

Abstract
Rationale:
Peutz–Jeghers syndrome (PJS) is an autosomal dominant genetic syndrome characterized by a unique type of gastrointestinal hamartomatous polyp associated with oral and anal mucocutaneous pigmentations. Peutz–Jeghers polyps occur most numerously in the small intestine but frequently in the colon and stomach, only a few cases have been reported in the duodenum.
Patient concern:
A further family history survey discovered 10 out of 14 members of the family (in 4 generations) had mucocutaneous pigmentations, but many of them were living in rural areas where they had no access to specialized medical services, so none were checked with endoscopy for polyps of hamartoma.
Diagnoses:
We report the case of a boy patient with mucocutaneous pigmentations over the lips, and a history of recurrent bouts of vomit and anemia over the preceding two years, no abdominal pain and mass. An upper gastrointestinal endoscopy revealed some small polyps in the stomach and multiple sessile polyps in the second part of the duodenum, but colonoscopy exam did not reveal any lesion.
Interventions:
A double polypectomy and duodenum segmentary resection with end-to-end anastomosis was performed. Histopathology of the resected duodenum polyps indicated it to be a typical hamartomatous polyp.
Outcomes:
The child was under regular follow-up and recovered well.
Lessons:
In this case, the patient was characteristic with pigmentations on his lips and intermittent upper intestinal obstruction (due to mass duodenal polyps), there are no definitive guidelines for the treatment to duodenal PJS hamartomatous polyp, each case requires tailor-made management.
Keywords: duodenal polyp, intestinal obstruction, Peutz–Jeghers syndrome, pigmentation
1. Introduction
Peutz–Jeghers syndrome (PJS) is an autosomal dominant genetic syndrome characterized by a unique type of gastrointestinal hamartomatous polyp associated with oral and anal mucocutaneous pigmentations.[1,2] Patients with PJS have an increased risk for common and unusual types of gastrointestinal and non-gastrointestinal tumors.[3] These polyps are also at risk of acute gastrointestinal bleeding, intussusceptions, and bowel obstruction. The genetic lesion responsible for the phenotypic expression is in 66% to 94% of the cases considered to be a mutation on the short arm of chromosome 19 (19p13.3) of STK11/LKB1.[4] Peutz–Jeghers polyps occur most numerously in the small intestine but frequently in the colon and stomach, only a few cases have been reported in the duodenum. We report a case of Peutz–Jeghers syndrome in a child patient with intermittent upper intestinal obstruction (due to mass duodenal polyps), and describes the clinical characteristics of PJS.
2. Case presentation
The institutional review board (The Second Affiliated Hospital of Shantou University Medical College) approved this work. A 7-year-old boy presented with history of recurrent bouts of vomit and anemia over the preceding two years. Physical examination revealed mucocutaneous pigmentations over the lips (Fig. 1A), no other lesions could be identified in the anal region or on the hands and feet, no abdominal pain and mass. Laboratory data showed anemia (hemoglobin 76 g/L), no other blood, urine, or stool changes were detected. We proceeded with an upper gastrointestinal endoscopy, which revealed some small polyps in the stomach and multiple sessile polyps in the second part of the duodenum (Fig. 1B). Additionally, colonoscopy was performed and no lesion was found. Tumor markers were checked: CA199 and CEA were normal. A double polypectomy and duodenum segmentary resection with end-to-end anastomosis was performed (Fig. 1C). Histopathology of the resected duodenum polyps indicated it to be a typical hamartomatous polyp (Fig. 1D). Postoperatively, the child was under regular follow-up and uneventful without recurrence.
Figure 1.
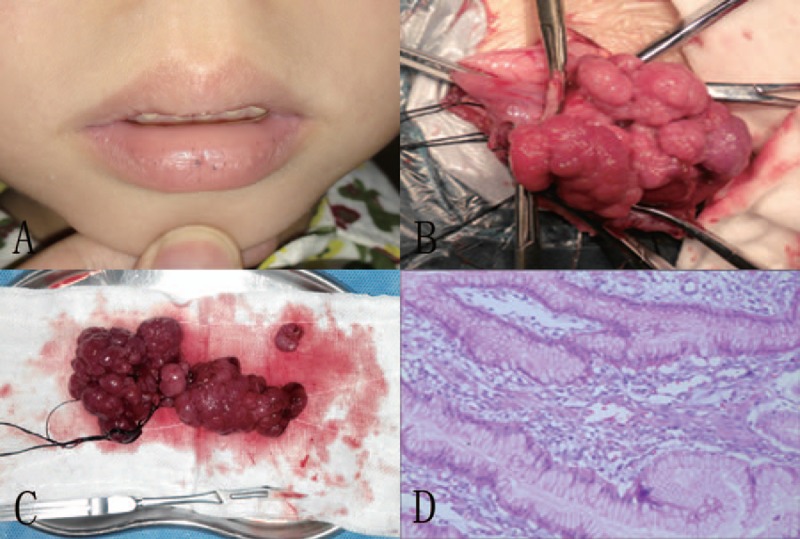
(A) Pigmentations on the patient's lips. (B) Mass polyps in the duodenum. (C) Multiple completely resected polyps. (D) Histopathological findings revealing hamartomatous characteristics of resected polyps (HE × 40). HE = hematoxylin-eosin.
A further detailed family history was obtained. The family survey included 14 family members from four generations. Ten members with mucocutaneous pigmentations (Fig. 2), 4 males and 6 females, they were not sure have hamartomatous polyp, because of many of them came from a rural area and did not evaluated endoscopically. Two of the 10 members appeared mucocutaneous pigmentations, the patient's maternal grandmother and great grandmother died of colorectal cancer.
Figure 2.
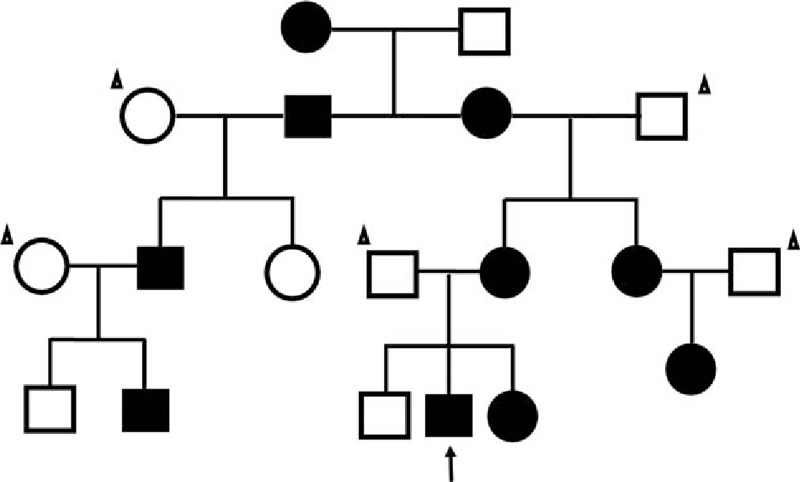
Pedigree of the family with PJS. Black symbols denote individuals with mucocutaneous pigmentations. Circles and squares indicate females and males, respectively, while the arrow indicates the reported patient and one with the triangle is Nonfamily members. PJS = Peutz–Jeghers syndrome.
3. Discussion
Clinical criteria for a definite diagnosis of PJS include the presence of a hamartoma associated with two of the following three signs: mucocutaneous pigmentation, polyposis of the small-bowel or family history of PJS.[1,3] Pigmentations usually appear in the first year of life but in adulthood may fade and in some cases even disappear.[4] Pigmentation on the lips and buccal mucosa are an essential feature, playing an important role in the early diagnosis. As far as this patient is concerned, lips spots and hamartomatous polyps in the gastrointestinal tract. But Some patients will only exhibit mucocutaneous pigmentation which is defined as incomplete PJS.[5]
Polyps found in PJS commonly present in adolescence and early adulthood. It grows during the first decade of life, and most patients become symptomatic between the ages of 10 to 30 years.[3,4] Patients usually present in the first decade due to polyp-related complications like abdominal pain, bowel obstruction, intussusception and overt or occult gastrointestinal bleeding.[6] Our patient presented with symptoms of intermittent upper intestinal obstruction and anemia for two years.
The etiology of PJS is unclear. Mutations of the STK11 gene seems to be responsible for PJS, while STK 11 is a tumor suppressor gene and encodes a serine/threonine kinase and maps to chromosome 19p13.3.[7,8] STK11 play a role of tumor suppressor in cells. Loss of STK11 protein kinase activity is associated with occurrence and development of tumor. So patients with PJS have an increasing incidence of malignant tumors compared with the general population. The most common malignancy associated with PJS is colorectal cancer, followed by breast, small bowel, gastric, and pancreatic cancers, with lifetime cumulative cancer risks up to 93%.[5,9] Malignancy in Peutz–Jeghers syndrome is considered to arise in adulthood, and occurs only rarely in children.[10] Patients should be instructed on the need for cancer surveillance, our patient is scheduled for follow-up abdominal ultrasound examination (liver gallbladder, pancreas, spleen) every six months and upper gastrointestinal endoscopy every three years according to the guidelines for PJS.
Combined endoscopic and surgical treatment has been reported and is considered to be the best in terms of quality of life.[11,12] Surgical resection may appropriately be used for the patients with giant or complication of polyp such as obstruction, intussusceptions or gastrointestinal bleeding. Considering the above mentioned boy patient, because of the count and size of the duodenum polyps, surgery was the preferred therapy. Recently, some experts have used double balloon enteroscopy removal small bowel PJS polyps, which may decrease the need for laparotomy.[3]
4. Conclusion
In summary, PJS is an autosomal dominant disease caused by germline mutation of the serine threonine kinase 11 and characterized by hamartomatous polyps in the gastrointestinal tract and mucocutaneous pigmentation. Patients with PJS have an increasing risk of developing cancer or transformation to malignant polyps in the gastroinstestinal tract and other organs. The main symptom of this patient is intermittent upper intestinal obstruction caused by mass duodenal polyps, which rarely obstruct the duodenum.[13,14] As there are no definitive guideline for the treatment to duodenal PJS hamartomatous polyp,[15] each case requires tailor-made management.
Footnotes
Abbreviations: CA199 = ca-antigen 199, CEA = carcino embryonie antigen, HE = hematoxylin-eosin, LKB1 = liver kinase B1, PJS = Peutz–Jeghers syndrome, STK11 = serine/threonine kinase 11.
Funding: the National Natural Science Foundation of China (Grant No. 81373745) supported this study.
The authors have no conflicts of interest to disclose.
References
- [1].Arber N, Moshkowitz M. Small bowel polyposis syndromes. Curr Gastroenterol Rep 2011;13:435–41. [DOI] [PubMed] [Google Scholar]
- [2].Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin North Am 2008;88:779–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Giardiello FM, Trimbath JD. Peutz–Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol 2006;4:408–15. [DOI] [PubMed] [Google Scholar]
- [4].Szanto P, Barbieru V, Badea R, et al. Unexpected Peutz–Jeghers syndrome in an adult presenting with intermittent upper intestinal obstruction. A case report. J Gastrointestin Liver Dis 2014;23:91–4. [PubMed] [Google Scholar]
- [5].Fan RY, Sheng JQ. A case of Peutz–Jeghers syndrome associated with high-grade intramucosal neoplasia. Int J Clin Exp Pathol 2015;8:7503–5. [PMC free article] [PubMed] [Google Scholar]
- [6].Kopacova M, Tacheci I, Rejchrt S, et al. Peutz–Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol 2009;15:5397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature 1998;391:184–7. [DOI] [PubMed] [Google Scholar]
- [8].Huang Z, Miao S, Wang L, et al. Clinical characteristics and STK11 gene mutations in Chinese children with Peutz–Jeghers syndrome. BMC Gastroenterol 2015;15:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].van Lier MG, Wagner A, Mathus-Vliegen EM, et al. High cancer risk in Peutz–Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 2010;105:1258–64. [DOI] [PubMed] [Google Scholar]
- [10].Wangler MF, Chavan R, Hicks MJ, et al. Unusually early presentation of small-bowel adenocarcinoma in a patient with Peutz–Jeghers syndrome. J Pediatr Hematol Oncol 2013;35:323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lin BC, Lien JM, Chen RJ, et al. Combined endoscopic and surgical treatment for the polyposis of Peutz–Jeghers syndrome. Surg Endosc 2000;14:1185–7. [DOI] [PubMed] [Google Scholar]
- [12].Seenath MM, Scott MJ, Morris AI, et al. Combined surgical and endoscopic clearance of small-bowel polyps in Peutz–Jeghers syndrome. J R Soc Med 2003;96:505–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Höchter W, Weingart J, Seib HJ, et al. Duodenal polyps. Incidence, histologic substrate and significance. Dtsch Med Wochenschr 1984;109:1183–6. [DOI] [PubMed] [Google Scholar]
- [14].De Silva WS, Pathirana AA, Gamage BD, et al. Extra-ampullary Peutz–Jeghers polyp causing duodenal intussusception leading to biliary obstruction: a case report. J Med Case Rep 2016;15:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Basford PJ, Bhandari P. Endoscopic management of nonampullary duodenal polyps. Therap Adv Gastroenterol 2012;5:127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]