

Abstract
BACKGROUND
Pompe disease (PD) is the first lysosomal storage disorder to be added to the Recommended Uniform Screening Panel for newborn screening. This condition has a broad phenotypic spectrum, ranging from an infantile form (IOPD), with severe morbidity and mortality in infancy, to a late-onset form (LOPD) with variable onset and progressive weakness and respiratory failure. Because the prognosis and treatment options are different for IOPD and LOPD, it is important to accurately determine an individual’s phenotype. To date, no enzyme assay of acid α-glucosidase (GAA) has been described that can differentiate IOPD vs LOPD using blood samples.
METHODS
We incubated 10 μL leukocyte lysate and 25 μL GAA substrate and internal standard (IS) assay cocktail for 1 h. The reaction was purified by a liquid–liquid extraction. The extracts were evaporated and reconstituted in 200 μL methanol and analyzed by LC-MS/MS for GAA activity.
RESULTS
A 700-fold higher analytical range was observed with the LC-MS/MS assay compared to the fluorometric method. When GAA-null and GAA-containing fibroblast lysates were mixed, GAA activity could be measured accurately even in the range of 0%–1% of normal. The leukocyte GAA activity in IOPD (n = 4) and LOPD (n = 19) was 0.44–1.75 nmol · h−1 · mg−1 and 2.0–6.5 nmol · h−1 · mg−1, respectively, with no overlap. The GAA activity of pseudodeficiency patients ranged from 3.0–28.1 nmol · h−1 · mg−1, showing substantial but incomplete separation from the LOPD group.
CONCLUSIONS
This assay allows determination of low residual GAA activity in leukocytes. IOPD, LOPD, and pseudodeficiency patients can be partially differentiated by measuring GAA using blood samples.
Pompe disease (PD),6 or glycogen storage disease type II, is an autosomal recessive disorder of glycogen metabolism caused by a deficiency of lysosomal acid α-glucosidase (GAA, EC 3.2.1.20). Classic infantile-onset Pompe disease (IOPD) presents in early infancy with hypertrophic cardiomyopathy, generalized muscle weakness, and failure to thrive. Patients with IOPD usually die within the first year of life from respiratory failure if not treated with enzyme replacement therapy. Late-onset Pompe disease (LOPD) is characterized by muscle weakness and respiratory insufficiency with onset ranging from childhood to adulthood, and a variable rate of disease progression. There is also a nonclassical infantile form with no cardiomyopathy (1–4).
Studies from the Taiwan newborn screening program showed that early initiation of enzyme replacement therapy in IOPD patients led to reversal of glycogen storage in muscle and restoration of cardiac size and function (5, 6). Long-term follow-up of these patients showed that they remained ventilator free with stable cardiac function and essentially normal motor development up to 90 months (7). Pompe disease was added to the Recommended Uniform Screening Panel in 2015. Measurement of GAA activity in dried blood spots by tandem mass spectrometry (MS/MS) (8), by fluorometry (9), or by digital microfluidic fluorometry (10) have been used for newborn screening of Pompe disease. In addition to detecting individuals with true Pompe disease, infants with low GAA due to pseudodeficiency, carrier status and variants of unknown significance (VOUS) are also detected. These screening assays cannot determine the clinical phenotype based on their enzyme activity measurements (11). A reliable and accurate post-newborn screening GAA enzyme analysis that can accurately measure low residual activity is needed. Such an assay with glucosidase alpha, acid (GAA) mutations and clinical findings would be important in guiding the management of screen positive individuals.
GAA hydrolyzes the terminal α-linked glucose residues in short polymers of glucose that are formed as remnants during breakdown of high molecular weight glycogen in tissues, especially in muscle and liver. Like GAA, maltase-glucoamylase present in leukocytes is also active at acidic pH on these substrates, and thus it was not initially possible to selectively measure GAA activity in whole blood. More recently, acarbose has been discovered as a selective inhibitor of maltase-glucoamylase, thus allowing newborn screening for Pompe disease using dried blood spots (8, 12). The classic GAA assay uses the fluorometric substrate 4-methylumbelliferyl-α-glucoside. Recent studies have shown that the analytical range of this assay, defined as the ratio of assay response measured with the normal control to that due to all GAA-independent processes, is relatively small compared to the corresponding value obtained with MS/MS assays (13), mainly due to the intrinsic fluorescence of the substrate itself (13). The low analytic range may be the reason that the fluorometric assay is unable to differentiate between IOPD vs LOPD in blood samples (14, 15). Evidence that residual GAA activity is correlated with the age of onset of Pompe disease comes from studies with skin fibroblasts (16, 17).
The analytical range of an LC-MS/MS assay was recently shown to be nearly 2 orders of magnitude higher than that for the fluorometric enzyme assay (13). Here we describe a new LC-MS/MS method to accurately measure residual GAA activity in leukocytes isolated from venous blood of healthy patients, carriers, patients carrying 1 or more pseudodeficiency alleles, and Pompe patients with a range of severity. We also studied GAA activity in immortalized B lymphocytes from Pompe patients with known age of onset of symptoms. Side-by-side comparative studies with the standard fluorometric assay were also carried out.
Methods
MATERIALS
Ammonium formate, acarbose, and formic acid were purchased from Sigma-Aldrich. CHAPS detergent was from Fisher Scientific. The substrate (S) and internal standard (IS) mixture for GAA was provided by the CDC. Reagents can now be obtained from PerkinElmer. Red blood cell lysis buffer for leukocyte extraction was from Qiagen.
PREPARATION OF ASSAY COCKTAIL
The S/IS mixture of GAA was dissolved in 0.1 mol/L ammonium formate buffer containing 12 μmol/L acarbose and 1 g/L CHAPS at pH 4.0 to a final concentration of 0.56 mmol/L for substrate and 5.6 μmol/L for IS.
SPECIMENS
Five to 10 mL blood collected in an acid citrate dextrose tube is the preferred specimen type for GAA enzyme analysis. Other anticoagulants such as EDTA or sodium heparin tubes are also acceptable. For an infant, 1–3 mL blood collection can be attempted. The normal controls, most of the carriers and individuals who were homozygous for the common pseudodeficiency allele [p.G675S_p.E689K] used for this study were de-identified samples from the Mount Sinai Genetic Testing Laboratory. Seven infant carriers were referred from newborn screening. A total of 4 patients with IOPD were included in this study. One was a known patient on enzyme replacement therapy treatment (IOPD-1). Blood was collected before the infusion. The other 3 patients were identified and confirmed from the New York State Pompe newborn screening. All had increased creatine kinase activity and cardiomyopathy. In the LOPD cohort (n = 19), 3 were adults (LOPD-4, 6, and 7). LOPD-4 and 6 were homozygous for the common splice site mutation c.-32-13T>G, the genotype associated with LOPD (18, 19). Both individuals had normal creatine kinase activity and no symptoms at the time of sampling. LOPD-7 had fatigue and increased creatine kinase activity and transaminases for a few years before diagnosis. Sixteen patients (including 1 sibling) were diagnosed from New York State Pompe newborn screening and predicted to be potential LOPD patients by GAA mutation status and reduced enzyme activity (Table 1). Ethical approval was obtained from the Icahn School of Medicine at Mount Sinai Institutional Review Board. Fibroblast and immortalized B-lymphocyte samples, including normal, IOPD, and LOPD samples, were from the Coriell Cell Repository (Camden, NJ).
Table 1.
GAA activities in patients with IOPD, LOPD, and pseudodeficiency alleles.
| ID | Age | GAAa (nmol · h−1 · mg−1) | Mean, % | Mutationa |
|---|---|---|---|---|
| IOPD-1 | 5.5 years | 0.9 | 1.2 | NAb |
| IOPD-2 | 10 days | 1.8 | 2.5 | // |
| IOPD-3 | 2 min | 0.4 | 0.6 | // |
| IOPD-4 | 3 min | 0.6 | 0.9 | _ // |
| LOPD-1 | 3 weeks | 3.2 | 4.5 | // |
| LOPD-2 | 2 weeks | 3.2 | 4.5 | // |
| LOPD-3 | 2 weeks | 2.7 | 3.7 | // |
| LOPD-4 | 28 years | 3.4 | 4.7 | // |
| LOPD-5 | 2 weeks | 3.1 | 4.4 | // |
| LOPD-6 | 32 years | 3.6 | 5.1 | // |
| LOPD-7 | 24 years | 4.0 | 5.6 | // |
| LOPD-8 | 6 weeks | 2.9 | 4.1 | // _ |
| LOPD-9 | 3 weeks | 4.1 | 5.8 | // |
| LOPD-10 | 2 weeks | 2.5 | 3.6 | // |
| LOPD-11 | 8.3 years | 2.0 | 2.8 | // |
| LOPD-12 | 2 weeks | 2.0 | 2.8 | // |
| LOPD-13 | 2 weeks | 2.6 | 3.7 | // |
| LOPD-14 | 2 weeks | 3.0 | 4.2 | // |
| LOPD-15 | 2 weeks | 3.6 | 5.1 | // |
| LOPD-16 | 2.5 weeks | 2.5 | 3.6 | // _ |
| LOPD-17 | 6 weeks | 6.5 | 9.2 | // |
| LOPD-18 | 2 weeks | 2.9 | 4.0 | // |
| LOPD-19 | 2 weeks | 2.7 | 3.8 | // |
| PD/Pathogenic-1 | 5 weeks | 16.6 | 23.2 | // |
| PD/Pathogenic-2 | 3 weeks | 8.8 | 12.4 | // |
| PD/Pathogenic-3 | 2 weeks | 4.9 | 6.9 | // |
| PD/Pathogenic-4 | 2 weeks | 5.0 | 7.0 | // |
| PD/Pathogenic-5 | 4 weeks | 6.5 | 9.1 | // |
| PD/Pathogenic-6 | 4 weeks | 8.8 | 12.3 | // |
| PD/Pathogenic-7 | 2 weeks | 3.0 | 4.2 | // |
| PD/Pathogenic-8 | 2 weeks | 5.0 | 7.0 | // |
| PD/Pathogenic-9 | 3 weeks | 8.4 | 11.7 | // |
| PD/Pathogenic-10 | 2 weeks | 5.5 | 7.8 | // _ |
| PD/VOUS-1 | 3 weeks | 3.6 | 5.0 | // |
| PD/VOUS-2 | 2 weeks | 16.6 | 23.3 | // |
| PD/VOUS-3 | 2 weeks | 11.3 | 15.8 | // |
| PD/VOUS-4 | 2 weeks | 5.3 | 7.5 | // |
| PD/VOUS-5 | 1.7 years | 9.16 | 12.9 | // |
| PD/VOUS-6 | 2 weeks | 8.26 | 11.6 | // |
| PD/VOUS-7 | 3 weeks | 3.43 | 4.8 | // |
| PD/PD-1 | Adult | 18.7 | 26.2 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-2 | Adult | 17.4 | 24.4 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-3 | Adult | 10.5 | 14.8 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-4 | Adult | 9.4 | 13.2 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-5 | Adult | 12.3 | 17.3 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-6 | Adult | 8.8 | 12.3 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-7 | Adult | 14.9 | 20.9 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-8 | Adult | 28.1 | 39.4 | p.G576S_p.E689K//p.G576S_p.E689K |
| PD/PD-9 | Adult | 21.6 | 30.3 | p.G576S_p.E689K//p.G576S_p.E689K |
| Normal range | 71.2 ± 38.2 (23.1–232.0) | |||
| Carrier range | 30.5 ± 17.3 (8.9–88.7) |
; ; .
NA, not applicable.
CALIBRATIONS AND QC
The CDC provided the calibration standard mixtures of product (P) and IS (P/IS) at ratios of 0, 0.05, 0.1, 0.5, 1, 2, and 5. Full calibration was performed every 6 months. The normal control pool was prepared by pooling the lysates from previously tested negative samples. The affected control pool was prepared by heating the pooled normal lysate at 70 °C for 2h to inactivate GAA activity. Included in every batch was the P/IS (1/1) calibrator, the normal control, the affected control, and a blank sample.
PREPARATION OF LEUKOCYTES PELLET
We added 3 parts of room temperature red blood cell (RBC) lysis solution to 1 part blood and mixed gently by inverting the tube 10 times. After incubating the sample for 10 min at ambient temperature, the samples were mixed again by inversion 10 times. The tubes were centrifuged for 5 min at 600g, the supernatant decanted, and the remaining pellet vortex-mixed for 5 s to disrupt the pellet. Then 5 mL of ice-cold deionized water was added and the tube vortex-mixed for 10 s to lyse the leftover RBC. Five milliliters of ice-cold 1.8% NaCl was added to restore isotonicity, the tube vortex-mixed for 10 s, and then the mixture centrifuged at 600g for 5 min. The supernatant was decanted and the pellet washed once more as above. After final centrifugation, the pellet was stored at −20 °C until analysis.
PREPARATION OF LEUKOCYTES LYSATE
On the day of enzyme assay, the frozen leukocyte pellet was thawed at room temperature and then kept on ice throughout the remainder of the process. We added 0.3–0.6 mL of ice-cold normal saline (0.9% NaCl) depending on the size of the pellet and then vortex-mixed to resuspend the pellet. Using a QSonica Q700 sonicator, set to amplitude 3% and process time 6 pulses (1 s/pulse), the pellet was sonicated with 6 – 8 pulses to disperse the pellet homogenously. Using a transfer pipette, lysates were transferred to a labeled 1.5 mL microfuge tube and centrifuged for 10 min at 400 × g.
DETERMINATION OF LYSATE PROTEIN CONCENTRATION
We pipetted 10 μL of blank, bovine serum albumin standards (0.2, 0.5, 0.75, 1.0, 1.25,1.5 g/L), quality controls, and sample lysates into 96-well plate. After adding 25 μL of protein assay reagent A (Bio-Rad 500-0113) to each well, the plate was vortex mixed for 60 s at 600 rpm. We then added 200 μL of protein assay reagent B (Bio-Rad 500-0114) to each well, followed by incubation at ambient temperature for 10 min. The absorbance of the blue color product was read at 750 nm on a Varioskan Flash microplate reader.
LEUKOCYTE LYSATE REACTION
Ten microliters of leukocyte lysate (containing 2–10 mg of protein) was incubated with 25 μL of assay cocktail at 37 °C for 1 h. The reaction was quenched with 100 μL of methanol/ethyl acetate (1:1, v/v) and followed by liquidliquid extraction with 400 μL ethyl acetate and 400 μL water. One hundred milliliters of the organic layer was transferred to a new 96-well plate and dried under nitrogen. The final sample was reconstituted in 200 μL of methanol.
LC-MS/MS ANALYSIS
An Agilent 6460 LC-MS/MS system was used. An Agilent Zorbax C18 reversed-phase column (50 mm × 2.1 mm internal diameter, 1.8-μm particle size) was maintained at 30 °C. Mobile phase A consisted of 5% acetonitrile with 0.1% formic acid and mobile phase B consisted of 1:1 methanol: acetonitrile with 0.1% formic acid. The LC conditions and MS parameters are described in the Supplemental Material file that accompanies the online version of this article at http://www.clinchem.org/content/vol63/issue4. The extracted ion chromatograms of P and IS of 6 enzymes including GAA can be viewed in online Supplemental Fig. 1.
ASSAY OF GAA BY FLUOROMETRY
Full details of fluorometric and LC-MS/MS GAA assays in leukocytes and B-lymphocytes are provided in the online Supplemental Material file.
Results
GAA STABILITY STUDIES
GAA stability was tested in blood samples stored in Vacutainer Tubes (BD Diagnostics) and in leukocyte lysates. Blood was collected into multiple tubes from 4 healthy donors and maintained at ambient temperature. The leukocytes were extracted immediately after sample collection, and 24, 48, 72, 96, and 120 h after blood collection. The mean (SD) GAA activity was 92.3% (10.2%) (24 h), 82.9% (16.4%) (48 h), 68.9% (14.0%) (72 h), 70.3% (15.2%) (96 h), and 49.0% (8.6%) (120 h) of the freshly processed leukocyte samples (Fig. 1A). Although this reduction would probably not cause a normal activity to erroneously enter into the affected range, it is important that the specimen be received and processed within 72 h of the blood draw. When an intermediate enzyme reduction is observed, a reference enzyme activity should be assessed. Random leukocyte lysates (n = 9) were assayed immediately after sonication and after storage at −80 °C. The lysates were tested again after 1 freeze-thaw or 2 freeze-thaw cycles to determine GAA stability. The GAA activities were 86.5 (8.9%) and 81.6 (9.4%) of the fresh sample activities after 1 and 2 freeze-thaw cycles, respectively (Fig. 1B). Long-term stability of normal control lysates stored at −80 °C was tested over a period of 12 months. The monthly mean of the normal control aliquots were stable, with an approximately 8.4% fluctuation and no trend of activity decline (Fig. 1C).
Fig. 1. GAA stability studies under different conditions.
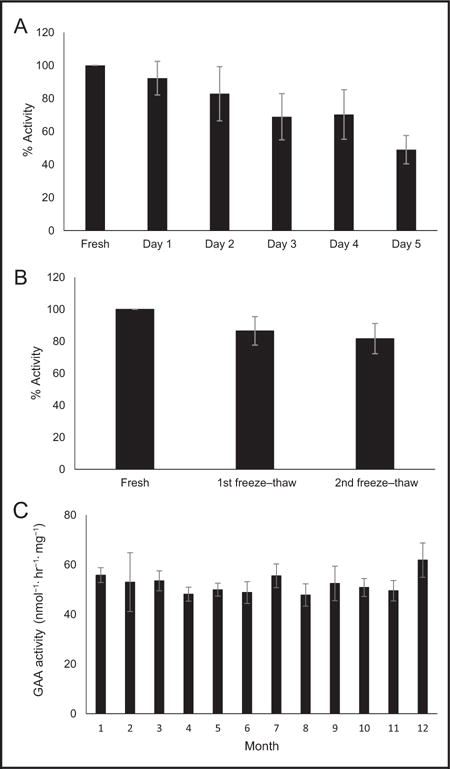
(A) GAA stability in blood stored at ambient temperature followed by leukocyte preparation on days 0, 1, 2, 3, 4, and 5 (n = 4); (B) GAA stability of lysate with 1 and 2 freeze thaw cycles (n = 9); (C) GAA long-term stability of lysate stored at −80 °C (n = 2–5 for each month). Error bars represent the SDs.
REACTION TIME COURSE, ASSAY PRECISION, AND ANALYTICAL RANGE
GAA product formation as a function of incubation time (15, 30, 60, 90, and 120 min) was studied with 5 leukocyte lysates. Results showed near linear increase in GAA product with incubation time (see online Supplemental Fig. 2). A 60-min incubation time was chosen for all subsequent assays, which was well within the linear range.
Ten aliquots of normal control pool were tested in the same assay, and the GAA activity was 61.1 (2.3) nmol · h−1 · mg−1 (CV of 3.7%). The same aliquots were tested in 8 different days, and the GAA activity was 57.3 (5.9) nmol · h−1 · mg−1 (CV of 10.3%).
The analytical range was defined as the enzyme-dependent assay response from the pooled normal control sample divided by the assay response for all GAA-independent processes (13). We anticipate that assays with a higher analytical range will be more accurate since the enzymatic activity values are spread over a larger range. In turn, we hypothesize that a high analytical range assay might be useful in distinguishing the small amounts of residual GAA activity in leukocyte lysates from IOPD, LOPD, and pseudodeficiency patients.
For MS/MS assays, the analytical range was calculated as: (GAA activity for normal control sample) − (GAA activity of the blank assay without leukocyte lysate)/(GAA activity of the assay without leukocyte lysate) (13). By using leukocyte lysates from non-Pompe pools, the analytical range for the LC-MS/MS assay of GAA was found to have a mean (SD) of 1135 (539) (see online Supplemental Table 3). The analytical range measured using fluorometry with 4-methylumbelliferyl-α-glucoside under identical assay conditions yielded a mean (SD) of 1.6 (0.1) (see online Supplemental Table 4). Thus, the analytical range for the LC-MS/MS assay was >700-fold higher than that for the fluorometric assay.
LINEARITY STUDIES WITH STANDARD REAGENTS AND CELL LYSATE MIXTURES
Calibration standard mixtures provided by the CDC were diluted 10-fold with methanol and analyzed directly by LC-MS/MS in triplicate. The P/IS ratio was linear from 0.05–5 with a value of R2 of 0.9998 (see online Supplemental Fig. 3). Linearity was also tested with cell lysate mixtures. A mixing study was designed to test whether small differences in GAA activity could be distinguished at the low activity range. A lysate from fibroblasts from an IOPD patient thought to be null in GAA activity because of homozygosity of a nonsense mutation (p.R854X) in the GAA gene (Coriell repository cell line GM00338) was mixed in various proportions with a lysate from normal fibroblasts of the same protein concentration to give 0%, 1%, 2%, 5%, 10%, and 50% normal fibroblast lysate.
The GAA activity was determined by LC-MS/MS in triplicate (Table 2). The GAA activity increased linearly with the increased percentage of normal lysate protein with a R2 = 0.9981 (Fig. 2). The mean ion counts for triplicate analysis of the blank, the 100% Pompe cell lysate, and the normal lysate were 74 [signal-to-noise ratio (S/N) ratio of 3.6], 658 (S/N ratio of 22.6) and 578 922, respectively. There was a widespread range of responses. Even the signal corresponding to a 0.1% of normal GAA activity in the Pompe cells was approximately 10-fold of the blank, showing that we could detect GAA well below 1% of normal. In contrast, a very high signal in blank was observed with fluorometric assay comparing to normal leukocyte control (10983 vs 17113, see online Supplemental Table 4).
Table 2.
Linear GAA activity responses in fibroblast mixtures (nmol · h−1 · mg−1).
| Expected proportion | Run 1 | Run 2 | Run 3 | Mean | SD | % of Normal |
|---|---|---|---|---|---|---|
| 100% Pompe (GM00338) | 0.1 | 0.1 | 0.2 | 0.1 | 0.0 | 0.12 |
| 99% Pompe + 1% normal | 1.4 | 1.4 | 1.4 | 1.4 | 0.0 | 1.1 |
| 98% Pompe + 2% normal | 2.6 | 2.4 | 2.7 | 2.6 | 0.1 | 2.1 |
| 95% Pompe + 5% normal | 6.4 | 6.7 | 6.7 | 6.6 | 0.2 | 5.4 |
| 90% Pompe + 10% normal | 12.6 | 13.9 | 13.6 | 13.3 | 0.6 | 11.0 |
| 50% Pompe + 50% normal | 61.6 | 69.0 | 69.3 | 66.6 | 3.5 | 54.8 |
| 100% Normal (GM23971) | 122.1 | 121.7 | 121.1 | 121.6 | 0.4 | 100.0 |
Fig. 2. Observed GAA activity as a function of the fraction of non-Pompe fibroblast protein added to IOPD fibroblasts.
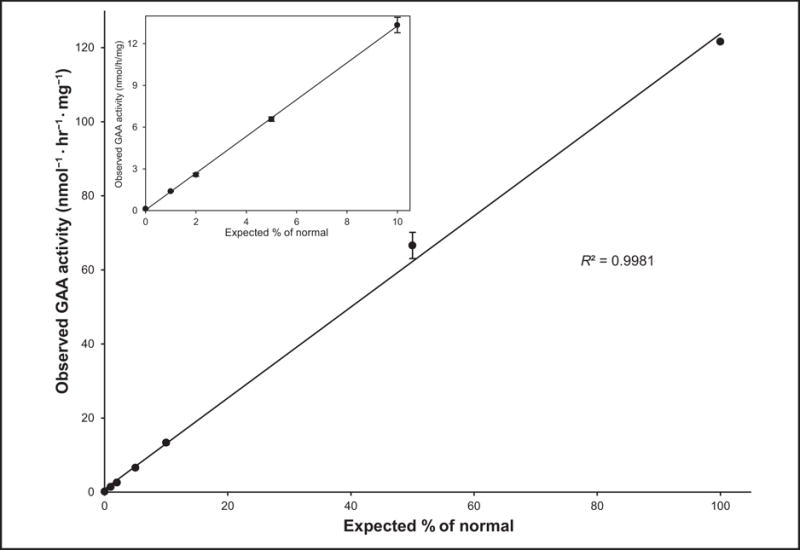
The x axis shows the expected percentage of normal or equivalently the percent of normal cell protein in the normal cell/IOPD cell mixture. The inset shows a blowup of the low-end range. Error bars are the SDs from triplicate analyses.
COMPARISON OF LC-MS/MS VS FLUOROMETRIC GAA ASSAY IN IMMORTALIZED B-LYMPHOCYTE CELL LINES
A further comparative study was performed in the lysates of immortalized lymphocytes from patients with early-onset Pompe disease (EOPD) and symptomatic LOPD patients (online Supplemental Table 7 lists the Coriell database information on the donors). These cells can be expanded to provide sufficient amounts that allowed us to carry out both LC-MS/MS and fluorometric assays on the same lysates under identical conditions.
The activities of the LOPD were all higher than those for the patients with EOPD by LC-MS/MS (Fig. 3, online Supplemental Tables 5 and 6). The separation between the EOPD and LOPD LC-MS/MS GAA activities was modest. In contrast, there is no correlation in GAA activity and severity of Pompe disease using the fluorometric assay.
Fig. 3. GAA activities of EOPD and LOPD in immortalized B-lymphocytes.
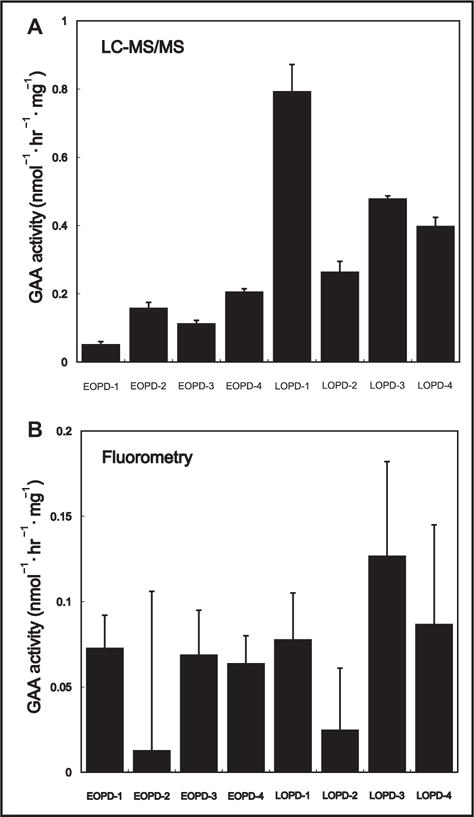
(A) Results analyzed by LC-MS/MS; (B) Results analyzed by fluorometry with 4-methylumbelliferyl-α-glucoside under identical assay conditions [same buffer and incubation conditions but with the MS/MS or the 4-methylumbelliferone substrates, appropriately (see the online Supplemental Material)]. Error bars are the SDs from triplicate analyses.
GAA ACTIVITY IN LEUKOCYTES FROM NORMAL CONTROLS, CARRIERS, PATIENTS WITH POMPE DISEASE, AND INDIVIDUALS WITH PSEUDODEFICIENCY ALLELES
The normal reference range for leukocytes was established with 160 normal controls. The GAA activity [mean (SD)] was 71.2 (38.2) nmol · h−1 · mg−1, ranging from 23.1–232.0 nmol · h−1 · mg−1. The mean value of normal controls was used to calculate the percentage of normal. The GAA activity of Pompe carriers (n = 59) was 42.8 (24.2%) of normal, ranging from 12.5–124.5 nmol · h−1 · mg−1. The GAA activity of IOPD (n = 4) was 1.3 (0.8%) of normal, ranging from 0.44–1.75 nmol · h−1 · mg−1. The GAA activity of LOPD (n = 19) was 4.5 (1.4%) of normal, ranging from 2.0-6.5 nmol · h−1 · mg−1. The GAA activity was significantly lower in IOPD patients compared to that of the LOPD patients (P < 0.001). No overlap between IOPD and LOPD patients was observed in our cohort.
GAA activities from individuals with pseudodeficiency alleles was summarized in 3 groups based on the combinations of the 2 alleles: homozygotes for the common pseudodeficiency allele [p.G576S_E689K] (PD/PD), compound heterozygotes of a pseudodeficiency allele and a pathogenic allele (PD/Pathogenic), and compound heterozygotes of a pseudodeficiency allele and a VOUS (PD/VOUS). The GAA activity of PD/PD (n = 9), PD/Pathogenic (n = 10), and PD/VOUS (n = 7) was 8.8–28.1, 3.0–16.6 and 3.4– 16.6 nmol · h−1 · mg−1, respectively. The mean value was 22.1%, 10.2%, and 11.6% of normal, respectively. Individuals who are homozygous of the pseudodeficiency allele clearly showed higher GAA activities than that seen in LOPD patients. However, compound heterozygotes of pseudodeficiency and a mutation (or VOUS) had some overlap with LOPD patients (Fig. 4) and most likely these patients will need careful clinical evaluation and further follow-up.
Fig. 4. Dot scatter plot of GAA activity in IOPD, LOPD, pseudodeficiency patients, and carriers.
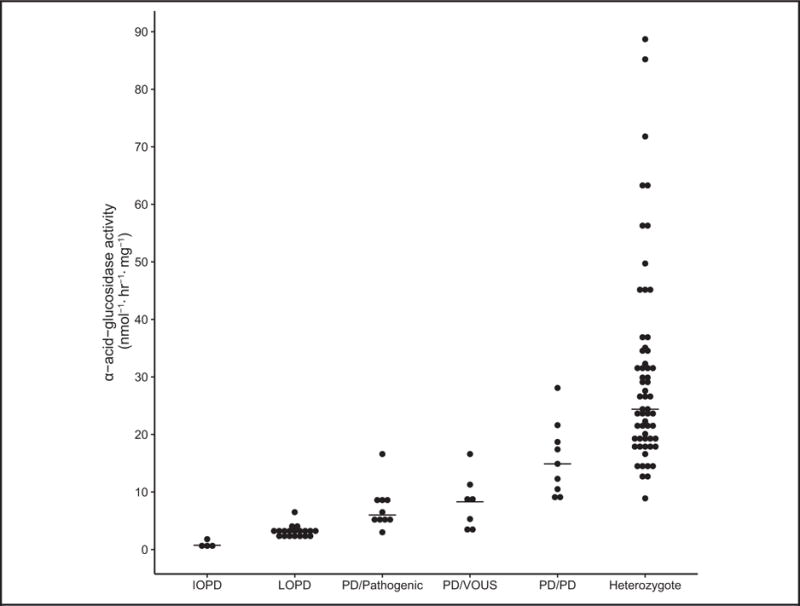
The pseudodeficiency patients are divided into 3 groups: homozygotes of the common pseudodeficiency allele [p.G576S_E689K] (PD/PD), compound heterozygotes of a pseudodeficiency allele and a pathogenic allele (PD/Pathogenic), and compound heterozygotes of a pseudodeficiency allele and a VOUS (PD/VOUS). In comparison, the normal GAA activity was 71.3 (38.2), ranging from 23.1–232.0 nmol · h−1 · mg−1.
We also tested fibroblast lysates, which used to be the gold standard specimen for establishing the enzymatic diagnosis of Pompe disease. The GAA activities of 3 IOPD patients (GM00338, GM00244, and GM20122) were 0.15, 0.08, and 0.08 nmol · h−1 · mg−1. The GAA activities of 2 patients with LOPD (GM11661 and GM00443) were 6.4 and 6.7 nmol · h−1 · mg−1. Normal fibroblast GAA was 92.8 (89.0) nmol · h−1 · mg−1 (n = 10). The IOPD GAA residual activity was lower in fibroblasts than in leukocytes, possibly due to the presence of residual interfering enzymes in the leukocytes that was not completely inhibited by acarbose, but other factors cannot be ruled out. Absolute GAA activity values should not be compared across different cell types (i.e., leukocytes, fibroblasts, B-lymphocyte cell line lymphoblasts), but relative comparison of GAA activities from different patients in the same cell type is meaningful. Clear trends of separations of GAA activities in patients with IOPD vs LOPD were demonstrated in all these cell types in our study.
Discussion
Here we report an LC-MS/MS method for measuring lysosomal enzyme activity in leukocytes. A key point is that the LC-MS/MS assay provides a much larger analytical range compared to the well-known fluorometric assay based on 4-methylumbelliferyl-α-glucose. The use of MS/MS vs fluorometry provides an advantage for newborn screening using dried blood spots in that a lower number of screen positives are found with equivalent cutoff values using the MS/MS platform (13). We hypothesize that this is due to the higher analytical range of the MS/MS assay. However, for accurate enzyme assays, the use of dried blood spots has a disadvantage that the measured activity cannot be normalized to the number of leukocytes (the major source of lysosomal enzymes in blood). By using purified leukocytes from blood, we can normalize the measured GAA activity to the total protein content (this is more convenient than normalizing to the number of leukocytes). This, together with the high analytical range, set the stage to explore the use of LC-MS/MS with leukocytes from patients with different Pompe disease severity.
A key finding (Fig. 2) is the demonstration that the LC-MS/MS assay can readily differentiate small changes in GAA activity at the low end because of low background noise and high dynamic range with real enzymatic response signals. This is not achievable with fluorometric assay (Fig. 3) since the background signal with fluorometry substrate was very high due to the intrinsic fluorescence of the substrate itself. Therefore the correlation of residual activity measured by fluorescence to the disease severity and age of onset could only be demonstrated in cultured fibroblast samples in earlier work. With our LC-MS/MS assay this correlation can be replicated in leukocytes extracted from routine blood samples. This improvement in the accuracy of low residual activity measurement is possibly due to the large analytical range of this method.
Close to 500 GAA mutations have been documented in the human gene mutation database and there is a lack of phenotypic data for many of the mutation combinations. Therefore, prediction of Pompe disease status based on genotype is often not possible. It seems reasonable to propose that screen-positive individuals either with 2 severe GAA mutations, or individuals who have LC-MS/MS–determined leukocyte GAA activity in the range typical of patients with IOPD, should be referred for immediate clinical examination in case it is necessary to start therapy immediately. However, following up patients with LOPD and screen-positive patients with VOUS or pseudodeficiency alleles is more challenging. Pseudodeficiency alleles are particularly high in Asian populations; 3.3%–3.9% of Taiwan Chinese and Japanese individuals are homozygous for the common pseudodeficiency allele alone (p.G576S; p.E689K) (20–22). Even our LC-MS/MS GAA assay does not show complete separation between the LOPD and some patients with pseudodeficiency (Fig. 4). Thus, the LC-MS/MS GAA is a useful tool to help determine the clinical phenotype but cannot be used alone to stratify screen-positive patients for planning clinical follow-up. The enzyme findings should always be interpreted together with biomarker findings, GAA mutation status and clinical findings.
Serum creatine kinase activity is increased in the majority of cases with Pompe disease (IOPD and LOPD), and the greatest increase is usually seen in patients with IOPD. However, the creatine kinase activity can be normal in patients with LOPD before disease manifestations (4). Recently, urinary glucose tetrasaccharide (Glc4), a breakdown product from glycogen, has been used to monitor disease burden and clinical response to treatment (23–26). All of these genetic and biochemical findings should be combined with the patient’s clinical findings in guiding patient care.
Fifteen of our patients in the LOPD cohort were infants identified from newborn screening and predicted to be LOPD based on nominally low residual GAA activity (approximately 5% of normal) and on 2 GAA mutations. These patients were asymptomatic at the time of blood collection. Among our cohort of patients with LOPD, only 1 adult patient (LOPD-7) was symptomatic, and the GAA activity was well within the range of our LOPD cohort. Additional study with symptomatic patients with LOPD is warranted to determine if they fall into the range reported in this study.
In conclusion, we report a LC-MS/MS assay of GAA enzymatic activity in leukocytes isolated from venous blood that displays a superior analytical range and analytical sensitivity compared to the well-known fluorometric GAA assay. The new assay differentiates patients with IOPD and LOPD in our cohort, and it also partially separates patients with LOPD from patients with pseudodeficiency.
Supplementary Material
Footnotes
Nonstandard abbreviations: PD, Pompe disease; IOPD, infantile-onset Pompe disease; LOPD, late-onset Pompe disease; GAA, lysosomal acid α-glucosidase; MS/MS, tandem mass spectrometry; VOUS, variants of unknown significance; S, substrate; IS, internal standard; P, product; RBS, red blood cell; S/N, signal-to-noise ratio; EOPD, early-onset Pompe disease; PD/PD, pseudodeficiency/pseudodeficiency; PD/Pathogenic, pseudodeficiency/pathogen mutation; PD/VOUS, pseudodeficiency/VOUS.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: M. Caggana, New York State Department of Health; L. DiAntonio, New York State Department of Health.
Consultant or Advisory Role: M.H. Gelb, PerkinElmer Corp.
Stock Ownership: None declared.
Honoraria: None declared.
Research Funding: J.J. Orsini, NIH/NICHD contract to perform screening for Pompe disease (to the institution); M. Caggana, Eunice Kennedy Shriver National Institute of Child Health and Human Development (HHSN275201400016C) and NICHD contract to conduct Pompe disease newborn screening (to the institution); E.E. Hughes, NYS Department of Health (to the institution); C. Stevens, Health Research Inc. (to the institution); L. DiAntonio, NIH/NICHD; M.P. Wasserstein, NICHD funded research to conduct pilot newborn screen for several conditions, including Pompe disease; M.H. Gelb, NIH grant.
Expert Testimony: M.P. Wasserstein, served as expert witness in patent litigation case related to therapeutics for Pompe disease.
Patents: None declared.
Role of Sponsor: The funding organizations played a direct role in the design of study, choice of enrolled patients, review and interpretation of data, preparation of manuscript, and final approval of manuscript.
References
- 1.Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J. Methods of diagnosis of patients with Pompe disease: data from the Pompe Registry. Mol Genet Metab. 2014;113:84–91. doi: 10.1016/j.ymgme.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: advances in the detection, under-standing of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genet. 2012;160:1–7. doi: 10.1002/ajmg.c.31324. [DOI] [PubMed] [Google Scholar]
- 3.Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–6. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–88. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chien YH, Hwu WL, Lee NC. Pompe disease: early diagnosis and early treatment make a difference. Pediatr Neonatol. 2013;54:219–27. doi: 10.1016/j.pedneo.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e1116–25. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- 7.Chien YH, Lee NC, Chen CA, Tsai FJ, Tsai WH, Shieh JY, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. 2015;166:985–91. e1–2. doi: 10.1016/j.jpeds.2014.10.068. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F, Gelb MH. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50:1785–96. doi: 10.1373/clinchem.2004.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiang SC, Hwu WL, Lee NC, Hsu LW, Chien YH. Algorithm for Pompe disease newborn screening: results from the Taiwan screening program. Mol Genet Metab. 2012;106:281–6. doi: 10.1016/j.ymgme.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Sista RS, Wang T, Wu N, Graham C, Eckhardt A, Winger T, et al. Multiplex newborn screening for Pompe, Fabry, Hunter, Gaucher, and Hurler diseases using a digital microfluidic platform. Clin Chim Acta. 2013;424:12–8. doi: 10.1016/j.cca.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dajnoki A, Muhl A, Fekete G, Keutzer J, Orsini J, Dejesus V, et al. Newborn screening for Pompe disease by measuring acid alpha-glucosidase activity using tandem mass spectrometry. Clin Chem. 2008;54:1624–9. doi: 10.1373/clinchem.2008.107722. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Kallwass H, Young SP, Carr C, Dai J, Kishnani PS, et al. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet Med. 2006;8:302–6. doi: 10.1097/01.gim.0000217781.66786.9b. [DOI] [PubMed] [Google Scholar]
- 13.Kumar AB, Masi S, Ghomashchi F, Chennamaneni NK, Ito M, Scott CR, et al. Tandem mass spectrometry has a larger analytical range than fluorescence assays of lysosomal enzymes: application to newborn screening and diagnosis of mucopolysaccharidoses types II, IVA, and VI. Clin Chem. 2015;61:1363–71. doi: 10.1373/clinchem.2015.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okumiya T, Keulemans JL, Kroos MA, Van der Beek NM, Boer MA, Takeuchi H, et al. A new diagnostic assay for glycogen storage disease type II in mixed leukocytes. Mol Genet Metab. 2006;88:22–8. doi: 10.1016/j.ymgme.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Yang CF, Liu HC, Hsu TR, Tsai FC, Chiang SF, Chiang CC, et al. A large-scale nationwide newborn screening pro-gram for Pompe disease in Taiwan: towards effective diagnosis and treatment. Am J Med Genet A. 2014;164A:54–61. doi: 10.1002/ajmg.a.36197. [DOI] [PubMed] [Google Scholar]
- 16.Umapathysivam K, Hopwood JJ, Meikle PJ. Correlation of acid alpha-glucosidase and glycogen content in skin fibroblasts with age of onset in Pompe disease. Clin Chim Acta. 2005;361:191–8. doi: 10.1016/j.cccn.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, et al. Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve. 2009;40:32–6. doi: 10.1002/mus.21376. [DOI] [PubMed] [Google Scholar]
- 18.Musumeci O, Thieme A, Claeys KG, Wenninger S, Kley RA, Kuhn M, et al. Homozygosity for the common GAA gene splice site mutation c.-32-13T>G in Pompe disease is associated with the classical adult phenotypical spectrum. Neuromuscul Disord. 2015;25:719–24. doi: 10.1016/j.nmd.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Montagnese F, Barca E, Musumeci O, Mondello S, Migliorato A, Ciranni A, et al. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): unusual features and response to treatment. J Neurol. 2015;262:968–78. doi: 10.1007/s00415-015-7664-0. [DOI] [PubMed] [Google Scholar]
- 20.Shigeto S, Katafuchi T, Okada Y, Nakamura K, Endo F, Okuyama T, et al. Improved assay for differential diagnosis between Pompe disease and acid alpha-glucosidase pseudodeficiency on dried blood spots. Mol Genet Metab. 2011;103:12–7. doi: 10.1016/j.ymgme.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Labrousse P, Chien YH, Pomponio RJ, Keutzer J, Lee NC, Akmaev VR, et al. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol Genet Metab. 2010;99:379–83. doi: 10.1016/j.ymgme.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 22.Kroos MA, Mullaart RA, Van Vliet L, Pomponio RJ, Amartino H, Kolodny EH, et al. p.[G576S; E689K]: pathogenic combination or polymorphism in Pompe disease? Eur J Hum Genet. 2008;16:875–9. doi: 10.1038/ejhg.2008.34. [DOI] [PubMed] [Google Scholar]
- 23.An Y, Young SP, Hillman SL, Van Hove JL, Chen YT, Millington DS. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem. 2000;287:136–43. doi: 10.1006/abio.2000.4838. [DOI] [PubMed] [Google Scholar]
- 24.An Y, Young SP, Kishnani PS, Millington DS, Amalfitano A, Corz D, Chen YT. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab. 2005;85:247–54. doi: 10.1016/j.ymgme.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Young SP, Piraud M, Goldstein JL, Zhang H, Rehder C, Laforet P, et al. Assessing disease severity in Pompe disease: the roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am J Med Genet C Semin Med Genet. 2012;160C:50–8. doi: 10.1002/ajmg.c.31320. [DOI] [PubMed] [Google Scholar]
- 26.Chien YH, Goldstein JL, Hwu WL, Smith PB, Lee NC, Chiang SC, et al. Baseline urinary glucose tetrasaccharide concentrations in patients with infantile- and late-onset Pompe disease identified by newborn screening. JIMD Rep. 2015;19:67–73. doi: 10.1007/8904_2014_366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.