

Abstract
DNA polymerase gamma (POLG) is the replicative polymerase responsible for maintaining mitochondrial DNA (mtDNA). Disorders related to its functionality are a major cause of mitochondrial disease. The clinical spectrum of POLG syndromes includes Alpers-Huttenlocher syndrome (AHS), childhood myocerebrohepatopathy spectrum (MCHS), myoclonic epilepsy myopathy sensory ataxia (MEMSA), the ataxia neuropathy spectrum (ANS) and progressive external ophthalmoplegia (PEO). We have collected all publicly available POLG-related patient data and analyzed it using our pathogenic clustering model to provide a new research and clinical tool in the form of an online server. The server evaluates the pathogenicity of both previously reported and novel mutations. There are currently 176 unique point mutations reported and found in mitochondrial patients in the gene encoding the catalytic subunit of POLG, POLG. The mutations are distributed nearly uniformly along the length of the primary amino acid sequence of the gene. Our analysis shows that most of the mutations are recessive, and that the reported dominant mutations cluster within the polymerase active site in the tertiary structure of the POLG enzyme. The POLG Pathogenicity Prediction Server (http://polg.bmb.msu.edu) is targeted at clinicians and scientists studying POLG disorders, and aims to provide the most current available information regarding the pathogenicity of POLG mutations.
Abbreviations: AHS, Alpers-Huttenlocher syndrome; ANS, Ataxia neuropathy spectrum; IP, Intrinsic processivity subdomain of POLGA spacer-domain; MCHS, Childhood myocerebrohepatopathy spectrum; MEMSA, Myoclonic epilepsy myopathy sensory ataxia; PDB ID, Four-character identification code for a protein structure in the RSCB PDB database; PEO, Progressive external ophthalmoplegia; POLG, DNA polymerase gamma; POLGA, Catalytic subunit of DNA polymerase gamma; POLGB, Accessory subunit of DNA polymerase gamma; PNF, Putatively non-functional enzyme; SNP, Single nucleotide polymorphism/non-pathogenic mutation
Keywords: POLG syndrome, DNA polymerase gamma, Mitochondrial disorder, Pathogenicity prediction, Patient database, Mutation database
Highlights
-
•
Multi-level access to crucial data supporting diagnosis/prognosis of POLG syndromes
-
•
Clustering protocol enables identification of novel neutral polymorphisms
-
•
Identical alleles displaying variable symptoms evidence unidentified components
-
•
POLG enzymes with premature stop codons, insertions/deletions group biochemically
-
•
Dominant POLG mutations all lie within a critical location in the structure
1. Introduction
DNA polymerase gamma is the enzyme responsible for replicating and maintaining mitochondrial DNA (reviewed in [15], [16]). The functional, holoenzyme form of POLG is a heterotrimer consisting of a catalytic subunit (POLGA) and a dimeric accessory subunit (POLGB). POLGA carries three catalytic activities: 5′− 3′ DNA polymerase, 3′− 5′ exonuclease and 5′-deoxyribose phosphate lyase. POLGB enhances DNA binding, catalysis and holoenzyme processivity. Mutations in POLG are associated with a wide range of human disorders that exhibit shared and progressive phenotypes (reviewed in [46]). The POLG disorders range from prenatally-fatal conditions and severe infantile onset disorders, such as Alpers disease (Alpers-Huttenlocher syndrome), to milder, late-onset conditions such as progressive external ophthalmoplegia, and are described collectively as POLG syndromes.
POLG mutations are relatively rare, with an estimated carrier frequency of 1/100 individuals in the Western world [11]. Most are recessive, and symptoms typically manifest only in compound heterozygous patients. Although the symptoms and severity of the conditions vary widely, all share a few common features: they largely affect tissues with high energy demand, such as in the nervous system, muscle and liver, and they are all progressive conditions that show direct correlation between age of onset and the severity of the condition [9].
Fig. 1.

Structural model of the POLG holoenzyme in complex with primer-template DNA. In the three-dimensional ternary complex of POLG, the pathogenic clusters in the catalytic core, POLGA, form five distinct functional regions [6]. Upper panel, the structure modeled after PDB structure 4ZTU[39] illustrates the pathogenic clusters of POLGA colored as green for Cluster 1, yellow for Cluster 2, red for Cluster 3, blue for Cluster 4 and cyan for Cluster 5. Lower panel, subclusters are distributed throughout the primary amino acid sequence of the POLGA polypeptide. Subclusters are assigned for each continuous block on the primary amino acid sequence. The accessory subunits, POLGB, are illustrated in light grey (proximal subunit) and dark grey (distal subunit). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Our clustering model demonstrates that the age of onset of numerous symptoms correlates strongly with the severity of the syndrome and the range of symptoms that are manifest (see Fig. 5, [9]). For example, the most severe form of POLG syndrome, MCHS (typical age of onset from birth to three years), includes patients that present with symptoms such as hypotonia, developmental delay, gastrointestinal dysfunction and hepatopathy. Patients diagnosed with AHS (typical age of onset from a few months after birth to 16 years) share most of these symptoms, but are also more likely to experience epileptic seizures. MEMSA patients (typical age of onset 13 to 25 years of age) are in most cases diagnosed with myopathies, neuropathies and ataxia. Patients diagnosed with ANS (typical age of onset from 15 to middle-age) show a wide variety in their symptoms, including symptoms from both the MEMSA and PEO patient groups. Finally, patients with PEO (typical age of onset from 30 years and above) represent the least severe end of the spectrum, and present with symptoms such as diplopia and ptosis.
Fig. 5.
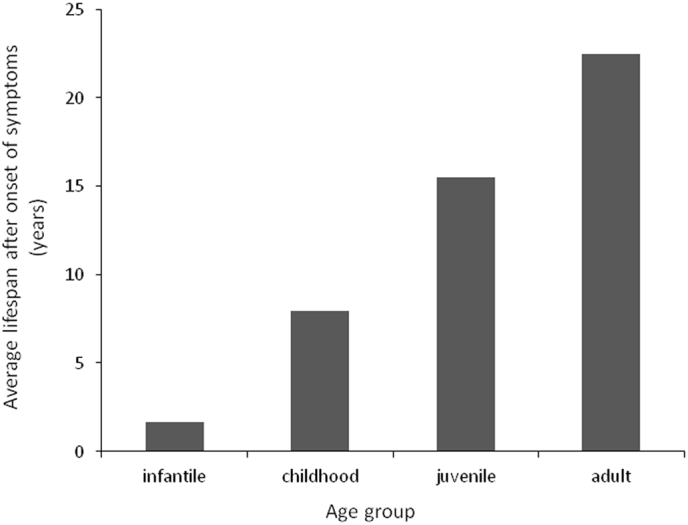
Average lifespan after onset of symptoms. POLG syndromes are progressive conditions, and age of onset correlates directly with the severity of the disease. Patient data were analyzed in cases for which both the age of onset of symptoms and age of death were reported, and included 144 patient cases. The use of valproic acid to treat patients with epileptic seizures may account for shortened lifespans in the data in the more severe cases of POLG syndromes.
2. Results
2.1. POLG pathogenicity prediction server
Table 1.
Contents of the POLG Pathogenicity Prediction Server and distribution of mutations. The majority of patient cases report compound heterozygous mutations.
| Total number of patient cases | 660 |
| Infantile onset cases | 181 |
| Childhood onset cases | 103 |
| Juvenile onset cases | 85 |
| Adult onset cases | 284 |
| Unknown age of onset cases | 7 |
| Unique missense mutations | 176 |
| Unique pathogenic mutation combinations | 215 |
| Compound heterozygous patients | 323 |
| Homozygous patients | 152 |
| Heterozygous patients | 128 |
| Reports referenced | 182 |
Fig. 2.

The mutation query interface of the POLG Pathogenicity Prediction Server. The server provides statistical predictions of age of onset, and shows the typical symptoms for patients with mutations that map within pathogenic clusters 1–5. Information about allelic configuration of mutations can be helpful in cases for which pedigrees are not available. The server also provides direct access to clinical descriptions of patient cases that have similar mutations.
Fig. 3.

Site navigation within the POLG Pathogenicity Prediction Server. The server provides access to collated data by various search interfaces, and provides statistical predictions of onset of symptoms based on the pathogenic clustering model.
2.2. Adjusting the cluster boundaries
The recently published POLG holoenzyme ternary structure makes it possible to identify all of the POLGA residues that interact directly with the DNA ligand. Of the 56 residues shown to interact with DNA, only six map outside of our previously defined pathogenic clusters. Because these 56 residues have a clear role in the functionality of POLG to bind and position the DNA ligand, they have a high risk for producing deleterious effects if mutated. By extending slightly the borders of four of our subclusters: 2B, 496–517 (previously 497–517); 2D, 752–769 (previously 752–767); 1F, 1098–1138 (previously 1104–1138); and 3C, 795–807 (previously 804–807), these residues are accommodated within the defined high-risk locations for mutations in the POLG structure. Full cluster definitions as defined previously based on biochemical, structural and genetic studies [9] are included as supplementary Table S2.
Fig. 4.

Positioning of Cluster 3D in the POLG apo-holoenzyme as compared to the holoenzyme ternary complex. Cluster 3D (residues 1047–1096, red surface representation) is located in a substantially different position between the apo-holoenzyme (panel A, PDB ID: 3IKM) and holoenzyme ternary complex (panel B, PDB ID: 4ZTU). The repositioning of Cluster 3D appears unlikely to represent a conformational change. Rather, this region of the tertiary structure is likely to contain disordered regions, rendering difficult its structural evaluation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.3. Common mutations in the patient data
Table 2.
Common POLG mutations. The three most commonly reported POLG mutations, p.A467T, p.W748S and p.G848S, have been reported in all possible compound heterozygous combinations, as well as in homozygous form. Heterozygous case reports are rare for p.A467T and p.W748S, and there are none for p.G484S. Comparatively, p.G848S appears to occur in more severe cases, and p.W748S consistently shows a slightly milder phenotype in comparison to p.A467T.
| Mutation | Clusters | Average age of onset (years) |
Standard deviation | Number of reported cases |
|---|---|---|---|---|
| W748S + E1143G/wt | 5 + SNP/– | 59.0 | 4.0 | 2 (1 outliera) |
| A467T/wt | 2/– | 42.8 | 11.2 | 5 (1 outlierb) |
| W748S/W748S | 5/5 | 21.4 | 10.4 | 27 (1 outlierc) |
| W748S + E1143G/W748S + E1143G | 5 + SNP/5 + SNP | 22.1 | 11.1 | 35 |
| A467T/A467T | 2/2 | 19.9 | 13.7 | 45 |
| G848S/G848S | 1/1 | 5.0 | 0.0 | 1 |
| W748S/A467T | 5/2 | 28.7 | 11.2 | 27 |
| W748S + E1143G/A467T | 5 + SNP/2 | 21.4 | 11.7 | 15 |
| A467T/W748S + K561M | 2/5 + 2 | 0.01 | 0.0 | 1 |
| A467T/G848S | 2/1 | 1.7 | 2.0 | 22 |
| W748S/G848S | 5/1 | 5.7 | 2.6 | 8 |
| A467T/PNF | 2/– | 1.5 | 1.0 | 14 (1 outlierd) |
| W748S/PNF | 5/– | 1.7 | 1.3 | 5 |
| W748S + Q497H + E1143G/ W748S + Q497H + E1143G |
5 + 2 + SNP/5 + 2 + SNP | 19.0 | 4.0 | 2 |
| W748S + Q497H + E1143G/A467T | 5 + 2 + SNP/2 | 17.0 | 0.0 | 1 |
Sarzi et al. [47], patient 36 (id: 404). [http://www.ncbi.nlm.nih.gov/pubmed/17452231].
Galassi et al. [48], (id: 409). [http://www.ncbi.nlm.nih.gov/pubmed/18504126].
Tzoulis et al. [49], (id: 552). [http://www.ncbi.nlm.nih.gov/pubmed/24841123].
Martikainen et al. [50], (id: 666). [http://www.ncbi.nlm.nih.gov/pubmed/27111573].
A comparison of the three mutations shows that p.G848S is associated with the most pathogenic phenotypes. When found in compound heterozygous form with p.A467T, the average age of onset is 1.7 years (± 2.0 years, infantile). By contrast, in compound heterozygous patients carrying p.G848S and p.W748S, the average age of onset is 5.7 years (± 2.6 years, childhood). A single patient carrying homozygous p.G848S has been reported with an age of five years at the time of examination [40]. Mutation p.G848S has been characterized biochemically to have a substantially reduced DNA-binding affinity and very low DNA polymerase activity [18]. A possible explanation for the lack of additional homozygous p.G848S patient case reports is that most of them result in prenatal mortality.
Patients carrying mutations p.A467T and p.W748S show similar ages of onset: the average age of onset for homozygous patients is 19.9 years (± 13.7) for p.A467T and 21.4 (± 10.4) years for p.W748S. These data corroborate the findings reported in a recent study on the extensive clinical heterogeneity of homozygous p.A467T patients [32]. Interestingly, compound heterozygous cases with genotype p.A467T/p.W748S manifest later, at ~25 years of age, suggesting that enzymes carrying deleterious mutations in different clusters could have compensatory properties. The heterozygous cases of p.A467T (five cases) and p.W748S (two cases) represent a small fraction of known carriers. The ExAC genome mapping project reports global population frequencies of 0.052% for p.A467T and 0.083% for p.W748S [7]. Moreover, in independent studies, p.A467T was found in the Belgian population at a particularly high frequency of 0.6% [43] and likewise, p.W748S was found in the Finnish population at a frequency of 0.8% [12]. By comparison, the frequency of p.W748S in the Finnish population is 0.57% in the ExAC dataset. These data alone argue that a dominant status for these mutations is unlikely in the absence of other genetic or environmental factors predisposing the patients to a mitochondrial disorder.
Biochemical studies show that in comparison to the wild type POLG, the p.A467T mutant enzyme exhibits moderate to substantially reduced DNA binding affinity resulting in similarly reduced polymerase processivity [25]. The p.W748S mutant enzyme was shown in one study to exhibit substantially reduced DNA binding affinity and polymerase processivity [3], whereas another reported enzymatic properties similar to the wild type enzyme [30]. A summary of the biochemical characterization of these mutations is included as Table S3.
2.4. Mutations yielding putatively non-functional POLG
Mutations that introduce frameshifts, premature stop codons, exon skipping or large deletions are likely to inactivate POLG function entirely and/or impact its folding, subunit interaction or stability. Structural perturbations would likely render the enzyme subject to cellular turnover. We have grouped together on the server mutations that would result in a putatively non-functional (PNF) POLG. Several of them have been characterized biochemically, corroborating such a non-functional status, though some may retain limited DNA binding capability or other partial functionality [17], [34]. Patients with compound heterozygous PNF mutations that carry the three most commonly reported POLG mutations typically manifest with very severe, infantile onset conditions. For example, 14 patients with a compound heterozygous p.A467T/PNF genotype manifested symptoms at an average age of onset of 1.5 years. For four reported patients with a p.W748S/PNF genotype, the average age of onset is 1.7 years. No patients have been reported with p.G848S/PNF mutations. These data argue that the enzyme level produced from the single, partially-functional allele is a crucial factor in these patients, as compared with the more moderate phenotypes exhibited by the compound heterozygous and homozygous cases in which pA467T, p.W748S or p.G848S are present. Several exceptions have been reported. A single patient carrying A467T + E873X/E873X (A467T + PNF/PNF) survived until 10 years of age [27]. One possible explanation suggested by the authors of the report is that the premature stop codons are leaky, and some functional enzyme is still produced by the PNF allele. Similarly, Roos et al. [34] reported two brothers with a compound heterozygous genotype p.T914P/c.3104 + 3A > T (Cluster 1 mutation/splice site mutation). Molecular analysis showed that skipping of POLG exon 19 caused by c.3104 + 3A > T was incomplete, such that some wild type POLG was still produced, leading to an ameliorated phenotype with the patients manifesting symptoms at 20 and 50 years of age. Similar molecular analyses are warranted to determine which specific PNF mutations might still produce some wild type enzyme.
PNF mutations are all recessive, and the PNF carrying enzymes are likely incapable of competing for DNA binding with wild type POLG at the mtDNA replication fork. This hypothesis is corroborated by the presence of asymptomatic carriers in the pedigrees of patient reports. As such, the PNF enzymes also serve to establish an approximate baseline for assessing the severity and dominant versus recessive status of a mutation. Because PNF enzymes are putatively incapable of contributing to mtDNA replication, a mutation that is present in an individual in compound heterozygous form with a PNF mutation is likely to be a recessive mutation that produces an enzyme that is to some extent capable of mtDNA replication. This assessment supports a recessive status for two of the most common mutations, p.A467T and p.W748S, because they have been found in compound heterozygous form with PNF mutations. The patient data corroborates the biochemical evidence that p.G848S is only marginally more processive than a PNF enzyme [18], and thus a p.G848S/PNF genotype is not viable.
The patient data supports a model in which each deleterious POLG mutation affects the overall capacity of the enzyme to replicate and maintain mtDNA. Although recessive mutations may exert deleterious effects on enzyme functionality, symptoms are not manifest during the lifetime of an individual because a single wild type allele is typically sufficient. In both compound heterozygous and homozygous cases, each mutation may reduce the overall cellular capacity to maintain mtDNA, such that the most energy-demanding and mitochondria-dense tissues are affected. The patient data show that homozygous mutations are similar to compound heterozygous mutations, such that each allele with one or more deleterious mutations renders the condition more severe as aging progresses. Moreover, mutations affecting different critical functionalities (and mapping to different clusters) of POLG may show either compensatory or exacerbating effects.
Effects of environmental and cellular stress factors such as infections, unhealthy lifestyle, malnutrition, sleep deprivation and other conditions could not be controlled in the patient data evaluated in this study. In addition to the inconsistent reporting of patient data (age of onset/age at examination/age of death) to explain the high standard deviation seen in the data, it is likely that other currently unidentified genetic factors contribute to disease progression. In homozygous cases, the possible effects of consanguinity could also not be controlled.
2.5. Dominant POLG mutations
The POLG Pathogenicity Prediction Server contains 50 unique missense mutations that have been reported as heterozygous POLG mutations in a total of 131 individual patients. Patient case reports are available for examination through the mutation query and patient data access interfaces on the server, and a list of all pathogenic heterozygous POLG mutations is also available with hyperlinks to the individual patient case reports. Based on the clinical data and in the absence of other explaining factors, these 50 mutations are tentatively assigned as pathogenic with a dominant inheritance mode. We have assessed the probability that they are the root cause of the symptoms described in the reported patient cases by taking into account the full patient dataset and their pathogenic cluster assignments.
We have subdivided the 50 heterozygous mutations into three groups as follows. Mutations in patient case reports that are not found at an elevated frequency in available population data and have been characterized as putatively-dominant in biochemical studies are categorized as most likely dominant. For 14 heterozygous mutations, only a single patient case has been reported, and additional data are required to confirm a putatively-dominant status. Mutations found in heterozygous patient cases without any other explanatory factors for the condition of the patients are categorized as having an unclear dominant pathogenicity. Mutations in which the case report indicated they are likely not the root cause for the condition of the patient have been categorized in the least likely dominant group. We have also taken into account biochemical data and reported asymptomatic carriers when assigning mutations into this last group. Our classification of the 50 heterozygous POLG mutations is discussed below, and is indicated on the list available on the POLG Pathogenicity Prediction Server.
Fig. 6.

Heterozygous POLG mutations. Panel A, model of the catalytic subunit of POLG illustrating mutations that have been reported to be heterozygous in the POLG Pathogenicity Prediction Server database. Mutations for which substantial corroborative evidence indicates dominant inheritance are shown in red spheres, and surround the polymerase active site (black arrow). Yellow spheres indicate mutations for which there is insufficient data to argue dominant status. Green spheres depict mutations least likely to be dominant. The color status is not indicative with respect to pathogenicity in compound heterozygous form. Two of the most common pathogenic POLG mutations, p.A467T and p.W748S, are shown in cyan spheres. Panel B, close-up of the active site of the polymerase domain of POLGA (black arrow). The putatively-dominant mutant residues (red spheres) interact with the incoming dNTP (orange), template DNA or the catalytic Mg-ions (black spheres). A hotspot for these is on the O-helix, in which every residue facing the polymerase active site has been reported in a heterozygous POLG patient. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Notably, it was shown in a murine model that mitochondria can be relatively tolerant of point mutations, and a 500-fold increase in point mutations did not limit the lifespan of the animals [45]. This would argue that neither the reduced nucleotide selectivity nor defective exonucleolytic editing observed for some putatively-dominant mutations are likely the sole contributors in dominant pathogenicity. In that regard, exonucleolytic proofreading has been reported to increase the fidelity of POLG 20–100 fold depending on the assay used [14], [21], [22], [29]. It also appears possible that proofreading activity is not the most critical function of the exonuclease domain; some mutations in the exonuclease domain result in reduced nucleotide polymerization rather than defects in proofreading [38], and it has been shown recently that exonucleolysis by POLGA is required for producing circular double-stranded DNA [26].
Though all mutations that are most likely to be dominant are located around the polymerase active site, not all mutations in close proximity to it are likely to be dominant. These include p.S433C and p.R1187W. There is only a single case report for p.S433C with an unspecified age of onset, and the mother of the patient was reported to be an asymptomatic carrier [13]. p.R1187W was reported in two cases in which the family histories do not support an assignment of dominant inheritance [33], [35].
Three mutations, p.G848S, p.T851A and p.R852C, located in a highly conserved β-hairpin structure (residues 844–856) that lies between the polymerase and exonuclease domains, were reported as heterozygous in one in 66, one in four and one in 16 heterozygous patient cases, respectively. This region of the POLG structure is important for mispair recognition [39], and is likely to be involved in facilitating correct DNA binding near the polymerase active site, thus affecting both the polymerization rate and fidelity of the enzyme. Because there are many asymptomatic carriers for each of the three mutations, they are categorized as unlikely-dominant mutations. Moreover, they appear in the available population data with frequencies of 0.016% for p.G848S, 0.008% for p.T851A and 0.0066% for p.R852C [7]. Though unlikely dominant, they are nonetheless highly pathogenic when found in compound heterozygous form.
Mutations that lie near the polymerase active site and are categorized as having an unclear dominant status include p.D930N, p.H945L and p.R953C. Only a single patient case has been reported for p.H945L with matching symptoms and age of onset typical of dominant POLG mutations, but with no available family history [4]. p.R953C has been reported as a heterozygous mutation in only one patient case of four, without any family background [24] and is reported at a population frequency of 0.0016% in the ExAC database [7]. p.D930N has been described as exhibiting dominant-like pathogenicity in a yeast model, without any corroborative patient data [1].
Fig. 7.
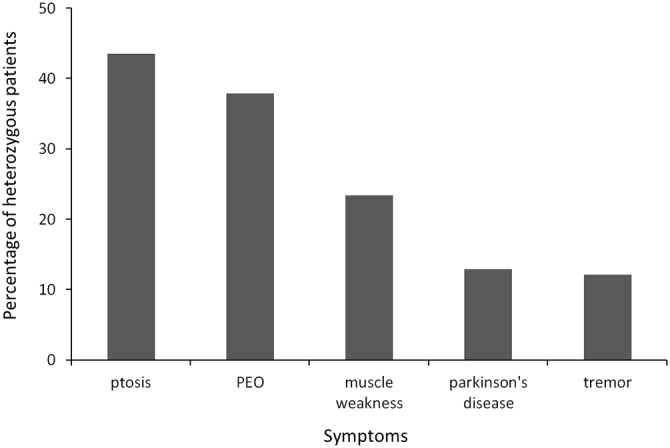
The most commonly reported symptoms of heterozygous POLG patients. Symptoms in order of commonness in heterozygous patients include ptosis, progressive external ophthalmoplegia (PEO), muscle weakness, Parkinson's disease and tremors.
The biochemical characterizations reported to date for the putatively-dominant mutations [2], [3], [10], [18] [25], [30], combined with the patient case reports, highlight the fact that the most critical properties of dominant pathogenic mutations in POLG appear to be severely-reduced polymerase activity with sufficient DNA binding affinity to compete with wild type enzyme at the mtDNA replication fork (Table S3).
3. Discussion
We report the development of an online POLG Pathogenicity Prediction Server. The server represents a comprehensive, interactive database that complements and extends substantially the online Human DNA Polymerase gamma Mutation Database (http://tools.niehs.nih.gov/polg/). POLG data and patient cases are accessible in numerous ways as described under Results, and additional content is provided by including analysis by the PON-P2 algorithm [28] and ExAC population frequency data [7]. Most notably, the POLG Pathogenicity Prediction Server incorporates our previously described clustering model for pathogenicity prediction, which was derived by evaluation of biochemical, structural and genetic data on POLG and the related family A group DNA polymerases, bacterial DNA polymerase I and bacteriophage T7 DNA polymerase [6], [9]. Because most POLG syndromes result from the incidence of compound heterozygous mutations, the clustering analysis of pairs of mutations based on the analysis of currently available mutations and mutational pairs gives insight into the likely pathogenicity of novel pairs, and of previously-identified alleles in combination with novel alleles. As such, we believe the prediction tool will be widely applicable as one parameter in the diagnosis of mitochondrial disorders.
Noteworthy challenges and/or limitations of the dataset include the varying methods and variety of tests employed in the examination of patients that have been reported and in particular, in publications prior to the generalized use of DNA sequencing to evaluate the spectrum of possible genes and genetic causes for mitochondrial disorders. Additionally, age of onset may not be reported as the first onset of symptoms; rather, the age of the patient at the time of examination has sometimes been reported. In view of these challenges, we highlight the importance of whole exome analysis (see for example [41]) and of reporting patient cases with full pedigrees and detailed descriptions of age of onset of symptoms. Though thorough clinical examination may not be possible for the immediate relatives of the patients, a description and overall assessment of their wellbeing would prove helpful. Finally, it is important to document the co-existence of potentially non-pathogenic polymorphisms such that any impact they might have in combination with a pathogenic mutation can be assessed appropriately.
Issues related to both intrinsic genetic and biochemical parameters must be evaluated further as new patient data on pathogenic mutations emerges. At present, the dataset of available mutations is characterized by the overrepresentation of the three common pathogenic mutations p.A467T, p.W748S and p.G848S that are present in composite in 71% of the 681 case reports. Whereas the relative frequency of new variants reported will likely remain similar, their absolute number will increase with streamlined reporting on an international scale. The POLG Pathogenicity Prediction Server features a contact form that can be used for pointing out errors in the existing data, notifying the database administrators of new publications and requesting confidential analysis of rare POLG mutations.
A potential shortcoming of the pathogenic clustering protocol is a direct reflection of the intrinsic structural and biochemical properties of POLG and indeed, of all enzymes. Whereas a new variant allele mapping to a pathogenic cluster is much more likely to be pathogenic than one that lies outside of a cluster, not all amino acid residues within a cluster or subcluster have a similar likelihood and/or level of pathogenicity. This inherent property is dependent strongly on both the biochemical function of the specific amino acid and the chemical nature of the amino acid change induced by the mutation. For example, an amino acid that is crucial to POLG function such as the catalytic residues in the pol motifs A, B and C that are conserved among family A DNA polymerases and which lie in POLG subclusters 1D, 1E and 1F, respectively, may be either underrepresented or absent in the dataset because amino acid substitutions are not tolerated in live individuals. On the other hand, conservative amino acid changes within cluster-mapping residues may show no clinical phenotypes. In such situations, use of the residue browser function of the server will provide relevant information for further evaluation. Nonetheless, in the ‘big picture’ sense, it is clear that combinations of mutations mapping to the pol domain cluster 1 in combination with those mapping in either the DNA-binding channel cluster 2 or the putative protein: protein interaction cluster 5 constitute the bulk of the patient cases (96) manifesting infantile onset, and involve one or more of the most common mutations reported. Additionally, nine of 10 putatively-dominant mutations that we have classified as most-likely dominant (56 of 57 patient cases reported) map within the catalytic subclusters of the pol domain, 1D and 1E. This includes for example, the pathogenic mutation p.Y955C [10], [20], [42]. The single mutation mapping outside of cluster 1 is P765T in cluster 2; though its location in the POLG structure suggests a high likelihood of dominance, there is only a single case report and no existing biochemical data.
Biochemical characterization of mutations in conjunction with the patient data suggests that mutations that impair nucleotide polymerization by POLG without affecting DNA binding carry the highest risk for dominant inheritance. The low number of reported patient cases for some of the putatively-dominant mutations and lack of consistent testing and reporting in patient pedigrees obscures the evaluation of a likely dominant status. Based on the available genetic, biochemical and clinical data in our database we have re-evaluated the status of 49 putatively-dominant mutations and divided them into three classes. We suggest that the 10 mutations that fall within the most-likely dominant group be considered strongly by clinicians as a root cause of mitochondrial dysfunction.
It is well documented that the symptoms and tissue specificity of pathogenicity in patients suffering from POLG syndromes varies widely, even among patients carrying the same POLG mutations (see for example [32]). Nonetheless our analysis shows a clear consistency in age of onset that does not differ with gender. We also observed an apparently stronger correlation of symptoms within families. This observation highlights the likelihood that other genetic components are contributory and the importance of whole exome analysis in the evaluation of POLG syndromes, and of mitochondrial disorders in general to identify such components. Efforts such as Mitochondrial Disease Sequence Data Resource (MSeqDR) are underway to address the issue of nuclear genetic modifiers [8], [36]. Future efforts should also address the possible contributions of mtDNA genetic background. At present, our understanding of the disease etiology of POLG syndromes together with the currently available diagnostic tools should be beneficial in family planning for couples undergoing genetic testing, even those with no familial history of mitochondrial disorders.
4. Materials and methods
Patient data deposited into the database and used in this study are anonymous and collected from publicly available journal articles. The source of each patient case is stated and available on the server. The data in the literature is reported in a non-standardized form and comprises a variety of different tests, examinations and details. The server categorizes each case based on the age of onset of the first symptoms reported into the following age groups: infantile, < 3 years; childhood, 3–13 years; juvenile, 13–20 years; or adult, ≥ 20 years. For reports in which only the age of the patient at the time of examination is reported, this age was used. If no age for the patient is reported, the age group is indicated as “unknown.”
Symptoms were categorized based only on the diagnoses provided in the report. For conditions such as Alpers syndrome, it might be the case that nearly all of the known symptoms for this condition have been reported in a patient case. However, if the diagnosis of Alpers has not been reported directly, the case has not been deposited as an Alpers entry. For other conditions such as ataxia, we have accepted terms such as “movement disorder,” “ataxic gait,” “gait disturbance” and “gait unsteadiness” as being synonymous.
The output of predictions of pathogenicity provided by the mutation query interface of the server are based on the pathogenic clustering model and statistics of existing patient cases with similar cluster-mapping mutations. The borders of the subclusters have been defined by available patient data, structural information and biochemical studies of mutations [6]. The statistical predictions reflect directly the contents of the database, and can be refined as new cases are added.
Transparency Document
Transparency document
Acknowledgments
This work was supported by National Institutes of Health Grant GM45295 to L.S.K. Anssi Nurminen was supported by the University of Tampere.
Footnotes
The Transparency document associated with this article can be found, in online version.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbacli.2017.04.001.
Appendix A. Supplementary data
Supplementary tables
References
- 1.Baruffini E., Horvath R., Dallabona C., Czermin B., Lamantea E., Bindoff L.…Lodi T. Predicting the contribution of novel POLG mutations to human disease through analysis in yeast model. Mitochondrion. 2011;11(1):182–190. doi: 10.1016/j.mito.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Chan S.S.L., Longley M.J., Copeland W.C. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J. Biol. Chem. 2005;280(36):31341–31346. doi: 10.1074/jbc.M506762200. [DOI] [PubMed] [Google Scholar]
- 3.Chan S.S.L., Longley M.J., Copeland W.C. Modulation of the W748S mutation in DNA polymerase gamma by the E1143G polymorphismin mitochondrial disorders. Hum. Mol. Genet. 2006;15(23):3473–3483. doi: 10.1093/hmg/ddl424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delgado-alvarado M., De P., Jiménez-urbieta H., Gago B., Gabilondo A., Bornstein B., Rodríguez-oroz M.C. Parkinsonism, cognitive deficit and behavioural disturbance caused by a novel mutation in the polymerase gamma gene. J. Neurol. Sci. 2015;350(1–2):93–97. doi: 10.1016/j.jns.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Estep P.A., Johnson K.A. Effect of the Y955C mutation on mitochondrial DNA polymerase nucleotide incorporation efficiency and fidelity. Biochemistry. 2011;50(29):6376–6386. doi: 10.1021/bi200280r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Euro L., Farnum G.A., Palin E., Suomalainen A., Kaguni L.S. Clustering of Alpers disease mutations and catalytic defects in biochemical variants reveal new features of molecular mechanism of the human mitochondrial replicase, Pol γ. Nucleic Acids Res. 2011;39(21):9072–9084. doi: 10.1093/nar/gkr618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ExAC Exome Aggregation Consortium (ExAC) browser. 2015. http://exac.broadinstitute.org Retrieved from.
- 8.Falk M.J., Shen L., Gonzalez M., Leipzig J., Lott M.T., Stassen A.P.M.…Wong L.J. Mitochondrial Disease Sequence Data Resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol. Genet. Metab. 2015;114(3):388–396. doi: 10.1016/j.ymgme.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farnum G.A., Nurminen A., Kaguni L.S. Mapping 136 pathogenic mutations into functional modules in human DNA polymerase γ establishes predictive genotype–phenotype correlations for the complete spectrum of POLG syndromes. Biochim. Biophys. Acta, Bioenerg. 2014;1837(7):1113–1121. doi: 10.1016/j.bbabio.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graziewicz M.A., Longley M.J., Bienstock R.J., Zeviani M., Copeland W.C. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat. Struct. Mol. Biol. 2004;11(8):770–776. doi: 10.1038/nsmb805. [DOI] [PubMed] [Google Scholar]
- 11.Hakonen A.H., Davidzon G., Salemi R., Bindoff L.A., Van Goethem G., Dimauro S.…Suomalainen A. Abundance of the POLG disease mutations in Europe, Australia, New Zealand, and the United States explained by single ancient European founders. Eur. J. Hum. Genet. 2007;15(7):779–783. doi: 10.1038/sj.ejhg.5201831. [DOI] [PubMed] [Google Scholar]
- 12.Hakonen A.H., Heiskanen S., Juvonen V., Lappalainen I., Luoma P.T., Rantamaki M.…Suomalainen A. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am. J. Hum. Genet. 2005;77(3):430–441. doi: 10.1086/444548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudson G., Deschauer M., Taylor R.W., Hanna M.G., Fialho D., Schaefer A.M., He L.P., Blakely E., Turnbull D.M., Chinnery P.F. POLG1, C10ORF2, and ANT1 mutations are uncommon in sporadic progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Neurology. 2006;66:1439–1441. doi: 10.1212/01.wnl.0000210486.32196.24. https://www.ncbi.nlm.nih.gov/pubmed/16682683. [DOI] [PubMed] [Google Scholar]
- 14.Johnson A.A., Johnson K.A. Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J. Biol. Chem. 2001;44613(512):38090–38096. doi: 10.1074/jbc.M106045200. [DOI] [PubMed] [Google Scholar]
- 15.Kaguni L.S. DNA polymerase γ, the mitochondrial replicase. Annu. Rev. Biochem. 2004;73(1):293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 16.Kaguni L.S., Oliveira M.T. Structure, function and evolution of the animal mitochondrial replicative DNA helicase. Crit. Rev. Biochem. Mol. Biol. 2016;9238:1–12. doi: 10.3109/10409238.2015.1117056. https://www.ncbi.nlm.nih.gov/pubmed/26615986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaliszewska M., Kruszewski J., Kierdaszuk B., Kostera-Pruszczyk A., Nojszewska M., Łusakowska A.…Tońska K. Yeast model analysis of novel polymerase gamma variants found in patients with autosomal recessive mitochondrial disease. Hum. Genet. 2015;134(9):951–966. doi: 10.1007/s00439-015-1578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kasiviswanathan R., Longley M.J., Chan S.S.L., Copeland W.C. Disease mutations in the human mitochondrial DNA polymerase thumb subdomain impart severe defects in mitochondrial DNA replication. J. Biol. Chem. 2009;284(29):19501–19510. doi: 10.1074/jbc.M109.011940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y.S., Kennedy W.D., Yin Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139(2):312–324. doi: 10.1016/j.cell.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis W., Day B.J., Kohler J.J., Hosseini S.H., Chan S.S.L., Green E.C.…Copeland W.C. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab. Invest. 2007;87(4):326–335. doi: 10.1038/labinvest.3700523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Longley M.J., Mosbaugh D.W. Properties of the 3′ to 5′ exonuclease associated with porcine liver DNA polymerase γ: Substrate specificity, product analysis, inhibition, and kinetics of terminal excision. J. Biol. Chem. 1991;36:24702–24711. https://www.ncbi.nlm.nih.gov/pubmed/1662214. [PubMed] [Google Scholar]
- 22.Longley M.J., Nguyen D., Kunkel T.a., Copeland W.C. The fidelity of human DNA polymerase γ with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 2001;276(42):38555–38562. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 23.Luo N., Kaguni L.S. Vol. 280. 2005. Mutations in the spacer region of Drosophila mitochondrial DNA polymerase affect DNA binding, processivity, and the balance between Pol and Exo function; pp. 2491–2497. https://www.ncbi.nlm.nih.gov/pubmed/1662214. [DOI] [PubMed] [Google Scholar]
- 24.Luoma P., Melberg A., Rinne J.O., Kaukonen J.A., Nupponen N.N., Chalmers R.M.…Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet. 2004;364(9437):875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 25.Luoma P.T., Luo N., Löscher W.N., Farr C.L., Horvath R., Wanschitz J.…Suomalainen A. Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum. Mol. Genet. 2005;14(14):1907–1920. doi: 10.1093/hmg/ddi196. [DOI] [PubMed] [Google Scholar]
- 26.Macao B., Uhler J.P., Siibak T., Zhu X., Shi Y., Sheng W.…Falkenberg M. The exonuclease activity of DNA polymerase γ is required for ligation during mitochondrial DNA replication. Nat. Commun. 2015;6:7303. doi: 10.1038/ncomms8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naviaux R.K., Nguyen K.V. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004;55(5):706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 28.Niroula A., Urolagin S., Vihinen M. PON-P2: prediction method for fast and reliable identification of harmful variants. PLoS One. 2015;10(2):1–17. doi: 10.1371/journal.pone.0117380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson M.W., Kaguni L.S. 3′ - 5′ exonuclease in Drosophila mitochondrial DNA polymerase. J. Biol. Chem. 1992;32:223136–223142. https://www.ncbi.nlm.nih.gov/pubmed/1429661. [PubMed] [Google Scholar]
- 30.Palin E.J.H., Lesonen A., Farr C.L., Euro L., Suomalainen A., Kaguni L.S. Functional analysis of H. sapiens DNA polymerase γ spacer mutation W748S with and without common variant E1143G. Biochim. Biophys. Acta. 2010;6:545–551. doi: 10.1016/j.bbadis.2010.02.003. https://www.ncbi.nlm.nih.gov/pubmed/20153822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian Y., Ziehr J.L., Johnson K.A. Alpers disease mutations in human DNA polymerase gamma cause catalytic defects in mitochondrial DNA replication by distinct mechanisms. Front. Genet. 2015;6:1–11. doi: 10.3389/fgene.2015.00135. https://www.ncbi.nlm.nih.gov/pubmed/25914719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajakulendran S., Pitceathly R.D.S., Taanman J.W., Costello H., Sweeney M.G., Woodward C.E.…Rahman S. A clinical, neuropathological and genetic study of homozygous A467T POLG-related mitochondrial disease. PLoS One. 2016;11(1):1–16. doi: 10.1371/journal.pone.0145500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reichenbach J., Schubert R., Horvàth R., Petersen J., Fütterer N., Malle E.…Zielen S. Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr. Res. 2006;60(3):321–326. doi: 10.1203/01.pdr.0000233252.60457.cf. [DOI] [PubMed] [Google Scholar]
- 34.Roos S., Macao B., Fusté J.M., Lindberg C., Jemt E., Holme E.…Falkenberg M. Subnormal levels of POLγA cause inefficient initiation of light-strand DNA synthesis and lead to mitochondrial DNA deletions and autosomal dominant progressive external ophthalmoplegia. Hum. Mol. Genet. 2013;22(12):2411–2422. doi: 10.1093/hmg/ddt094. [DOI] [PubMed] [Google Scholar]
- 35.Rouzier C., Chaussenot A., Serre V., Fragaki K., Bannwarth S., Ait-El-Mkadem S.…Paquis-Flucklinger V. Quantitative multiplex PCR of short fluorescent fragments for the detection of large intragenic POLG rearrangements in a large French cohort. Eur. J. Hum. Genet. 2014;22(4):542–550. doi: 10.1038/ejhg.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen L., Diroma M.A., Gonzalez M., Navarro-Gomez D., Leipzig J., Lott M.T.…Gai Xiaowu. MSeqDR: a centralized knowledge repository and bioinformatics web resource to facilitate genomic investigations in mitochondrial disease. Hum. Mutat. 2016;37(6):540–548. doi: 10.1002/humu.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stumpf J.D., Copeland W.C. Mitochondrial DNA replication and disease: Insights from DNA polymerase γ mutations. Cell. Mol. Life Sci. 2011;68:219–233. doi: 10.1007/s00018-010-0530-4. https://www.ncbi.nlm.nih.gov/pubmed/20927567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szczepanowska K., Foury F. A cluster of pathogenic mutations in the 3′–5′ exonuclease domain of DNA polymerase gamma defines a novel module coupling DNA synthesis and degradation. Hum. Mol. Genet. 2010;19(18):3516–3529. doi: 10.1093/hmg/ddq267. [DOI] [PubMed] [Google Scholar]
- 39.Szymanski M.R., Kuznetsov V.B., Shumate C., Meng Q., Lee Y.-S., Patel G.…Yin Y.W. Structural basis for processivity and antiviral drug toxicity in human mitochondrial DNA replicase. EMBO J. 2015;34(14):1959–1970. doi: 10.15252/embj.201591520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang S., Wang J., Lee N.-C., Milone M., Halberg M.C., Schmitt E.S.…Wong L.-J.C. Mitochondrial DNA polymerase gamma mutations: an ever expanding molecular and clinical spectrum. J. Med. Genet. 2011;48(10):669–681. doi: 10.1136/jmedgenet-2011-100222. [DOI] [PubMed] [Google Scholar]
- 41.Taylor R.W., Pyle A., Griffin H., Blakely E.L., Duff J., He L.…Chinnery P.F. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2016;312:68–77. doi: 10.1001/jama.2014.7184. https://www.ncbi.nlm.nih.gov/pubmed/25058219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Goethem G., Dermaut B., Löfgren A., Martin J.-J., Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 43.Van Goethem G., Martin J.J., Dermaut B., Löfgren A., Wiball A., Ververken D.…Van Broeckhoven C. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul. Disord. 2003;13:133–142. doi: 10.1016/s0960-8966(02)00216-x. http://dx.doi.org/10.1016/S0, https://www.ncbi.nlm.nih.gov/pubmed/12565911. [DOI] [PubMed] [Google Scholar]
- 44.Stewart J.D., Horvath R., Baruffini E., Ferrero I., Bulst S., Watkins P.B., Fontana R.J., Day C.P., Chinnery P.F. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. 2010;52:1791–1796. doi: 10.1002/hep.23891. https://www.ncbi.nlm.nih.gov/pubmed/21038416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vermulst M., Bielas J.H., Kujoth G.C., Ladiges W.C., Rabinovitch P.S., Prolla T.A., Loeb L.A. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007;39(4):540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 46.Young M.J., Copeland W.C. Human mtochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016;38:52–62. doi: 10.1016/j.gde.2016.03.005. https://www.ncbi.nlm.nih.gov/pubmed/27065468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarzi E., Bourdon A., Chrétien D., Zarhrate M., Corcos J., Slama A., Cormier-Daire V., de Lonlay P., Munnich A., Rötig A. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J. Pediatr. 2007;150:531–534. doi: 10.1016/j.jpeds.2007.01.044. http://www.ncbi.nlm.nih.gov/pubmed/17452231. [DOI] [PubMed] [Google Scholar]
- 48.Galassi G., Lamantea E., Invernizzi F., Tavani F., Pisano I., Ferrero I., Palmieri L., Zeviani M. Additive effects of POLG1 and ANT1 mutations in a complex encephalomyopathy. Neuromuscul. Disord. 2008;18:465–470. doi: 10.1016/j.nmd.2008.03.013. http://www.ncbi.nlm.nih.gov/pubmed/18504126. [DOI] [PubMed] [Google Scholar]
- 49.Tzoulis C., Tran G.T., Coxhead J., Bertelsen B., Lilleng P.K., Balafkan N., Payne B., Miletic H., Chinnery P.F., Bindoff L.A. Molecular pathogenesis of polymerase γ-related neurodegeneration. Ann. Neurol. 2014;76:66–81. doi: 10.1002/ana.24185. http://www.ncbi.nlm.nih.gov/pubmed/24841123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martikainen M., Ng Y., Gorman G., Alston C., Blakely E., Schaefer A., Chinnery P., Burn D., Taylor R., McFarland R., Turnbull D. Clinical, genetic, and radiological features of extrapyramidal movement disorders in mitochondrial disease. JAMA Neurol. 2016;73:668–674. doi: 10.1001/jamaneurol.2016.0355. http://www.ncbi.nlm.nih.gov/pubmed/27111573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document
Supplementary tables