preparation and submission of this work.
References
- 1. Cholesterol Treatment Trialists Collaboration , Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670‐1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown MS, Herz J, Goldstein JL. LDL‐receptor structure. Calcium cages, acid baths and recycling receptors. Nature. 1997;388(6643):629‐630. [DOI] [PubMed] [Google Scholar]
- 3. Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;(50 Suppl):S172‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154‐156. [DOI] [PubMed] [Google Scholar]
- 5. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161‐165. [DOI] [PubMed] [Google Scholar]
- 6. Cohen JC, Boerwinkle E, TH Mosley Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264‐1272. [DOI] [PubMed] [Google Scholar]
- 7. Kotowski IK, Pertsemlidis A, Luke A, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low‐density lipoprotein cholesterol. Am J Hum Genet. 2006;78(3):410‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benjannet S, Hamelin J, Chretien M, Seidah NG. Loss‐ and gain‐of‐function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J Biol Chem. 2012;287(40):33745‐33755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lopez D. PCSK9: an enigmatic protease. Biochim Biophys Acta. 2008;1781(4):184‐191. [DOI] [PubMed] [Google Scholar]
- 10. Zhao Z, Tuakli‐Wosornu Y, Lagace TA, et al. Molecular characterization of loss‐of‐function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445‐448. [DOI] [PubMed] [Google Scholar]
- 12. Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high‐dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403‐414. [DOI] [PubMed] [Google Scholar]
- 13. Koren MJ, Lundqvist P, Bolognese M, et al. Anti‐PCSK9 monotherapy for hypercholesterolemia: the MENDEL‐2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2531‐2540. [DOI] [PubMed] [Google Scholar]
- 14. Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate‐ or high‐intensity statin therapy on LDL‐C lowering in patients with hypercholesterolemia: the LAPLACE‐2 randomized clinical trial. JAMA. 2014;311(18):1870‐1882. [DOI] [PubMed] [Google Scholar]
- 15. Stroes E, Colquhoun D, Sullivan D, et al. Anti‐PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS‐2 randomized, placebo‐controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541‐2548. [DOI] [PubMed] [Google Scholar]
- 16. Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD‐2): a randomised, double‐blind, placebo‐controlled trial. Lancet. 2015;385(9965):331‐340. [DOI] [PubMed] [Google Scholar]
- 17. Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double‐blind, placebo‐controlled trial. Lancet. 2015;385(9965):341‐350. [DOI] [PubMed] [Google Scholar]
- 18. Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low‐density lipoprotein cholesterol levels: results from 2 randomized, double‐blind, placebo‐controlled, ascending‐dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60(19):1888‐1898. [DOI] [PubMed] [Google Scholar]
- 19. Colbert A, Umble‐Romero A, Prokop S, Xu R, Gibbs J, Pederson S. Characterization of a quantitative method to measure free proprotein convertase subtilisin/kexin type 9 in human serum. mAbs. 2014;6(4):1103‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user's guides (1989‐2009). Ellicott City, MD: Icon Development Solutions; 2009. [Google Scholar]
- 21. Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35(4):401‐421. [DOI] [PubMed] [Google Scholar]
- 22. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481‐504. [DOI] [PubMed] [Google Scholar]
- 23. Byon W, Fletcher CV, Brundage RC. Impact of censoring data below an arbitrary quantification limit on structural model misspecification. J Pharmacokinet Pharmacodyn. 2008;35(1):101‐116. [DOI] [PubMed] [Google Scholar]
- 24. Levy G. Pharmacologic target‐mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248‐252. [DOI] [PubMed] [Google Scholar]
- 25. Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507‐532. [DOI] [PubMed] [Google Scholar]
- 26. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic‐pharmacodynamic modeling of biologics. Expert Opin Drug Metab Toxicol. 2009;5(7):803‐812. [DOI] [PubMed] [Google Scholar]
- 27. Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target‐mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35(5):573‐591. [DOI] [PubMed] [Google Scholar]
- 28. Sharma A, Jusko WJ. Characterization of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1996;24(6):611‐635. [DOI] [PubMed] [Google Scholar]
- 29. Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21(4):457‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holford N. The visual predictive check—superiority to standard diagnostic (Rorschach) plots. Population Approach Group Europe 2005:June 16, 2005; Abstract 2738.
- 31. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krause A, Lowe PJ. Visualization and communication of pharmacometric models with berkeley madonna. CPT Pharmacometrics Syst Pharmacol. 2014;3:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicholls SJ, Brandrup‐Wognsen G, Palmer M, Barter PJ. Meta‐analysis of comparative efficacy of increasing dose of atorvastatin versus rosuvastatin versus simvastatin on lowering levels of atherogenic lipids (from VOYAGER). Am J Cardiol. 2010;105(1):69‐76. [DOI] [PubMed] [Google Scholar]
- 34. Dong JQ, Salinger DH, Endres CJ, et al. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin Pharmacokinet. 2011;50(2):131‐142. [DOI] [PubMed] [Google Scholar]
- 35. Mousavi SA, Berge KE, Leren TP. The unique role of proprotein convertase subtilisin/kexin 9 in cholesterol homeostasis. J Intern Med. 2009;266(6):507‐519. [DOI] [PubMed] [Google Scholar]
- 36. Grefhorst A, McNutt MC, Lagace TA, Horton JD. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res. 2008;49(6):1303‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nassoury N, Blasiole DA, Tebon Oler A, et al. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic. 2007;8(6):718‐732. [DOI] [PubMed] [Google Scholar]
- 38. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. 2008;105(6):1820‐1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmidt RJ, Beyer TP, Bensch WR, et al. Secreted proprotein convertase subtilisin/kexin type 9 reduces both hepatic and extrahepatic low‐density lipoprotein receptors in vivo. Biochem Biophys Res Commun. 2008;370(4):634‐640. [DOI] [PubMed] [Google Scholar]
- 40. Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49(2):394‐398. [DOI] [PubMed] [Google Scholar]
- 41. Mayne J, Dewpura T, Raymond A, et al. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim J, Ahn BJ, Chae HS, et al. A population pharmacokinetic‐pharmacodynamic model for simvastatin that predicts low‐density lipoprotein‐cholesterol reduction in patients with primary hyperlipidaemia. Basic Clin Pharmacol Toxicol. 2011;109(3):156‐163. [DOI] [PubMed] [Google Scholar]
- 43. Faltaos DW, Urien S, Carreau V, et al. Use of an indirect effect model to describe the LDL cholesterol‐lowering effect by statins in hypercholesterolaemic patients. Fundam Clin Pharmacol. 2006;20(3):321‐330. [DOI] [PubMed] [Google Scholar]
- 44. Hazra A, Pyszczynski NA, DuBois DC, Almon RR, Jusko WJ. Modeling of corticosteroid effects on hepatic low‐density lipoprotein receptors and plasma lipid dynamics in rats. Pharm Res. 2008;25(4):769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Turley SD, Spady DK, Dietschy JM. Role of liver in the synthesis of cholesterol and the clearance of low density lipoproteins in the cynomolgus monkey. J Lipid Res. 1995;36(1):67‐79. [PubMed] [Google Scholar]
- 46. Oh ES, Lee SH, Park MS, Park K, Chung JY. Modeling of the LDL cholesterol‐lowering effect of atorvastatin in Korean dyslipidemic patients and non‐patient volunteers. Int J Clin Pharmacol Ther. 2012;50(9):647‐656. [DOI] [PubMed] [Google Scholar]
- 47. Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low‐density lipoprotein binds to proprotein convertase subtilisin/kexin type‐9 (PCSK9) in human plasma and inhibits PCSK9‐mediated LDL receptor degradation. J Biol Chem. 2013;288(12):8279‐8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplemental Table S1 Baseline Characteristics in Integrated Phase 1a and Phase 1b Studies