

Abstract
Transforming growth factor-β (TGF-β) plays an important role in the development and maintenance of embryonic dopaminergic (DA) neurons in the midbrain. To study the function of TGF-β signaling in the adult nigrostriatal system, we generated transgenic mice with reduced TGF-β signaling in mature neurons. These mice display age-related motor deficits and degeneration of the nigrostriatal system. Increasing TGF-β signaling in the substantia nigra through adeno-associated virus expressing a constitutively active type I receptor significantly reduces 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration and motor deficits. These results suggest that TGF-β signaling is critical for adult DA neuron survival and that modulating this signaling pathway has therapeutic potential in Parkinson disease.
SIGNIFICANCE STATEMENT We show that reducing Transforming growth factor-β (TGF-β) signaling promotes Parkinson disease-related pathologies and motor deficits, and increasing TGF-β signaling reduces neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a parkinsonism-inducing agent. Our results provide a rationale to pursue a means of increasing TGF-β signaling as a potential therapy for Parkinson's disease.
Keywords: AAV, MPTP, Parkinson's disease, TGF-β
Introduction
The progressive loss of dopaminergic (DA) neurons in the substantia nigra (SN) is a hallmark of Parkinson's disease (PD; Dauer and Przedborski, 2003). Transforming growth factor β (TGF-β) has multiple associations with the nigrostriatal system and with PD (Roussa et al., 2009; Hegarty et al., 2014). TGF-β is upregulated in PD brains (Mogi et al., 1995), and genetic association studies suggest that variation in the TGFB2 gene may influence susceptibility to idiopathic PD (Goris et al., 2007). TGF-β is essential for the development and survival of embryonic DA neurons (Roussa et al., 2009; Hegarty et al., 2014). TGF-β2 or TGF-β3 knock-out mouse embryos display significantly reduced numbers of DA neurons (Roussa et al., 2006; Zhang et al., 2007). TGF-β promotes the survival of embryonic DA neurons in culture and protects them against toxicity from the parkinsonism-inducing toxin N-methylpyridinium ion (MPP+; Krieglstein and Unsicker, 1994; Poulsen et al., 1994; Krieglstein et al., 1995). Due to the poor postnatal health and subsequent premature death of TGF-β2 or TGF-β3 knock-out mice, it remained unclear whether TGF-β plays a role in adult DA neurons. TGF-β2 haplodeficiency (TGF-β2+/−) mice are viable and have fewer DA neurons in the SN at 6 weeks of age and display significantly reduced dopamine concentration in the striatum in adulthood (6 months of age; Andrews et al., 2006). The progressive loss of DA neurons and the aggregation of α-synuclein are observed in Smad3 (a key component of the TGF-β signaling pathway) null mice at 2–3 months of age and in Smad3 haplodeficiency mice at 19–20 months of age (Tapia-González et al., 2011). These results suggest that deficiency in TGF-β signaling may increase the risk of developing PD and that modulating this pathway may have therapeutic effects. However, several studies failed to show protective effects of TGF-β in animal models of PD. Continuous administration of TGF-β3 over substantia nigra failed to prevent 6-hydroxydopamine-induced neuronal cell death in rat (Sauer et al., 1995). In mice, overexpression of TGF-β1 in the striatum by adenovirus exacerbates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced loss of DA neurons and enhances dopamine depletion in the striatum (Sánchez-Capelo et al., 2003). These findings are in striking contrast to the in vitro observation that TGF-β protects embryonic DA neurons against MPP+-induced degeneration (Krieglstein and Unsicker, 1994; Poulsen et al., 1994; Krieglstein et al., 1995). The cause of these contradictory results is not known and could be due to the fact that in vivo delivery of the TGF-β ligands may initiate TGF-β signaling in many cell types in vivo, since essentially all cell types in the brain express TGF-β receptors. Given the highly cell type-specific and context-dependent nature of TGF-β signaling, it is important to study the role of TGF-β signaling in disease progression in a cell type-specific manner to determine whether the TGF-β signaling pathway may serve as a potential therapeutic target for promoting the survival of DA neurons. In this study, we used transgenic mice and viral-mediated gene transfer to drive the expression of mutant TGF-β receptors to achieve cell type-specific manipulation of TGF-β signaling in neurons. We show that reducing TGF-β signaling in mature neurons in transgenic mice causes age-related motor deficits and degeneration of the nigrostriatal system. Importantly, increasing TGF-β signaling in the substantia nigra reduces MPTP-induced dopaminergic neurodegeneration and motor deficits.
Materials and Methods
Mice
Transgenic mice expressing tTA (tetracycline-controlled transactivator) under control of the CaMKII promoter (CaMKII-tTA mice; Fan et al., 2001) and transgenic mice expressing a truncated, kinase-defective TGF-β type II receptor (TBRIIΔk) under the control of tetO regulatory sequences (tetO-TBRIIΔk mice; Tesseur et al., 2006) were crossed to generate CaMKII-tTA+/TBRIIΔk+ mice. Littermate CaMKII-tTA+/TBRIIΔk− mice were used as controls. TBRIIΔk functions as a potent inhibitor of TGF-β signaling in neurons and is tagged with a flag epitope (Tesseur et al., 2006). Male, wild-type C57BL/6 mice were purchased from The Jackson Laboratory. All mice were kept under a 12 h light/dark cycle with ad libitum access to food and water. All animal care and procedures complied with the Animal Welfare Act and were performed in accordance with institutional guidelines and approved by the VA Palo Alto institutional animal care and use committee.
Behavior analysis
Rotarod test.
Mice were tested on a rotarod apparatus to assess coordination and balance, according to published procedures (Luo et al., 2014). The mice were placed on the rotating rotarod for 1 min at 3 rpm, after which the speed was accelerated to 40 rpm over 5 min, and the latency (in seconds) to fall was recorded. Each mouse underwent three test sessions, and the best time was chosen for analysis.
Pole test.
For the pole test, the mouse was placed head upward on the top of a vertical wooden pole (diameter, 1 cm; height, 50 cm) with a rough surface (Ogawa et al., 1985). Each mouse was trained to habituate to the pole for two trials and then allowed to descend five times on a single session. The time in which the mouse reached the floor with all four paws was recorded.
Morris water maze.
Spatial learning and memory was assessed by the Morris water maze (MWM; Chin et al., 2005; Roberson et al., 2007). The water maze pool (diameter, 152 cm) contained opaque water (22–23°C) with a platform (15 × 15 cm) submerged 1.5 cm below the surface. Mice were trained to locate a cued (days 1–3) or hidden (days 4–8) platform. A black and white-striped mast (15 cm height) was mounted on the platform for cued training sessions and was removed for hidden platform sessions. Each day the mice were tested for two sessions (3.5 h apart), and each session consisted of two (cued training) or three (hidden platform) 60 s trials with a 15 min intertrial interval. The platform location was changed for each cued training session but remained constant in the hidden platform sessions. A 60 s probe trial (with platform removed) was performed at 24 h after completion of the hidden platform sessions. The entry point for the probe trials was in the quadrant opposite to the target quadrant. Performance was analyzed using the EthoVision Automated Tracking System (Noldus Information Technology).
Gait analysis.
Forelimb and hindlimb paws were painted with red and blue nontoxic paint (Brooks and Dunnett, 2009). Mice were placed on a brightly lit platform (15 × 15 cm) from which they could escape into a dim hut along a 100-cm-long and 7-cm-wide runway lined with white paper. Each mouse was tested in three trials per day with an intertrial interval of 5 min over 3 consecutive days. Six steps from the middle portion of each run were analyzed for stride length, hind-base width (the distance between the right and left hindlimb strides), front-base width (the distance between the right and left frontlimb strides), and paw angle (paws central axis relative to its walking direction). Mean values were used for statistical analysis.
Tissue processing
Mice were perfused transcardially with saline, and one hemi-brain was fixed for 24 h in 4% paraformaldehyde and equilibrated in 30% sucrose for histological analysis, as previously described (Luo et al., 2007). The other hemi-brain was snap frozen and stored at −80°C for biochemical analysis. For stereological analysis of DA neurons in the substantia nigra, the whole brain was fixed and sectioned.
Immunohistochemistry
Immunohistochemistry was performed on free-floating sections following standard procedures (Luo et al., 2006, 2007). The following primary antibodies were used: flag (1:1000; catalog #F1804, Sigma-Aldrich; RRID: AB_262044); CD68 (1:50; catalog #MCA1957, Bio-Rad; RRID: AB_322219; Luo et al., 2013); c-Fos (1:1000; catalog #PC38, Millipore; RRID: AB_2106755); calbindin (1:10,000; catalog #CB 38, Swant; RRID: AB_10000340); NeuN (1:1000; catalog #MAB377, Millipore; RRID: AB_2298772); and p-Smad2 (1:1000; catalog #AB3849, Millipore; RRID: AB_11213091). After overnight incubation, primary antibody staining was revealed using biotinylated secondary antibodies and the ABC Kit (Vector Laboratories) with diaminobenzidine (DAB; Sigma-Aldrich). Photographs were acquired using a BX51 Microscope (Olympus) and a SPOT Flex shifting pixel CCD camera with SPOT Advanced software (SPOT Imaging Solutions). The number of Purkinje cells was evaluated on calbindin-immunostained sections, according to a previously established method (Buffo et al., 1997). Briefly, the number of Purkinje cells in five serial sections were counted for each mouse. The length of the Purkinje cell layer was measured using MetaMorph Microscopy Automation and Image Analysis Software (Molecular Devices; RRID: SCR_002368), and the density was determined by dividing the number of Purkinje cells by this length. The number of NeuN+ neurons in the spinal cord was estimated according to the study by Ross et al. (2010), using the cell count function of MetaMorph and validated by manual counts.
Cresyl Violet staining
Brain sections were mounted on Superfrost Plus slides (Fisher Scientific), air dried, rehydrated, and stained with 0.02% Cresyl Violet (Sigma-Sigma) in acetate buffer, pH 3.2, then were dehydrated through a series of alcohols, cleared in xylene, and coverslipped. The number of neurons in the CA1 pyramidal cell layers was quantified with MetaMorph Imaging software (Luo et al., 2006).
Real-time quantitative PCR
The striata were dissected from saline-perfused mice, flash frozen on dry ice, and stored at −80°C. RNA was extracted using an RNeasy Mini Kit (Qiagen) according to the manufacturer instructions. A total of 1000 ng of RNA was treated with DNase I (Invitrogen) and then converted to cDNA using the SuperScript III First Strand Synthesis System (Invitrogen). cDNA was diluted 1:10 in water. Real-time quantitative PCR (qPCR) was performed using SYBR Green I Master Mix (Roche) on a LightCycler 480 (Roche). The following primers were used: for inducible nitric oxide synthase (iNOS) 2: forward primer, GTTCTCAGCCCAACAATACAAGA; reverse primer, GTGGACGGGTCGATGTCAC; for tumor necrosis factor-α (TNF-α): forward primer, TGGAACTGGCAGAAGAG; reverse primer, CCATAGAACTGATGAGAGG; for monocyte chemoattractant protein 1 (MCP-1): forward primer, TTA AAA ACC TGG ATC GGA ACC AA; reverse primer, GCA TTA GCT TCA GAT TTA CGG GT; and for actin: forward primer, GGC TGT ATT CCC CTC CAT CG; reverse primer, CCA GTT GGT AAC AAT GCC ATG T.
Melting curves were used to confirm the purity of the amplified product. Cycle threshold (Ct) values were normalized to actin. ΔΔCt values were used to yield the fold change over control in each experiment (Villeda et al., 2014; Kirby et al., 2015).
Western blot analysis
Brain regions were dissected after perfusion of the animal, snap frozen, and lysed in RIPA lysis buffer (500 mm Tris, pH 7.4, 150 mm NaCl, 0.5% Na deoxycholate, 1% NP40, 0.1% SDS, and complete protease inhibitors; Roche; Villeda et al., 2014; Kirby et al., 2015). The total protein concentration was quantified using a bicinchoninic acid kit (Pierce). Tissue lysates were mixed with 4× NuPage LDS loading buffer (Invitrogen), loaded on a 4–12% SDS polyacrylamide gradient gel (Invitrogen) and subsequently transferred onto a nitrocellulose membrane. The blots were blocked in 5% milk in Tris-buffered saline with Tween and incubated with anti-actin (1:5000; catalog #A5060, Sigma-Aldrich; RRID:AB_476738) and anti-flag (1:1000; catalog #F1804, Sigma-Aldrich; RRID: AB_262044). Flag signals were detected by horseradish peroxidase-conjugated secondary antibodies and an ECL kit (GE Healthcare/Pharmacia Biotech). For c-Fos analysis, the striata were dissected from the brain sections under a microscope, and proteins were extracted using the Qproteome FFPE Tissue Kit (Qiagen). The proteins were visualized and quantified on a LI-COR Odyssey IR Imaging System (Odyssey CLx; RRID:SCR_014579).
Adeno-associated virus preparation
Adeno-associated virus (AAV) expressing ALK5CA-T2A-eGFP under a cytomegalovirus (CMV) promoter (AAV-ALK5CA) was generated at the Stanford Neuroscience Gene Vector and Virus Core. ALK5CA, a constitutively active mutant of ALK5 containing a T204D substitution (Wieser et al., 1995), was isolated from pCMV5-rALK5CA-HA (obtained from Joan Massague, Memorial Sloan Kettering Cancer Center, New York, NY). AAV expressing T2A-eGFP (AAV-GFP) was generated as a control. AAVs were generated with AAV-DJ capsids (Grimm et al., 2008) for high-efficiency in vivo neuronal infection (Xu et al., 2012; Villeda et al., 2014).
Stereotaxic injection of AAV
Male, wild-type C57BL/6 mice (8 weeks old; The Jackson Laboratory) were used. Stereotaxic injections were performed under isoflurane anesthesia with a 5 μl Hamilton syringe. After each mouse was placed in a stereotaxic frame, 1 μl of the vector suspended in PBS (5 × 108 infectious U/ml) was injected into the substantia nigra over 5 min. The following coordinates were used for substantia nigra (2.9 mm posterior to the bregma, 1.3 mm lateral to the midsagittal suture, and 4.2 mm ventral to the skull; Bensadoun et al., 2000).
MPTP injury
Two weeks after AAV stereotaxic injection, MPTP (M103, Sigma-Aldrich) was injected intraperitoneally based on a published protocol (8 mg/kg in saline, 4 times/d, 2 h apart, for 2 d; Martens et al., 2012). Seven days after the last MPTP injection, mice were perfused and brains were collected for immunohistochemistry and stereological estimation of dopaminergic neurons.
Stereological estimation of dopaminergic neurons in the substantia nigra
The number of dopaminergic neurons in the substantial nigra was estimated stereologically (Zhang et al., 2007; Martens et al., 2012). Briefly, whole-brain sections containing substantia nigra were stained with an antibody against tyrosine hydroxylase (TH; 1:200; catalog #AB152, Millipore; RRID: AB_390204) with the ABC kit and DAB. Stereological counting of bregma −2.8 to −4.04 mm (six serial sections) was used to quantify the number of TH+ neurons in the substantia nigra, according to previously described procedures (Zhang et al., 2007; Martens et al., 2012).
Data and statistical analysis
Data are presented as the mean ± SEM. Data were analyzed using a two-tailed Student's t test for comparing two groups or ANOVA for comparing multiple groups. A Bonferroni's or Turkey's post hoc test was used to compare pairs of groups following ANOVA. Statistical analysis was performed with Prism software (GraphPad; RRID:SCR_002798). A p value of <0.05 was considered to be statistically significant.
Results
Mice with reduced TGF-β signaling in mature neurons display age-related memory and motor deficits
We have previously shown that reducing neuronal TGF-β signaling in mice resulted in age-dependent neurodegeneration and promoted amyloid-β accumulation and dendritic loss in a mouse model of Alzheimer's disease (Tesseur et al., 2006). To further study the function of TGF-β signaling specifically in neurons, we generated double transgenic CaMKII-tTA+/TBRIIΔk+ mice by crossing CaMKII-tTA+ mice (Mayford et al., 1996), with tet-O-TBRIIΔk+ mice expressing kinase-deficient TGF-β type II receptor (TBRIIΔk; Tesseur et al., 2006). TBRIIΔk functions as a potent inhibitor of TGF-β signaling and has been used in other transgenic mice to specifically inhibit signaling in pancreatic or skin cells (Böttinger et al., 1997; Wang et al., 1997). Immunostaining with a flag antibody showed expression of the flag epitope (presented in TBRIIΔk) in the hippocampus, cortex, and striatum (Fig. 1A), which is consistent with previous reports about the CaMKII-tTA+ mice (Mayford et al., 1996; Kholodilov et al., 2004). Weak, scattered flag immunoreactivity was also observed in the brainstem and cerebellum (<10% of cells). No flag immunoreactivity was observed in the substantia nigra. Among the forebrain regions, the striatum showed the most intense flag immunoreactivity (Fig. 1B). In addition, Western blot analysis of the flag epitope confirmed the expression of flag and TBRIIΔk in the above brain regions (Fig. 1C).
Figure 1.
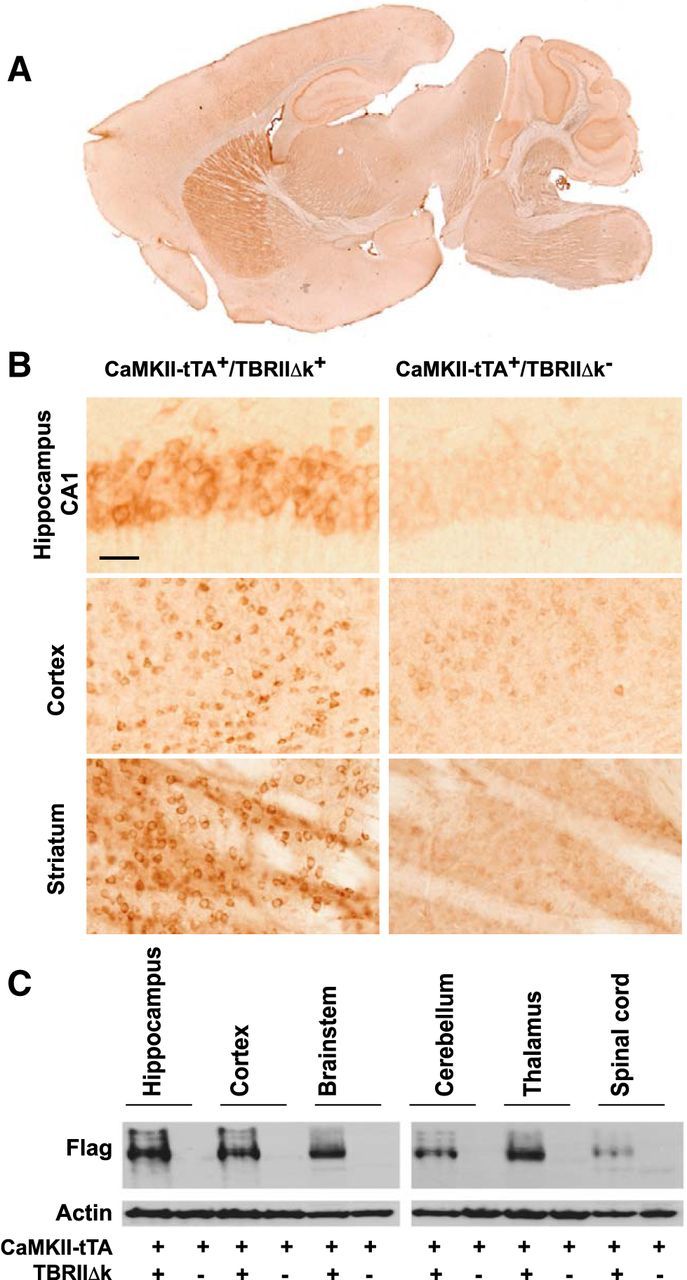
Expression of the transgene in CaMKII-tTA+/TBRIIΔk+ mice. CaMKII-tTA+/TBRIIΔk+ mice were generated by crossing CaMkII-tTA+ mice with tet-O-TBRIIΔk+ mice expressing a kinase-deficient TGF-β type II receptor. CaMKII-tTA+/TβRIIΔk+ and CaMKII-tTA+/TBRIIΔk− (as control) mice were killed at 2 months of age and their brains were sectioned sagittally. The transgene expression was visualized by immunostaining with an antibody against a flag epitope (present in TBRIIΔk). A, A representative image of flag immunoreactivity in different brain regions. Note the strong flag immunoreactivity in the striatum. B, Higher-magnification images showing flag immunoreactivity in the hippocampus, cortex, and striatum in the CaMKII-tTA+/TBRIIΔk+ mice (left panels), compared with CaMKII-tTA+/TBRIIΔk− mice (right panels). Scale bar, 50 μm. C, Tissue lysates from different brain regions were subjected to Western blot analysis using anti-flag and β-actin antibodies.
The CaMKII-tTA+/TBRIIΔk+ mice (the TBRIIΔk was left on throughout the experiment) displayed moderate learning and memory deficits, as assessed by the MWM, and severe motor deficits, as assessed by gait analysis (Fig. 2), at 18 months of age. In the Morris water maze test, the CaMKII-tTA+/TBRIIΔk+ mice showed significantly longer escape latencies in the hidden platform tests (Fig. 2A) and spent less time in target quadrant in the probe trial (Fig. 2B,C). Footprint analysis showed both hindlimb and forelimb stride length was significantly reduced compared with CaMKII-tTA+/TBRIIΔk− control mice (Fig. 2D–F). The distance between the forelimbs during stance was also significantly reduced in the CaMKII-tTA+/TBRIIΔk+ mice, whereas there was a tendency toward smaller hindlimb stance, but this was not significantly different (data not shown). In addition, ∼38% of the CaMKII-tTA+/TBRIIΔk+ mice (n = 7) displayed a series of severe symptoms, including tremor of the head in the resting position, impairments in righting reflex, a hunched back with kyphosis, a prolapsed rectum, and loss of hair and excoriated skin. They were not able to complete the rotarod and pole tests and showed more profound gait impairments in footprint analysis.
Figure 2.

Mice with reduced TGF-β signaling in neurons display learning, memory, and motor deficits. A–C, CaMKII-tTA+/TBRIIΔk+ and CaMKII-tTA+/TBRIIΔk− (as control) mice were tested for spatial learning and memory function using MWM. A, The average escape latency of each session in the visible platform (1–6 sessions) and hidden platform (7–16 sessions) parts of the MWM test. The mice underwent a probe test 24 h later. B, C, Travel routes (B) and the time spent in the target and opposite quadrants (C) were analyzed using the EthoVision Automated Tracking System. D–F, Footprint analysis was performed at 18 months of age. Representative paw prints of CaMKII-tTA+/TBRIIΔk+ mice and CaMKII-tTA+/TBRIIΔk− mice (as a control) are shown in D. E, F, Quantification of hindlimb (blue prints, E) and forelimb (red prints, F) stride length. C, E, F, Bars represent the mean ± SEM and were analyzed by ANOVA (C) or unpaired t test (E, F). *p < 0.05; **p < 0.01. n = 10–12 mice/group.
CaMKII-tTA+/TBRIIΔk+ mice display degeneration of the nigrostriatal system
To determine whether the motor deficits in the CaMKII-tTA+/TBRIIΔk+ mice are related to the disruption of the nigrostriatal system, brain sections were immunostained for TH to label DA neurons and subjected to stereological quantification, according to published methods (Zhang et al., 2007; Martens et al., 2012). The CaMKII-tTA+/TBRIIΔk+ mice showed a 20% decrease in the number of TH+ DA neurons in the SN pars compacta (SNpc) and a 40% reduction in TH+ fiber density in the striatum, compared with CaMKII-tTA+/TBRIIΔk− mice (Fig. 3A,B). To determine whether there is cell loss in other brain regions, we performed Cresyl Violet staining and immunostaining with NeuN and calbindin. Compared with the CaMKII-tTA+/TBRIIΔk− mice, the CaMKII-tTA+/TBRIIΔk+ mice did not show obvious abnormal brain morphology or cell loss in the hippocampus (Fig. 3D), cerebellum, or spinal cord. The mean number of calbindin+ Purkinje cells was 52.92 ± 13.68 and 50.95 ± 3.89/mm in the CaMKII-tTA+/TBRIIΔk− and CaMKII-tTA+/TBRIIΔk+ mice, respectively (p = 0.892, by two-tailed t test). The mean number of NeuN+ neuron cells in the spinal cord was 210.8 ± 19.29 and 167.9 ± 17.72/ventral horn in the CaMKII-tTA+/TBRIIΔk− and CaMKII-tTA+/TBRIIΔk+ mice, respectively (p = 0.129, by two-tailed t test). However, the calbindin immunoreactivity was significantly reduced in the striatum (Fig. 3C) and hippocampus (Fig. 3E) of the CaMKII-tTA+/TBRIIΔk+ mice. These results suggested that reduced TGF-β signaling in the forebrain led to damage of DA nerve terminals in the striatum and loss of DA neurons in the SN and that the motor deficits observed in CaMKII-tTA+/TBRIIΔk+ mice may be related to dysfunction of the nigrostriatal system.
Figure 3.
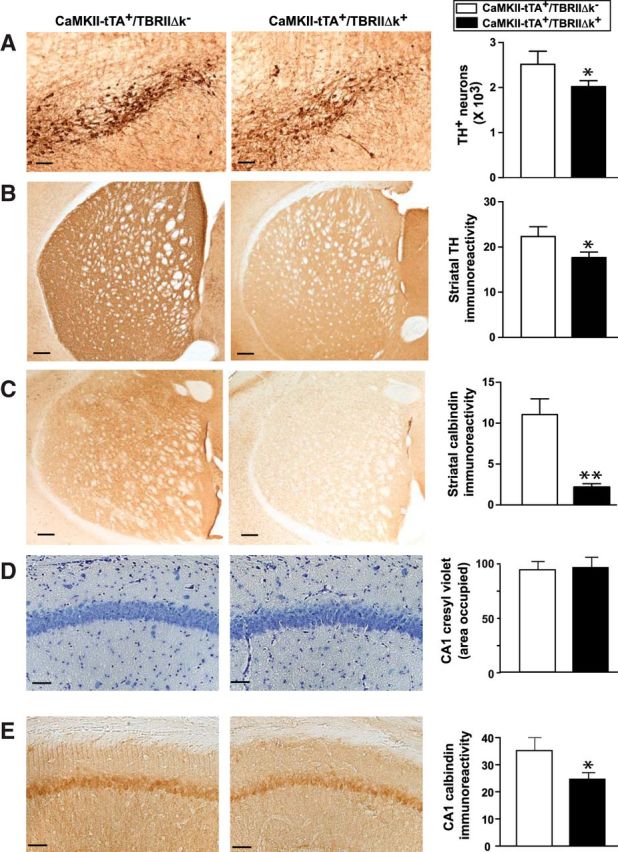
Mice with reduced TGF-β signaling in neurons display degeneration of the nigrostriatal system. CaMKII-tTA+/TBRIIΔk+ mice were generated by crossing CaMkII-tTA mice with tet-O-TBRIIΔk mice expressing a kinase-deficient TGF-β type II receptor. CaMKII-tTA+/TBRIIΔk− mice were used as controls. A, B, Brain sections from these mice were immunostained with an antibody against TH and were subjected to stereological analysis of TH+ DA neurons in substantia nigra pars compacta (A) and TH+ nerve terminals in the striatum (B). C, E, Brain sections were immunostained with an antibody against calbindin, and calbindin immunoreactivity was measured in the striatum (C) and hippocampal CA1 (E). D, Brain sections were stained with 0.02% Cresyl Violet. Scale bar, 50 μm. Bars represent the mean ± SEM and were analyzed by unpaired t test. *p < 0.05; **p < 0.01. n = 10–12 mice/group.
Expression of the immediate early gene c-fos increases in response to depolarization and is therefore an in vivo marker for neuronal activation (Sagar et al., 1988). c-fos and Fos protein have been widely used to investigate basal ganglia responses to changes in dopaminergic neurotransmission. Decreased DA input to the striatum induces increased Fos expression in the striatal neurons (Cole and Di Figlia, 1994). The activity of c-fos has also been used to assess the response of striatal neurons to levodopa following the destruction of substantia nigra (Robertson et al., 1989). To determine whether reduced TGF-β signaling affected the expression of c-Fos, we performed immunostaining with an antibody against c-Fos. The CaMKII-tTA+/TBRIIΔk+ mice showed significant increase of c-Fos expression in the striatum and hippocampus (Fig. 4A,B). In agreement with the immunohistochemistry data, Western blot analysis of striatal lysates revealed significantly increased expression of c-Fos in the CaMKII-tTA+/TBRIIΔk+ mice (Fig. 4C).
Figure 4.

Mice with reduced TGF-β signaling display increased expression of c-Fos. A, B, Brain sections from CaMKII-tTA+/TBRIIΔk+ or CaMKII-tTA+/TBRIIΔk− mice were immunostained with an antibody against c-Fos, and c-Fos+ cells were analyzed in the hippocampus (dentate gyrus; A) and striatum (B). C, Representative Western blot analysis of striatal lysates probed with anti-c-Fos and anti-actin antibodies. c-Fos expression was quantified and normalized to actin with a LI-COR Odyssey IR Imaging System. Scale bar, 50 μm. Bars represent the mean ± SEM and were analyzed by unpaired t test. *p < 0.05. n = 10–12 mice/group.
Increasing TGF-β signaling in the substantia nigra through AAV-Alk5CA
Since reducing TGF-β signaling in CaMKII-tTA+/TBRIIΔk+ mice led to the dysfunction of the nigrostriatal system, we hypothesized that increasing TGF-β signaling would rescue DA neurons and restore functional deficits. We made use of a constitutively active form of TGF-β type I receptor Alk5 (Alk5CA) and generated AAV that encodes GFP/ALK5CA (termed AAV-Alk5CA; Fig. 5A) or encodes only GFP (AAV-GFP) as a control. Alk5CA, a constitutively active mutant of ALK5 containing a T204D substitution, activates TGF-β signaling in a ligand-independent manner (Wieser et al., 1995) and has been used in transgenic mice to activate TGF-β signaling in a cell type-specific manner (Bartholin et al., 2008). AAVs were generated with AAV-DJ capsids (Grimm et al., 2008) for high-efficiency in vivo neuronal infection (Xu et al., 2012; Villeda et al., 2014). We injected these viruses into the substantia nigra of wild-type mice by stereotaxic surgery. Two weeks after injection, the mice were killed to analyze GFP expression (Fig. 5B,D) and TGF-β signaling (Fig. 5C). The AAV-Alk5CA side showed increased phospho-Smad 2 immunoreactivity, suggesting the activation of TGF-β signaling. The colocalization of GFP (green) and TH (red) immunoreactivity demonstrated the expression of the reporter GFP (and Alk5CA) in dopaminergic neurons (Fig. 5D). The majority of infected cells (GFP+) are neurons (81 ± 11%; n = 4 mice).
Figure 5.

Generating AAV-encoding Alk5CA to activate TGF-β signaling in dopaminergic neurons. A, Schematic structure of AAV-encoding Alk5CA and GFP (termed AAV-Alk5CA). In addition, AAV-GFP was generated as a control (data not shown). B–D, AAV-Alk5CA or AAV-GFP was injected into the substantia nigra of wild-type mice, and 2 weeks later the mice were killed and analyzed for GFP expression (B, D) and TGF-β signaling (C). The substantia nigra is outlined in B and C, with insets in C showing higher magnification. Dopaminergic neurons were immunolabeled with an antibody against TH (red), and the reporter gene (GFP, green) and Alk5CA expressing cells appear yellow after superimposition (D). Scale bars: B, C, 50 μm; D, 20 μm.
Delivery of AAV-Alk5CA significantly reduces MPTP-induced dopaminergic neurodegeneration and motor deficits
To test our hypothesis above, we investigated whether local delivery of AAV-Alk5CA might exert protection against the degeneration of DA neurons induced by neurotoxin MPTP, which is commonly used in experimental parkinsonism (Meredith and Rademacher, 2011). We first injected AAV-Alk5CA stereotaxically into the right substantia nigra and AAV-GFP into the left substantia nigra of each wild-type mouse. Two weeks later, mice received MPTP to induce dopaminergic degeneration, according to a published dosing regimen (Martens et al., 2012). Five days after the last MPTP injection, mice were killed and analyzed for dopaminergic neurodegeneration and microgliosis (Fig. 6). The right substantia nigra (received AAV-Alk5CA) showed significantly more TH+ dopaminergic neurons than the left side (received AAV-GFP as a control; Fig. 6A). Consistent with this observation, the right striatum showed significantly higher intensity of TH immunoreactivity (Fig. 6B). The reduced neurodegeneration in the AAV-Alk5CA side was mirrored by microglial response. Reactive microglia measured by CD68 immunoreactivity was markedly reduced in the AAV-Alk5CA side compared with the AAV-GFP control side (Fig. 6C). In addition, the expression of proinflammatory mediators TNF-α, iNOS, and MCP-1 in the striata were all significantly reduced in the AAV-Alk5CA side compared with the AAV-GFP control side (Fig. 6D–F).
Figure 6.
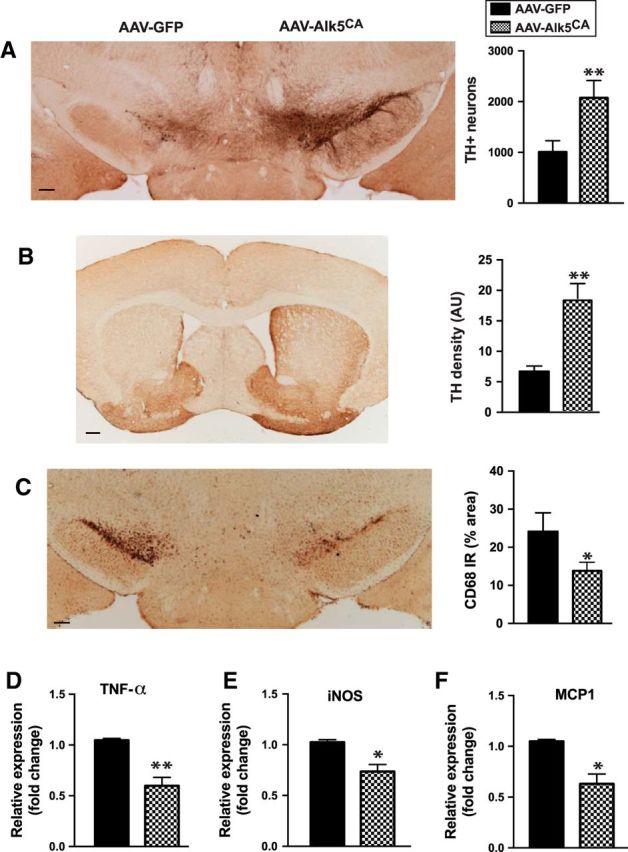
Delivery of AAV encoding Alk5CA significantly reduces MPTP-induced dopaminergic neurodegeneration and microgliosis. AAV-Alk5CA was stereotaxically injected into the right substantia nigra, and AAV-GFP was injected as a control into the left substantia nigra of the same wild-type C57BL/6 mice (male, 2 months of age). Two weeks later, mice were injected with MPTP and were killed 5 d later. A–C, MPTP-induced dopaminergic neurodegeneration was assessed by TH immunostaining in the substantia nigra pars compacta (A, stereological estimation of TH+ neurons) and striatum (B, intensity of TH immunoreactivity), and reactive microglia were assessed by CD68 immunostaining (C). D–F, mRNA was isolated from the striata, and the expression of TNF-α, iNOS, and MCP-1 was analyzed by qPCR. Scale bars: A–C, 200 μm. Bars indicate the mean ± SEM. *p < 0.05; **p < 0.01, by paired t test. n = 7 mice/group.
To further study the effects of AAV-Alk5CA, we injected AAV-Alk5CA stereotaxically into both sides of the substantia nigra of wild-type C57BL/6 mice (male, 2 months of age). AAV-GFP as a control was injected in another set of animals. Two weeks later, half of the mice from both groups was injected with MPTP (Martens et al., 2012) and the other half was injected with PBS. We used a battery of behavioral tests to assess motor function 5 d after the last MPTP injection (Fig. 7). Home-cage behavior monitored with the Smart-Homecage platform (Luo et al., 2014) revealed no difference between the mice receiving AAV-GFP and those receiving AAV-Alk5CA after being injected with PBS (Fig. 7A–C). MPTP caused significant impairments in locomotor behavior in mice receiving AAV-GFP, but AAV-Alk5CA mice showed significantly improved locomotor behavior, as shown by moving distance (Fig. 7A), activity counts (Fig. 7B), and active time (Fig. 7C). In addition, the animals receiving AAV-Alk5CA performed significantly better in the pole and wire hang tests (Fig. 7D,E) than those receiving AAV-GFP after MPTP administration. Thus, the delivery of AAV-Alk5CA significantly reduced motor deficits associated with MPTP injury. Postmortem stereology revealed that the mice receiving AAV-GFP and those receiving AAV-Alk5CA had similar estimated numbers of TH+ DA neurons in the SNpc after being injected with PBS. MPTP caused a significant loss of DA neurons in the SNpc, but the number of TH+ DA neurons in the SNpc was significantly higher in the mice receiving AAV-Alk5CA than in those receiving AAV-GFP (Fig. 7F). Similarly, a higher intensity of TH immunoreactivity in the striatum (Fig. 7G) and reduced CD68 immunoreactivity in the SNpc (Fig. 7H) were observed in the mice receiving AAV-Alk5CA compared with those receiving AAV-GFP after MPTP injection. In summary, stereotaxically delivering AAV-Alk5CA into the substantia nigra provides potent neuroprotective effects against MPTP.
Figure 7.

Delivery of AAV encoding Alk5CA significantly reduces MPTP-induced locomotor deficits and neurodegeneration. AAV-Alk5CA was stereotaxically injected into both sides of the substantia nigra of wild-type C57BL/6 mice (male, 2 months of age). AAV-GFP, as a control, was injected into both sides of the substantia nigra in another set of animals. Two weeks later, mice were injected with MPTP or PBS (as a control). A–E, Motor function was assessed by Smart-Homecage (A–C), a pole test (D), and a wire hang test (E) 5 d after MPTP injection. A–C, Measurements of Smart-Homecage, travel distance (A), activity counts (B), and active time (C) were analyzed by Cagescore. F–H, Mice were then killed, and MPTP-induced DA neurodegeneration was assessed by TH immunostaining in the substantia nigra pars compacta (F) and striatum (G), and reactive microglia were assessed by CD68 immunostaining (H). Bars indicate the mean ± SEM. *p < 0.05; **p < 0.01, two-way ANOVA. n = 6–7 mice/group.
Discussion
Our results demonstrate that deficiency in neuronal TGF-β signaling promotes a parkinsonian phenotype and that TGF-β signaling activation through AAV-Alk5CA protects mice from MPTP neurotoxicity. They highlight the essential role of TGF-β signaling in adult DA neurons and support TGF-β signaling as a potential therapeutic target for PD. Our results are in line with studies showing that mice with reduced TGF-β signaling (through TGF-β2 or Smad3 haplodeficiency) show fewer DA neurons in the substantia nigra in adulthood (Andrews et al., 2006; Tapia-González et al., 2011). While TGF-β signaling was reduced globally in those studies, our genetic study using CaMKII-tTA mice allowed us to specifically reduce TGF-β signaling postnatally in neurons, thus linking TGF-β signaling in terminally differentiated, mature neurons to PD. By using a double-transgenic approach, we achieved regionally selective expression of TBRIIΔk in projection targets of the mesencephalic dopaminergic system. The expression of TBRIIΔk in the striatum (but not in the substantia nigra) and the loss of DA neurons in the substantia nigra in our model suggest that reduced TGF-β signaling in the striatum contributes to the loss of DA neurons in the substantia nigra. This observation appears to support the “dying-back” hypothesis (Dauer and Przedborski, 2003), that is, the degeneration of striatal terminals caused by reduced TGF-β signaling may lead to the loss of neuronal cell bodies in the substantia nigra. These results also suggest that the DA neurons may be particularly sensitive to a reduction of TGF-β signaling. It has been shown that TGF-β signaling is reduced in the aged brain (Tichauer et al., 2014). Therefore, our findings may have implications for the pathogenesis of sporadic Parkinson's disease. In addition, our findings support the notion that the motor deficits observed in the CaMKII-tTA+/TBRIIΔk+ mice are likely due to the degeneration of nigrostriatal system. However, we are not able to exclude the contribution from other brain areas. It would be of particular interest to investigate in the future whether the injection of AAV-Alk5CA into the striatum or substantia nigra will rescue the motor deficits and nigrostriatal degeneration in the CaMKII-tTA+/TBRIIΔk+ mice.
In contrast to previous attempts, which failed to protect DA neurons through the administration of TGF-β3 or the overexpression of TGF-β1 (Sauer et al., 1995; Sánchez-Capelo et al., 2003), our approach through AAV-Alk5CA significantly reduced MPTP-induced DA neurodegeneration, neuroinflammation, and motor deficits. While every cell type in the brain may respond to TGF-β1 or TGF-β3 in those earlier studies (Sauer et al., 1995; Sánchez-Capelo et al., 2003), our viral approach allowed us to increase TGF-β signaling more specifically in neurons. Given the cell type-specific nature of TGF-β signaling, we believe that cell type-specific manipulation of TGF-β signaling contributes, at least partially, to the potent protective effects we observed in our studies. During the past 30 years, AAV has been widely explored as a gene therapy tool (Kotterman and Schaffer, 2014; Samulski and Muzyczka, 2014). AAV-mediated delivery of the TGF-β superfamily proteins, such as glial cell line-derived neurotrophic factor, neurturin, and growth/differentiation factor 5, has been examined for the ability to protect and/or restore degenerating dopaminergic neurons in animal models and in clinical trials (Bartus et al., 2014; Kelly et al., 2015). While these approaches involve delivering the ligands, our approach takes advantage of a constitutively active form of the receptor, thus offering an alternative and effective approach.
Altogether, our results support a critical role of TGF-β signaling in mature neurons. They suggest that TGF-β signaling may present a therapeutic target for PD and provide a strong proof of concept for testing AAV-Alk5CA and related approaches in additional preclinical studies for PD toward possible use in patients.
Footnotes
This work was supported by grants from the National Institutes of Health (NS-092868, to J.L; AG23708 and AG20603, to T.W.-C.) and by a grant from the Michael J. Fox Foundation for Parkinson's Research (to J.L). We thank Dr. Jiasheng Zhang, Dr. Eric J. Huang, Sneha Krishna, and Suzannah Rhin for technical assistance.
The authors declare no competing financial interests.
References
- Andrews ZB, Zhao H, Frugier T, Meguro R, Grattan DR, Koishi K, McLennan IS (2006) Transforming growth factor beta2 haploinsufficient mice develop age-related nigrostriatal dopamine deficits. Neurobiol Dis 21:568–575. 10.1016/j.nbd.2005.09.001 [DOI] [PubMed] [Google Scholar]
- Bartholin L, Cyprian FS, Vincent D, Garcia CN, Martel S, Horvat B, Berthet C, Goddard-Léon S, Treilleux I, Rimokh R, Marie JC (2008) Generation of mice with conditionally activated transforming growth factor beta signaling through the TbetaRI/ALK5 receptor. Genesis 46:724–731. 10.1002/dvg.20425 [DOI] [PubMed] [Google Scholar]
- Bartus RT, Weinberg MS, Samulski RJ (2014) Parkinson's disease gene therapy: success by design meets failure by efficacy. Mol Ther 22:487–497. 10.1038/mt.2013.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensadoun JC, Déglon N, Tseng JL, Ridet JL, Zurn AD, Aebischer P (2000) Lentiviral vectors as a gene delivery system in the mouse midbrain: cellular and behavioral improvements in a 6-OHDA model of Parkinson's disease using GDNF. Exp Neurol 164:15–24. 10.1006/exnr.2000.7409 [DOI] [PubMed] [Google Scholar]
- Böttinger EP, Jakubczak JL, Roberts IS, Mumy M, Hemmati P, Bagnall K, Merlino G, Wakefield LM (1997) Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J 16:2621–2633. 10.1093/emboj/16.10.2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB (2009) Tests to assess motor phenotype in mice: a user's guide. Nat Rev Neurosci 10:519–529. 10.1038/nrn2652 [DOI] [PubMed] [Google Scholar]
- Buffo A, Holtmaat AJ, Savio T, Verbeek JS, Oberdick J, Oestreicher AB, Gispen WH, Verhaagen J, Rossi F, Strata P (1997) Targeted overexpression of the neurite growth-associated protein B-50/GAP-43 in cerebellar Purkinje cells induces sprouting after axotomy but not axon regeneration into growth-permissive transplants. J Neurosci 17:8778–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L (2005) Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci 25:9694–9703. 10.1523/JNEUROSCI.2980-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Di Figlia M (1994) Reserpine increases Fos activity in the rat basal ganglia via a quinpirole-sensitive mechanism. Neuroscience 60:115–123. 10.1016/0306-4522(94)90207-0 [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R (2001) DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci 21:788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris A, Williams-Gray CH, Foltynie T, Brown J, Maranian M, Walton A, Compston DA, Barker RA, Sawcer SJ (2007) Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson's disease. J Neurol 254:846–848. 10.1007/s00415-006-0414-6 [DOI] [PubMed] [Google Scholar]
- Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, Kay MA (2008) In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol 82:5887–5911. 10.1128/JVI.00254-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty SV, Sullivan AM, O'Keeffe GW (2014) Roles for the TGFbeta superfamily in the development and survival of midbrain dopaminergic neurons. Mol Neurobiol 50:559–573. 10.1007/s12035-014-8639-3 [DOI] [PubMed] [Google Scholar]
- Kelly MJ, O'Keeffe GW, Sullivan AM (2015) Viral vector delivery of neurotrophic factors for Parkinson's disease therapy. Expert Rev Mol Med 17:e8. 10.1017/erm.2015.6 [DOI] [PubMed] [Google Scholar]
- Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, Dauer W, Burke RE (2004) Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci 24:3136–3146. 10.1523/JNEUROSCI.4506-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ED, Kuwahara AA, Messer RL, Wyss-Coray T (2015) Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc Natl Acad Sci U S A 112:4128–4133. 10.1073/pnas.1422448112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman MA, Schaffer DV (2014) Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15:445–451. 10.1038/nrg3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein K, Unsicker K (1994) Transforming growth factor-beta promotes survival of midbrain dopaminergic neurons and protects them against N-methyl-4-phenylpyridinium ion toxicity. Neuroscience 63:1189–1196. 10.1016/0306-4522(94)90583-5 [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K (1995) TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J 14:736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Lin AH, Masliah E, Wyss-Coray T (2006) Bioluminescence imaging of Smad signaling in living mice shows correlation with excitotoxic neurodegeneration. Proc Natl Acad Sci U S A 103:18326–18331. 10.1073/pnas.0605077103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Ho PP, Buckwalter MS, Hsu T, Lee LY, Zhang H, Kim DK, Kim SJ, Gambhir SS, Steinman L, Wyss-Coray T (2007) Glia-dependent TGF-beta signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J Clin Invest 117:3306–3315. 10.1172/JCI31763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, Zhu L, Alabsi H, Getachew R, Narasimhan R, Wabl R, Fainberg N, James ML, Wong G, Relton J, Gambhir SS, Pollard JW, Wyss-Coray T (2013) Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med 210:157–172. 10.1084/jem.20120412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, Bieri G, Castellano JM, Beaupre GS, Wyss-Coray T (2014) Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol 5:12. 10.3389/fneur.2014.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, Farese RV Jr (2012) Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest 122:3955–3959. 10.1172/JCI63113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274:1678–1683. 10.1126/science.274.5293.1678 [DOI] [PubMed] [Google Scholar]
- Meredith GE, Rademacher DJ (2011) MPTP mouse models of Parkinson's disease: an update. J Parkinsons Dis 1:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Narabayashi H, Riederer P, Nagatsu T (1995) Transforming growth factor-beta 1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson's disease. Neurosci Lett 193:129–132. 10.1016/0304-3940(95)11686-Q [DOI] [PubMed] [Google Scholar]
- Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y (1985) A simple quantitative bradykinesia test in MPTP-treated mice. Res Commun Chem Pathol Pharmacol 50:435–441. [PubMed] [Google Scholar]
- Poulsen KT, Armanini MP, Klein RD, Hynes MA, Phillips HS, Rosenthal A (1994) TGF beta 2 and TGF beta 3 are potent survival factors for midbrain dopaminergic neurons. Neuron 13:1245–1252. 10.1016/0896-6273(94)90062-0 [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316:750–754. 10.1126/science.1141736 [DOI] [PubMed] [Google Scholar]
- Robertson GS, Herrera DG, Dragunow M, Robertson HA (1989) L-dopa activates c-fos in the striatum ipsilateral to a 6-hydroxydopamine lesion of the substantia nigra. Eur J Pharmacol 159:99–100. 10.1016/0014-2999(89)90049-6 [DOI] [PubMed] [Google Scholar]
- Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, Hu L, Mok SI, Shah A, Savner EM, Tolias C, Corfas R, Chen S, Inquimbert P, Xu Y, McInnes RR, Rice FL, Corfas G, Ma Q, Woolf CJ, et al. (2010) Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron 65:886–898. 10.1016/j.neuron.2010.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussa E, Wiehle M, Dünker N, Becker-Katins S, Oehlke O, Krieglstein K (2006) Transforming growth factor beta is required for differentiation of mouse mesencephalic progenitors into dopaminergic neurons in vitro and in vivo: ectopic induction in dorsal mesencephalon. Stem Cells 24:2120–2129. 10.1634/stemcells.2005-0514 [DOI] [PubMed] [Google Scholar]
- Roussa E, von Bohlen und Halbach O, Krieglstein K (2009) TGF-beta in dopamine neuron development, maintenance and neuroprotection. Adv Exp Med Biol 651:81–90. 10.1007/978-1-4419-0322-8_8 [DOI] [PubMed] [Google Scholar]
- Sagar SM, Sharp FR, Curran T (1988) Expression of c-fos protein in brain: metabolic mapping at the cellular level. Science 240:1328–1331. 10.1126/science.3131879 [DOI] [PubMed] [Google Scholar]
- Samulski RJ, Muzyczka N (2014) AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol 1:427–451. 10.1146/annurev-virology-031413-085355 [DOI] [PubMed] [Google Scholar]
- Sánchez-Capelo A, Colin P, Guibert B, Biguet NF, Mallet J (2003) Transforming growth factor beta1 overexpression in the nigrostriatal system increases the dopaminergic deficit of MPTP mice. Mol Cell Neurosci 23:614–625. 10.1016/S1044-7431(03)00081-2 [DOI] [PubMed] [Google Scholar]
- Sauer H, Rosenblad C, Björklund A (1995) Glial cell line-derived neurotrophic factor but not transforming growth factor beta 3 prevents delayed degeneration of nigral dopaminergic neurons following striatal 6-hydroxydopamine lesion. Proc Natl Acad Sci U S A 92:8935–8939. 10.1073/pnas.92.19.8935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-González S, Giráldez-Pérez RM, Cuartero MI, Casarejos MJ, Mena MÁ, Wang XF, Sánchez-Capelo A (2011) Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol Neurodegener 6:72. 10.1186/1750-1326-6-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wyss-Coray T (2006) Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest 116:3060–3069. 10.1172/JCI27341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichauer JE, Flores B, Soler B, Eugenín-von Bernhardi L, Ramírez G, von Bernhardi R (2014) Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav Immun 37:187–196. 10.1016/j.bbi.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, Zou B, Simmons DA, Xie XS, Longo FM, Wyss-Coray T (2014) Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med 20:659–663. 10.1038/nm.3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Greenhalgh DA, Bickenbach JR, Jiang A, Bundman DS, Krieg T, Derynck R, Roop DR (1997) Expression of a dominant-negative type II transforming growth factor beta (TGF-beta) receptor in the epidermis of transgenic mice blocks TGF-beta-mediated growth inhibition. Proc Natl Acad Sci U S A 94:2386–2391. 10.1073/pnas.94.6.2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massagué J (1995) GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J 14:2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Südhof TC (2012) Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron 73:990–1001. 10.1016/j.neuron.2011.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J, Tang S, Janak PH, Tecott LH, Huang EJ (2007) Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat Neurosci 10:77–86. 10.1038/nn1816 [DOI] [PMC free article] [PubMed] [Google Scholar]