

Abstract
For infants with SCID, the ideal conditioning regimen before allogeneic HCT would omit cytotoxic chemotherapy to minimize short-and long-term complications. We performed a prospective pilot trial with G-CSF plus plerixafor given to the host to mobilize HSC from their niches. We enrolled six patients who received CD34-selected haploidentical cells and one who received T-replete matched unrelated BM. All patients receiving G-CSF and plerixafor had generally poor CD34+ cell and Lin−CD34+CD38−CD90+CD45RA−HSC mobilization, and developed donor T cells, but no donor myeloid or B-cell engraftment. Although well tolerated, G-CSF plus plerixafor alone failed to overcome physical barriers to donor engraftment.
Keywords: severe combined immunodeficiency, plerixafor, haploidentical, engraftment
SCID is a heterogeneous group of genetic disorders with the shared phenotype of profoundly deficient T cells and absent B lymphocyte function, which if untreated leads to early mortality from severe infections. Although progress has been made toward gene therapy for certain SCID genotypes, the vast majority of patients with SCID require an allogeneic HCT for curative therapy (1). When an HLA-matched relative is not available, T-cell-depleted haploidentical-related donors are often used due to their rapid availability (2). However, in certain SCID subtypes, transplantation rarely results in significant myeloid engraftment or donor B-cell lineage chimerism unless myeloablative chemotherapy is administered, necessitating lifelong immunoglobulin replacement (2). In very young infants with non-malignant diseases, such as SCID, avoidance of alkylating forms of chemotherapy is desirable, as these agents can cause significant short- and long-term toxicities. These toxicities are magnified in patients with the T−B−NK+ forms of SCID associated with defects in genes required to repair breaks in DNA (3).
Plerixafor (also called AMD3100) is a reversible antagonist of CXCR4, a chemokine receptor on the surface of normal HSCs that binds to SDF-1 and is essential for HSC retention in the BM. Mouse models demonstrate that plerixafor prior to HCT can lead to donor myeloid engraftment (4). However, it has never been utilized in the absence of chemotherapy as conditioning for an HCT in humans.
To investigate whether these agents could allow HCT for SCID to be successful without chemotherapy, we performed a prospective pilot trial, which tested the hypothesis that G-CSF and plerixafor as the sole-conditioning agents prior to allogeneic HCT would facilitate donor myeloid and B-cell engraftment in children with “graft-permissive” SCID (those with an NK− phenotype; an NK+ phenotype with a 10/10 HLA-matched related or URD; or an NK+ phenotype with maternal engraftment and a maternal haploidentical graft).
Patients and methods
Study population
Eligible patients presented to the UCSF Benioff Children’s Hospital between December 2010 and September 2011 with a new diagnosis of SCID, as defined by accepted criteria (5, 6), or previously transplanted SCID but with poor T-cell function (absolute CD4 count <200 × 106/L and PHA <30%) and ongoing clinical problems more than a year following initial HCT, and who therefore met our institutional criteria for a “boost” HCT (7, 8). The initial HCTs for the patients undergoing boost HCT were previously reported: UPN237 (9) & UPN1056 & 1057 (2). All patients had genotypic confirmation of their diagnosis. All but one patient treated during this time period consented to enroll in one of the trials. Newly diagnosed patients had PBMCs tested for evidence of TME, using a quantitative PCR-based method involving amplification of STR sequences, as previously described (10, 11). The trial was approved by the UCSF Committee on Human Research, and informed consent was obtained from the related donors and the parents of the patients in accordance with the Declaration of Helsinki and was registered at www.clinicaltrials.gov as NCT01182675.
Donor hematopoietic stem cell collection and manipulation
Newly diagnosed patients and potential donors were tested for HLA compatibility at HLA-A, -B, -C, and –DR, with – DQ and –DP added in later years. If a patient was uninfected and had a readily available 10 of 10 matched URD, then stem cell source was utilized. Otherwise, for patients with evidence of TME, the mother was the preferred HSC donor due to presumed host tolerance toward maternal cells. Boost HCT was performed using cells from the original donor.
For matched donors, unmanipulated (excepting ABO depletion, if required) BM was the HSC source. For mismatched donors, mobilized PBSCs were prepared by CD34 selection using the CliniMACs (Miltenyi Biotec, Bisley, UK) System, as previously described (12). Cell counting and gating was as previously described (2). The goal for the infused cell dose was 5 × 108 total nucleated cells/kg of recipient body weight for matched BM, and >10 × 106 CD34+ cells/kg with ≤6 × 104 CD3+ T cells/kg for haploidentical PBSC. Excess cells were cryopreserved.
Transplant regimen
Patients with graft-permissive SCID received G-CSF (5 mcg/kg/dose intravenously every 12 h for eight doses) beginning on Day −4 before HCT, and on Day −0, plerixafor (240 mcg/kg subcutaneous) was given 9–12 h prior to scheduled HSCs infusion, analogous to how these agents are used prior to stem cell mobilization for apheresis (13). Daily WBC counts were performed during the regimen to monitor for hyperleukocytosis. If the WBC count rose to >50 000 cells/mcl, subsequent doses of G-CSF were lowered to 3 mcg/kg, and if the WBC rose to >75 000 cells/mcl despite G-CSF dose modifications, G-CSF was discontinued, but no patients reached this safety threshold. Plerixafor was administered under IND exemption #110 775. For patients with pre-HCT, evidence of maternal GVHD or high-level TME (absolute T cells ≥1000 × 106/L), fludarabine (1 mg/kg/day intravenously for three days), was administered on Days −4 to −2 to treat or prevent GVHD flares, per our institutional practice (2). Patients who received unmanipulated BM were given GVHD prophylaxis with cyclosporine (Day −1 through four months post-HCT, tapering over the last month), mycophenolate mofetil (Day +1 through Day +30, then tapered over two wk), and rabbit ATG (Thymoglobulin®; 2.5 mg/kg/day for three days on Days −7 through −5). Patients who received CD34-selected PBSCs were not given pharmacologic GVHD prophylaxis.
Analysis of mobilization, engraftment, and immunologic parameters
The efficacy of the HSC mobilization was measured by daily WBC counts from prior to administration of G-CSF until Day +1 following HCT. In addition, in the four patients with newly diagnosed SCID, the percentage of CD34+ cells in the patient’s peripheral blood was measured immediately prior to administration of plerixafor, 6 and 9 h after the dose, and immediately prior to HCT (12–14 h after the dose). In three of these patients, the mobilization of host Lin−CD34+CD38−CD90+CD45RA− HSCs was measured by flow cytometry using previously established fluorescent antibodies to known HSC markers (14).
Post-transplant donor chimerism was determined using sorted CD3-, CD19-, and CD14/15-positive cells and STR markers, as above (10, 11). NK cell chimerism was not tested. Sorted cells were tested for purity by flow cytometry, and the interassay variation was +/− 1%.
Lymphocyte subsets, including naïve and memory markers CD45RA and CD45RO, were assessed by flow cytometry and compared to normal ranges for age (15, 16). T-cell function was assessed by 3H-thymidine incorporation in response to PHA, and reported as a percentage of stimulated immunologically competent control lymphocytes tested simultaneously (Mayo Medical Laboratories, Rochester, MN, USA). TRECs and T-cell receptor spectra-typing were performed on a subset of patients (17). B-cell function was measured by the ability to produce IgM and IgA within the normal range for age, presence of appropriate IgM ISH at ≥1:8 dilution, and specific antibodies following vaccination, if performed.
Supportive care
Active infections at diagnosis were treated with appropriate antimicrobial therapies that continued until evidence of infection resolution. These included high-dose cotrimoxazole (5 mg/kg/dose of trimethoprim component four times daily) for patients with PCP. Rotavirus infections were managed with supportive care. Other anti-infective prophylaxis was administered as previously described (2). Acute and chronic GHVD was graded on standard criteria (18, 19).
Statistical considerations
EFS and OS were estimated by the Kaplan–Meier method using log-rank tests (NCSS8, Kaysville, UT, USA). Events were defined as a conditioned second HSC infusion or death.
Results
Patient characteristics
Seven patients were enrolled, four with newly diagnosed SCID and three had SCID with poor post-HCT T-cell function requiring boost transplants (Table 1). All but two of the newly diagnosed patients were born prior to the implementation of newborn screening for SCID (20) and were identified due to infections, including vaccine-strain rotavirus (21). One patient with X-linked SCID (UPN 1533) also had an unusually high number of transplacental maternally engrafted T cells that were primarily (89%) CD4/CD8 double negative. This was the only patient to receive fludarabine prior to HCT (Table 2).
Table 1.
Patient characteristics at time of HCT
| UPN | Age at HCT | Gender | Ethnicity | Gene mutation | CD3 | CD4 | CD8 | CD19 | CD16/56 | PHA (%) | TME | Indication for HCT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| (× 106/L) | ||||||||||||
| Newly diagnosed | ||||||||||||
| 1509 | 4 month | M | Hispanic | RAG-1 (c.1181G>A + c.1951T > C) | 14 | 14 | 0 | 0 | 258 | 0 | 0 | Rotavirus |
| 1533 | 6 month | M | Caucasian | IL2RG (c.858 Splice Site) | 8898* | 214 | 750 | 1822 | 214 | 0 | 45 | PCP, rotavirus |
| 1535 | 2 month | F | Hispanic | IL-7R (c.212T>C + c.539A > C) | 111 | 95 | 0 | 340 | 308 | 0 | 1 | None (NBS) |
| 1591 | 2 month | M | Hispanic | IL2RG (c.822_840del19) | 0 | 0 | 0 | 1096 | 45 | 0 | 0 | None (NBS) |
| Boost transplants | ||||||||||||
| 1056† | 6 yr | M | Caucasian | IL2RG (c.31T > A) | 334 | 188 | 127 | 103 | 28 | 43 | NA | Severe molluscum |
| 1057† | 6 yr | M | Caucasian | IL2RG (c.31T > A) | 437 | 90 | 325 | 106 | 17 | 51 | NA | Severe molluscum |
| 237 | 15 yr | F | Navajo | Artemis (c.597C > A) | 451 | 209 | 242 | 0 | 99 | 34 | NA | FTT |
UPN, unique patient number; PHA, phytohemagglutinin; TME, transplacental maternal engraftment; RAG, recombinase-activating gene; PCP, Pneumocystis jiroveci pneumonia; NBS, newborn screen; FTT, failure to thrive; NA, not applicable.
T cells primarily of maternal origin.
UPN 1056 and 1057 were identical twins.
Table 2.
HCT features and clinical outcomes
| UPN | Conditioning | Donor | HSC source | CD34 dose (× 106/kg) | CD3 dose (× 104/kg) | Additional cells | Clinical outcome | F/U (years) |
|---|---|---|---|---|---|---|---|---|
| Newly diagnosed | ||||||||
| 1509 | G-CSF + Plerixafor (240 mcg/kg × 1) |
6/10 Mother | PBSC (CD34 Selected) | 30 | 3 | None | aGVHD, now alive and well | 3.7 |
| 1533 | G-CSF + Plerixafor (240 mcg/kg × 1) + Fludarabine (1 mg/kg × 3) |
5/10 Mother | PBSC (CD34 Selected) | 20 | 0.8 | Full HCT D+133 | alive and well | 2.5 |
| 1535 | G-CSF + Plerixafor (240 mcg/kg × 1) |
5/10 Mother | PBSC (CD34 Selected) | 40 | 0.1 | None | alive and well | 2.5 |
| 1591 | G-CSF + Plerixafor (240 mcg/kg × 1) + rATG (8 mg/kg) |
12/12 URD | BM (RBC Depleted) | 5.9 | 7200 | Boost D+48 | alive and well | 1.8 |
| Boost Transplants | ||||||||
| 1056 | G-CSF + Plerixafor (240 mcg/kg × 1) |
5/8 Mother | PBSC (CD34 Selected) | 16.4 | 0.2 | None | alive and improved | 2.3 |
| 1057 | G-CSF + Plerixafor (240 mcg/kg × 1) |
5/8 Mother | PBSC (CD34 Selected) | 16.4 | 0.2 | None | alive and improved | 2.3 |
| 237 | G-CSF + Plerixafor (240 mcg/kg × 1) |
6/8 Brother | PBSC (CD34 Selected) | 10.1 | 0.5 | None | alive and improved | 2.0 |
UPN, unique patient number; HSC, hematopoietic stem cells; F/U, follow-up; G-CSF, granulocyte-colony-stimulating factor; PBSC, peripheral blood stem cells; BM, bone marrow; RBC, red blood cell; URD, unrelated donor; DLI, donor lymphocyte infusion; aGVHD, acute graft-versus-host disease.
Adverse events to plerixafor and G-CSF
No patient developed clinical signs of bone pain, injection site erythema, abdominal bloating, nausea, diarrhea, or perioral paresthesias following plerixafor and G-CSF. One patient (UPN 1533) had unusual post-HCT features that were not felt to be a side effect of G-CSF or plerixafor, including neutropenia, eosinophilia, eosinophilic enteritis, and failure to thrive. This patient had incomplete elimination of maternal double-negative CD3+ cells on the day of HCT (CD3 cells = 3049 × 106/L; CD4 = 79 × 106/L; CD8 = 277 × 106/L), which persisted for the next four months. The maternal donor did not have detectable circulating CD4/CD8 double-negative T cells. We concluded that the patient’s double-negative T cells were derived from the maternal population present prior to HCT and responsible for his unusual symptoms and therefore performed a second HCT from a new matched URD with conditioning intended to eliminate this population of cells (see below).
Myelomobilization kinetics with plerixafor and G-CSF
Most patients had low absolute WBC counts prior to conditioning, with a median of 3.2 × 109/L (range, 1.4–4.8 × 109/L), as shown in Fig. 1a. After treatment with G-CSF, the WBC count rose to a median of 19.5 × 109/L by the day of HCT (range, 3.8–34.6 × 109/L) and then promptly began to fall after the G-CSF was stopped; however, overall responses to G-CSF were variable. Interestingly, the percentage of CD34+ cells in the peripheral blood rose minimally in the four patients with newly diagnosed SCID, from a median baseline of 0.52% (range, 0.34–1.14%) of PBMCs to a median peak of 1.01% (range, 0.89–1.62%) at the time of HCT, with two patients showing a fall in peripheral blood CD 34% between 9 h post-plerixafor and time of HCT (Fig. 1b). Data were not available for the three boost patients. Furthermore, mobilization of HSCs (Lin−CD34+CD38−CD90+CD45RA− cells) following plerixafor demonstrated a median increase of 3.9-fold (range, 2.55–4.31) from baseline to time of HCT, again with two patients showing a fall in circulating HSCs between 9 h post-plerixafor and time of HCT (Fig. 1c).
Fig. 1.
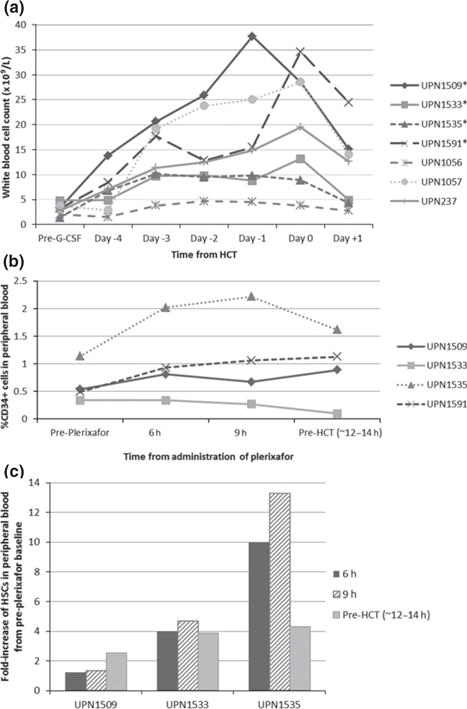
(a) WBC following G-CSF and plerixafor *Infants at time of treatment with G-CSF and plerixafor. (b) CD34+ cell% following G-CSF and plerixafor. (c) HSC (Lin−CD34+CD38−CD90+CD45RA−) fold increase following plerixafor. (b, c) Note: Analysis not performed in all patients.
T-cell engraftment and reconstitution
As expected, the four newly diagnosed patients engrafted with donor T cells, and the three boost patients who started with adequate donor T-cell percentages had at least temporary improvement in their T-cell numbers and function (Table 3). Engraftment of donor T cells was accompanied by Grade II skin GVHD in only one patient (UPN 1509), who also received the highest dose of haploidentical T cells. The patient who received URD BM had evidence of donor cells by STRs (18% of CD 3% cells), but very low T-cell numbers for a prolonged period post-HCT, possibly due to the use of pre-HCT rATG. On Day +48, he received a boost of donor BM (4 × 108 total nucleated cells/kg) without GVHD prophylaxis and subsequently had a rise in his T-cell numbers and function (Table 3). Over time, TREC numbers improved and T-cell receptor V-beta diversity by spectratyping demonstrated less absent/oligoclonal peaks in the analyzed patients (Table 4).
Table 3.
Immunologic outcomes of engrafted patients
| UPN | Time to reach (months)
|
At last follow-up (or prior to second HCT*)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| CD4 > 200 × 106/L | CD4 > 400 × 106/L | PHA >50% | CD4 (× 106/L) | CD4/CD45RA+ (%) | PHA (%) | CD19 (× 106/L) | Donor CD19 (%) | Donor CD14/15 (%) | |
| Newly diagnosed | |||||||||
| 1509 | 2.8 | NR | 5.8 | 65 | 6 | 81 | 0 | 0 | 0 |
| 1533 | 2.4 | 2.4 | NR | 404* | 2* | 56* | 51* | 1* | 0* |
| 1535 | 1 | 3.9 | 2 | 1114 | 51 | 100 | 845 | 0 | 0 |
| 1591 | 4.8 | 5.8 | 4.8 | 1535 | 96 | 100 | 205 | 0 | 0 |
| Boost transplants | |||||||||
| 1056 | 1 | 1 | 2 | 179 | 0 | 100 | 96 | 0 | 0 |
| 1057 | 8.5 | NR | NA | 347 | 56 | 100 | 143 | 1 | 0 |
| 237 | NA | NR | 3.8 | 227 | 21 | 100 | 0 | 0 | 0 |
UPN, unique patient number; PHA, phytohemagglutinin.
Table 4.
Extended immunologic outcomes over time
| UPN | Test | Baseline | +100 Days | +6 months | +1 yr | +2 yr |
|---|---|---|---|---|---|---|
| 1509 | TRECs (#/μL) | 0 | 0 | 0 | 5 | 16 |
| TCR V-beta* | ND | ND | 1/4/19 | 4/6/14 | 1/8/15 | |
| 1535 | TRECs (#/μL) | 0 | 0 | 0 | 83 | 66 |
| TCR V-beta* | ND | 2/5/17 | 1/2/21 | 0/1/23 | 2/0/22 | |
| 1591 | TRECs (#/μL) | 0 | 0 | 0 | 58 | ND |
| TCR V-beta* | ND | 4/3/17 | 3/1/20 | 0/3/21 | 2/1/21 |
TRECs, T-cell receptor excision circles; TCR V-beta, T-cell receptor V-beta spectratyping.
Reported as # of monoclonal or absent/oligoclonal/polyclonal peaks (with normal ≥20/24 polyclonal peaks); ND, Not done; bold = normal.
Myeloid and B-cell engraftment and B-cell reconstitution
Significant myeloid engraftment was not seen in any patient (Table 3). Two patients who had BM aspirations for other indications had 5% (UPN1509) and 15% (UPN 1533) of the BM CD34+ selected cells of donor origin, but the purities of the bead-selected cells were low and these values may have therefore been falsely high due to contamination with donor T cells.
Although no detectable donor myeloid cells were present in the blood, it was still possible that a very small number of engrafted donor HSCs in the BM might be sufficient to produce donor B cells. Unfortunately, in all four patients with B− SCID, including two who had received plerixafor, no B cells were detectable in the patient’s blood post-HCT. In the five patients with B+ SCID, two had very low numbers of donor B cells detected; however, this was at the limit of detection of the assay.
All surviving patients continue to require routine immunoglobulin replacement, except one patient who received a conditioned second HCT (UPN1533) and one patient with IL-7R deficiency (UPN1535), both of whom have made specific antibody responses to vaccines.
Clinical outcomes
All seven patients are alive with follow-up time of 1.8–3.7 years. One patient required a conditioned second transplant for treatment of unusual eosinophilia and BM hypocellularity. This with the patient with unusual maternal double-negative T cells (UPN1533), who received half-dose busulfan (1.1 mg/kg/dose every 12 h for eight doses, targeted to a CSS of 300 ng/mL), fludarabine (1.33 mg/kg/day × four days), and alemtuzumab (0.5 mg/kg/day × three days), followed by unmodified BM from a 9 of 10 (A- antigen) mismatched URD. He developed Grade III acute GVHD, which was eventually controlled with corticosteroids and MSCs. He achieved 100% engraftment from his second donor and is now thriving with normal T- and B-cell function.
Discussion
In our prospective pilot trial, the use of G-CSF plus plerixafor in children as the sole-conditioning agent prior to allogeneic HCT in children with graft-permissive SCID did not facilitate donor myeloid and B-cell engraftment. This is in contrast to the mouse model where a dose of 5 mg/kg of plerixafor followed by infusion of 16 × 108 total donor BM cells/kg 2 h later resulted in increased donor cell engraftment (from ~1% to ~5%) (4). One possible explanation is that there may be preferential rebinding of host HSCs to unoccupied niches. Alternatively, it may be that the dose of plerixafor utilized was too low for the children enrolled on this trial or that the mouse biology does not fully translate to humans. We administered the FDA-approved dose for stem cell mobilization prior to collection via apheresis in adults, as pediatric-specific pharmacokinetics has not been published. However, with this dosing, we saw relatively poor CD34+ cell and Lin−CD34+CD38−CD90+CD45RA−HSC mobilization into the peripheral blood, as compared to what is normally seen in patients following chemotherapy and G-CSF (22). Preliminary data in mouse models suggest that much higher doses of plerixafor improve HSC engraftment (A. Czechowicz, unpublished data). Given the absence of apparent side effects at this dosage, a plerixafor dose-escalation trial would be a potential next step. Another possibility would be repeated dosages, either in multiple sequential days (up to four daily doses have been used for HSC mobilization before apheresis (23) prior to a single HSC infusion, or through repetitive weekly injections of plerixafor followed by rein-fusions of additional HSCs, as shown in the mouse model to improve donor engraftment (4). Alternatively, it is possible that the use of G-CSF in combination with the plerixafor directly stimulated HSC proliferation (24), so that any vacated niches were soon filled by a new host HSC, thereby preventing donor HSCs from attaching. Therefore, further attempt to utilize plerixafor for pre-HCT “myelomobilization” should likely be undertaken without concomitant G-CSF.
Ultimately, even an optimal dose of plerixafor in children with SCID may be insufficient to open enough BM niches to allow for adequate donor HSC engraftment to produce enough donor B cells to support production of protective antibodies. Future efforts utilizing plerixafor to enhance donor HSC engraftment may need to combine it with low doses of chemotherapy, which might enhance the effects of the chemotherapy by liberating the host HSCs from the anti-apoptotic effects of the BM stroma. Preliminary reports support the feasibility of this approach in patients with acute leukemia (25). Ideally, non-chemotherapeutic methods of HSC elimination may eventually be available, such as anti-CD45 (26) or anti-c-kit (27) monoclonal antibodies, and their efficacy may also be potentially enhanced via prior treatment with plerixafor.
In conclusion, “myelomobilization” of HSCs with plerixafor and G-CSF at these doses and timing is insufficient to create enough marrow niches to allow sufficient donor HSC engraftment for production of donor myeloid and B cells. However, these initial data demonstrate that the administration of one standard adult dose of plerixafor to young infants is safe, and should serve as the basis of future trials that combine plerixafor with other agents. Given the rarity of SCID, efforts to perform these trials via a multicenter consortium, such as the Primary Immune Deficiency Treatment Consortium (6), may generate more rapid results that improve the outcomes of these patients.
Acknowledgments
This publication was supported by NIH/NCRR UCSF-CTSI Grant Number UL1 RR024131 and the UCSF Jeffrey Modell Foundation Diagnostic Center for Primary Immunodeficiencies. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Abbreviations
- ATG
anti-thymocyte globulin
- BM
bone marrow
- CSS
concentration steady state
- EFS
event-free survival
- FDA
Food and Drug Administration
- G-CSF
granulocyte-colony-stimulating factor
- GVHD
graft-versus-host disease
- HCT
hematopoietic cell transplantation
- HSC
hematopoietic stem cells
- IND
investigational new drug
- ISH
isohemagglutinins
- MSCs
mesenchymal stromal cells
- OS
overall survival
- PBMC
peripheral blood mononuclear cells
- PBSC
peripheral blood stem cells
- PCP
Pneumocystis jiroveci pneumonia
- PHA
phytohemagglutinin
- SCID
severe combined immunodeficiency
- SDF-1
stromal cell-derived factor-1
- STR
short tandem repeat
- TME
transplacental maternal engraftment
- TREC
T-cell receptor excision circles
- UCSF
University of California San Francisco
- UPN
unique patient number
- URD
unrelated donor
- WBC
white blood cell
Footnotes
Authors’ contributions
All of the authors have made significant contributions to this study: Christopher Dvorak and Morton Cowan designed the trial, with input from Jennifer Puck and Biljana Horn. Jennifer Puck performed patient genotyping and TREC and spectratyping analysis. Agnieszka Czechowicz, Judith Shizuru, and Rose Ko performed and analyzed laboratory data. Christopher Dvorak collected and analyzed clinical data, and wrote the manuscript with input from all of the authors.
Conflict of interest
All the authors disclose they have no conflict of interest.
References
- 1.Dvorak C, Cowan M. Hematopoietic stem cell transplantation for primary immunodeficiency disease. Bone Marrow Transplant. 2008;41:119–126. doi: 10.1038/sj.bmt.1705890. [DOI] [PubMed] [Google Scholar]
- 2.Dvorak C, Hung G, Horn B, Dunn E, Oon C, Cowan M. Megadose CD34(+) cell grafts improve recovery of T cell engraftment but not B cell immunity in patients with severe combined immunodeficiency disease undergoing haplocompatible nonmyeloablative transplantation. Biol Blood Marrow Transplant. 2008;14:1125–1133. doi: 10.1016/j.bbmt.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Dvorak C, Cowan M. Radiosensitive severe combined immunodeficiency disease. Immunol Allergy Clin North Am. 2010;30:125–142. doi: 10.1016/j.iac.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Larochelle A, Fricker S, Bridger G, Dunbar CE, Abkowitz JL. Mobilization as a preparative regimen for hematopoietic stem cell transplantation. Blood. 2006;107:3764–3771. doi: 10.1182/blood-2005-09-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffith L, Cowan M, Notarangelo L, et al. Improving cellular therapy for primary immune deficiency diseases: Recognition, diagnosis, and management. J Allergy Clin Immunol. 2009;124:1152–1160. doi: 10.1016/j.jaci.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dvorak C, Cowan M, Logan B, et al. The natural history of children with severe combined immunodeficiency: Baseline features of the first fifty patients of the Primary Immune Deficiency Treatment Consortium prospective study 6901. J Clin Immunol. 2013;33:1156–1164. doi: 10.1007/s10875-013-9917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kline R, Stiehm E, Cowan M. Bone marrow ‘boosts’ following T cell-depleted haploidentical bone marrow transplantation. Bone Marrow Transplant. 1996;17:543–548. [PubMed] [Google Scholar]
- 8.Booth C, Ribeil J, Audat F, et al. CD34 stem cell top-ups without conditioning after initial haematopoietic stem cell transplantation for correction of incomplete haematopoietic and immunological recovery in severe congenital immunodeficiencies. Br J Haematol. 2006;135:533–537. doi: 10.1111/j.1365-2141.2006.06333.x. [DOI] [PubMed] [Google Scholar]
- 9.O’Marcaigh AS, Desantes K, Hu D, et al. Bone marrow transplantation for T-B- severe combined immunodeficiency disease in Athabascan-speaking Native Americans. Bone Marrow Transplant. 2001;27:703–709. doi: 10.1038/sj.bmt.1702831. [DOI] [PubMed] [Google Scholar]
- 10.Ozyurek E, Cowan M, Koerper M, Baxter-Lowe L, Dvorak C, Horn B. Outcomes of children with unstable mixed chimerism after reduced intensity hematopoietic stem cell transplantation for non-malignant disorders. Bone Marrow Transplant. 2008;42:83–91. doi: 10.1038/bmt.2008.89. [DOI] [PubMed] [Google Scholar]
- 11.Scharf S, Smith A, Hansen J, McFarland C, Erlich H. Quantitative determination of bone marrow transplant engraftment using fluorescent polymerase chain reaction primers for human identity markers. Blood. 1995;85:1954–1963. [PubMed] [Google Scholar]
- 12.Dvorak C, Gilman A, Horn B, et al. Haploidentical related-donor hematopoietic cell transplantation in children using megadoses of CliniMACs-selected CD34(+) cells and a fixed CD3(+) dose. Bone Marrow Transplant. 2013;48:508–513. doi: 10.1038/bmt.2012.186. [DOI] [PubMed] [Google Scholar]
- 13.Flomenberg N, Devine SM, Dipersio JF, et al. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood. 2005;106:1867–1874. doi: 10.1182/blood-2005-02-0468. [DOI] [PubMed] [Google Scholar]
- 14.Pang WW, Price EA, Sahoo D, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comans-Bitter W, DE Groot R, van den Beemd R, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997;130:388–393. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 16.Huenecke S, Behl M, Fadler C, et al. Age-matched lymphocyte subpopulation reference values in childhood and adolescence: Application of exponential regression analysis. Eur J Haematol. 2008;80:532–539. doi: 10.1111/j.1600-0609.2008.01052.x. [DOI] [PubMed] [Google Scholar]
- 17.Sarzotti M, Patel D, Li X, et al. T cell repertoire development in humans with SCID after nonablative allogeneic marrow transplantation. J Immunol. 2003;170:2711–2718. doi: 10.4049/jimmunol.170.5.2711. [DOI] [PubMed] [Google Scholar]
- 18.Przepiorka D, Weisdorf D, Martin P, et al. Consensus conference on acute GVHD grading. Bone Marrow Transplant. 1995;15:825–828. [PubMed] [Google Scholar]
- 19.Filipovich A, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11:945–956. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Puck J. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: The winner is T-cell receptor excision circles. J Allergy Clin Immunol. 2012;129:607–616. doi: 10.1016/j.jaci.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bakare N, Menschik D, Tiernan R, Hua W, Martin D. Severe combined immunodeficiency (SCID) and rotavirus vaccination: Reports to the Vaccine Adverse Events Reporting System (VAERS) Vaccine. 2010;28:6609–6612. doi: 10.1016/j.vaccine.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 22.Van Epps D, Bender J, Lee W, et al. Harvesting, characterization, and culture of CD34 + cells from human bone marrow, peripheral blood, and cord blood. Blood Cells. 1994;20:411–423. [PubMed] [Google Scholar]
- 23.Dipersio J, Micallef I, Stiff P, et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27:4767–4773. doi: 10.1200/JCO.2008.20.7209. [DOI] [PubMed] [Google Scholar]
- 24.Gibbs KJ, Gilbert P, Sachs K, et al. Single-cell phospho-specific flow cytometric analysis demonstrates biochemical and functional heterogeneity in human hematopoietic stem and progenitor compartments. Blood. 2011;117:4226–4233. doi: 10.1182/blood-2010-07-298232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uy G, Rettig M, Motabi I, et al. A phase 1/2 study of chemo-sensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–3924. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straathof K, Rao K, Eyrich M, et al. Haemopoietic stem-cell transplantation with antibody-based minimal-intensity conditioning: A phase 1/2 study. Lancet. 2009;374:912–920. doi: 10.1016/S0140-6736(09)60945-4. [DOI] [PubMed] [Google Scholar]
- 27.Czechowicz A, Kraft D, Weissman I, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science. 2007;318:1296–1299. doi: 10.1126/science.1149726. [DOI] [PMC free article] [PubMed] [Google Scholar]