

Abstract
Genomic studies in acute myeloid leukemias (AML) have identified mutations which drive altered DNA methylation, including TET2 and IDH2. Here we show that models of AMLs resulting from TET2 or IDH2 mutations combined with FLT3ITD mutations are sensitive to 5-Azacytidine or to the IDH2 inhibitor AG-221, respectively. 5-Azacytidine and AG-221 treatment induced an attenuation of aberrant DNA methylation and transcriptional output, and resulted in a reduction in leukemic blasts consistent with anti-leukemic activity. These therapeutic benefits were associated with restoration of leukemic cell differentiation, and the normalization of hematopoiesis was derived from mutant cells. By contrast, combining AG-221 or 5-Azacytidine with FLT3 inhibition resulted in a reduction in mutant allele burden, progressive recovery of normal hematopoiesis from non-mutant stem-progenitor cells, and reversal of dysregulated DNA methylation and transcriptional output. Together, our studies suggest combined targeting of signaling and epigenetic pathways can increase therapeutic response in AML.
Keywords: leukemia, IDH, targeted therapy, epigenetics, FLT3
Introduction
Gene discovery studies in acute myeloid leukemias (AML), lymphomas, sarcomas, brain tumors, and epithelial tumors have identified recurrent somatic mutations in genes with a role in DNA methylation (1, 2). The methylation pathway is most commonly disrupted in AML by mutations in TET2, IDH1, IDH2, DNMT3A, and WT1 (3–7). TET2 mutations abrogate 5′-methylcytosine hydroxylase activity, which is required for DNA de-methylation, whereas neomorphic IDH1 and IDH2 mutations through 2-hydroxyglutarate (2HG) production, competitively inhibit TET enzymatic activity (8–10). Mutations in this pathway occur in more than 30–35% of AML patients, and correlative studies have shown that these disease alleles are associated with distinct transcriptional, epigenetic, and prognostic signatures (11–14). Taken together, these findings demonstrate an important role for mutations in epigenetic modifiers in AML pathogenesis and risk stratification.
Murine and human studies have shown that mutations in TET2 or in IDH1 or IDH2 are most commonly initiating events in malignant transformation that promote hematopoietic stem cell self-renewal and myeloid expansion (15–17). These data suggest that transformation requires the acquisition of additional somatic mutations, which cooperate with mutations in epigenetic regulators to promote leukemogenesis. Consistent with this hypothesis, we and others have shown that Tet2 loss and Idh1 or Idh2 mutations can cooperate with known-occurring disease alleles, including Flt3ITD, to induce AML in vivo (18–20). The resulting leukemic cells continue to bear evidence of epigenetic reprogramming, including extensive hypermethylation and transcriptional silencing of key target loci, including Gata2, which is required for normal hematopoietic differentiation. A feature of Flt3ITD;Tet2-mutant leukemia is resistance to traditional AML chemotherapy and to FLT3 kinase inhibitors, thus antiproliferative therapy is insufficient to effectively target leukemias marked by mutations that disrupt DNA demethylation (18, 21). As a consequence, in subsets of patients, mutations in these genes are associated with inferior survival (22–24). Towards the goal of developing epigenetic agents, we therefore sought to characterize therapies that can potentially restore the balance of DNA methylation and demethylation alone and in combination with anti-proliferative therapies.
RESULTS
TET2-mutant AML is specifically responsive to DNA hypomethylating agents
Retrospective studies in myelodysplastic syndrome (MDS) have suggested that TET2-mutant patients are more likely to respond to DNA hypomethylating agents (HMA) (25, 26). However, the mechanisms by which 5-Azacytidine (5-Aza) and other HMA induce response in MDS and AML have not been fully delineated. We therefore first sought to assess the therapeutic efficacy of 5-Aza in a transplantable model of TET2-mutant AML. We engrafted CD45.2+ Flt3ITD;Tet2-mutant AML cells and CD45.1+ support marrow into CD45.1+ congenic recipients, and allowed recipient mice to develop AML with engraftment of 80–90% CD45.2+ Flt3ITD;Tet2-mutant cells and expansion of leukemic blasts in the peripheral blood, bone marrow (BM), and spleen. We then treated mice with vehicle or 5-Aza using a dose and schedule (5 mg/kg days 1–5 of 21 day cycles) similar to the clinical context. This allowed us to assess the impact of 5-Aza on Flt3ITD;Tet2-mutant AML cells relative to wild-type cells in vivo. Four cycles of 5-Aza therapy resulted in a marked therapeutic response, with near-elimination of leukemic blasts in the bone marrow and spleen, and restoration of megakaryocytes in the BM and germinal centers in the spleen (Fig. 1A). We observed normalization of peripheral blood cell counts and splenomegaly with 5-Aza therapy, with a significant reduction in the total white blood cell count (WBC) and spleen weight and an increase in platelet count and hematocrit (Fig. 1B–D). Morphologic examination and flow cytometric analysis of the peripheral blood confirmed that 5-Aza therapy reduced the proportion of morphologic blasts (Fig. 1C) and significantly reduced the proportion of cKit+Mac1− immature cells in the peripheral blood (Fig. 1D and Supplementary Fig. 1A).
Figure 1. Response of Tet2−/− Flt3ITD leukemia model to 5-Aza therapy.
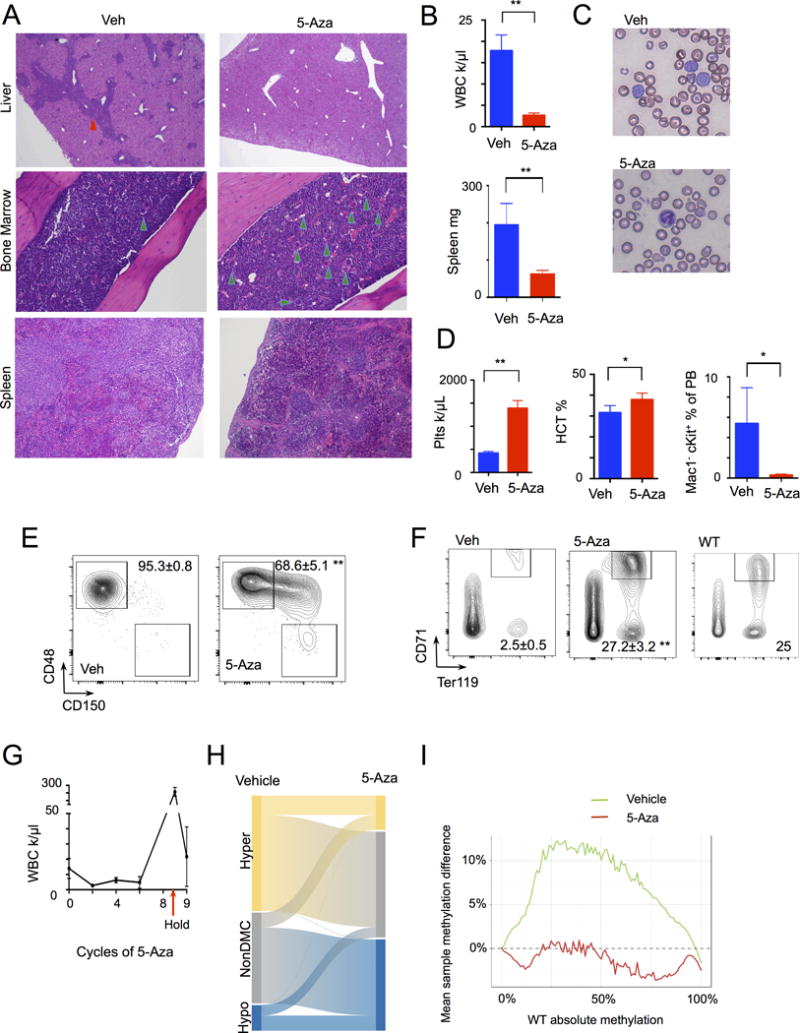
Tet2−/− Flt3ITD leukemia bone marrow transplant model mice were treated with vehicle (n=5) or 5-Aza (n=5) therapy. A, Histology of treatment response in liver, bone marrow, and spleen, infiltrating leukemic cells (red arrowhead), megakaryocytes (green arrowheads). B–F, Response to treatment in (B) WBC and spleen weight, (C) peripheral blood (PB) smears, (D) platelet, hematocrit, and PB Mac1−cKit+ population, (E) bone marrow stem cell differentiation with percentage of multipotent progenitors (MPP, CD48+CD150− gated on LSK, lin−Sca+cKit+), and (F) bone marrow erythroid differentiation with percentage of CD71+Ter119+ erythroid progenitors, WT- representative wild-type sample. Graphs and numbers indicate mean ± sem. G, WBC of mice following treatment with cycles of 5-Aza (n=5), holding of therapy (arrow), and resumption of 5-Aza. H, Graph of ERRBS analysis comparing vehicle (n=4) and 5-Aza treatment (n=4), indicating proportion of hypo-, unchanged, and hyper-methylated differentiated methylated cytosines (DMCs). I, Graph of overall genomic methylation proportion comparing WT LSKs to Tet2−/− Flt3ITD vehicle (green) and 5-Aza (red) treated LSKs. **p<.01, *p<.05 by t-test.
5-Azacytidine reverses aberrant stem-progenitor expansion and attenuates DNA hypermethylation in TET2-mutant AML
The lineage−Sca+cKit+ (LSK) population in Flt3ITD;Tet2-mutant AML is comprised of monomorphic CD48+CD150− Multipotent Progenitors (MPPs), which serve as leukemia stem cells capable of propagating the disease in serial transplantation studies (18). 5-Aza therapy tended to normalize the BM LSK compartment, with a significant reduction in CD48+CD150− MPPs and partial restoration of the long-term hematopoietic stem cell (LT-HSC, CD48− CD150+) compartment over time (Fig. 1E). TET2 has an important role in erythroid differentiation and Tet2 loss or mutation leads to a reduction in erythroid precursors (27, 28), with near-complete loss of megakaryocyte and erythroid progenitors (MEPs) in the Flt3ITD;Tet2-mutant model (18). 5-Aza treatment led to a reversal of anemia and thrombocytopenia and helped to restore CD71+Ter119+ erythroid precursors and MEPs (Fig. 1F and Supplementary Fig. S1B). Based on these data, we hypothesized that 5-Aza therapy might induce reversible differentiation of Flt3ITD;Tet2-mutant AML cells, such that therapeutic efficacy is only maintained with continued treatment. Prolonged therapy (8 cycles, 24wks) with 5-Aza induced a durable response, with a sustained reduction in the WBC (Fig. 1G). However, following suspension of therapy, we observed recurrent AML in all treated mice, with an increase in WBC 4wks after the last treatment dose and expansion of blasts in the blood that could, however, again be reduced with resumption of 5-Aza therapy (Fig. 1G).
As we previously showed that the leukemic phenotype induced by Tet2 loss and Flt3ITD is driven by aberrant hypermethylation, we next examined the impact of 5-Aza on the DNA methylation status of CD45.2+ Flt3ITD;Tet2-mutant LSK cells from mice treated with vehicle or 5-Aza, using enhanced reduced representation bilsulfite sequencing (ERRBS) (29). These studies showed that 5-Aza therapy reversed many of the sites with aberrant DNA hypermethylation induced by Tet2 and Flt3ITD in leukemia derived stem-progenitor cells (Fig. 1H, 1I, and Supplementary Fig. S1C–E). Taken together, these data show that 5-Aza restored a significant component of the epigenetic patterning of normal LSK cells, although we observed additional, 5-Aza induced hypomethylation of cytosine residues beyond those affected by the Tet2 and Flt3itd epigenetic program (Fig. 1H, I). These data suggest that DNA hypomethylating agents promote the differentiation of Tet2-mutant AML cells while reversibly restoring normal DNA methylation patterns.
Efficacy of AG-221, a specific IDH2 inhibitor, in IDH2-mutant AML in vivo
We next investigated the efficacy of targeted epigenetic therapies in IDH2-mutant AML. Preclinical studies in cell lines and patient samples have shown that small molecule, targeted inhibition of mutant IDH1 and IDH2 can block the production of the oncometabolite 2HG and thereby induce in vitro differentiation (30, 31). Despite these important insights, the impact of small molecule inhibition of mutant IDH enzymes in vivo, alone and in combination with other targeted AML therapies have not been characterized in detail. We crossed a conditional mouse model that expresses the Idh2R140Q AML disease allele from the endogenous locus (Fig. 2A and Supplementary Fig. S2A) to mice with the inducible Mx1-Cre allele and the Flt3ITD knock-in allele. This allowed us to generate Mx1-Cre Idh2R140QFlt3ITD mice in which both mutant disease alleles are expressed from the endogenous loci. Consistent with previous retroviral and transgenic models (19, 20), Mx1-Cre Idh2R140QFlt3ITD mice developed AML. The disease was characterized by expansion of blasts in the blood, BM, and spleen, increased levels of 2HG in the serum, leukocytosis, and splenomegaly, (Fig. 2B and Supplementary Fig. S2B–C), similar to the phenotype of Flt3ITD;Tet2-mutant AML model. The leukemia was characterized by expansion of cKit+ cells in the peripheral blood, reduction in erythroid precursors and MEPs, and replacement of the LSK compartment by CD48+CD150− MPPs (Fig. 2C and Supplementary Fig. S2D). Importantly Idh2R140QFlt3ITD murine leukemias exhibited a similar hypermethylation phenotype to human IDH2-mutant AML, with similar alterations in locus-specific DNA methylation and gene expression (Fig. 2D and Supplementary Fig. S2E).
Figure 2. Idh2R140QFlt3ITD mouse model of leukemia.
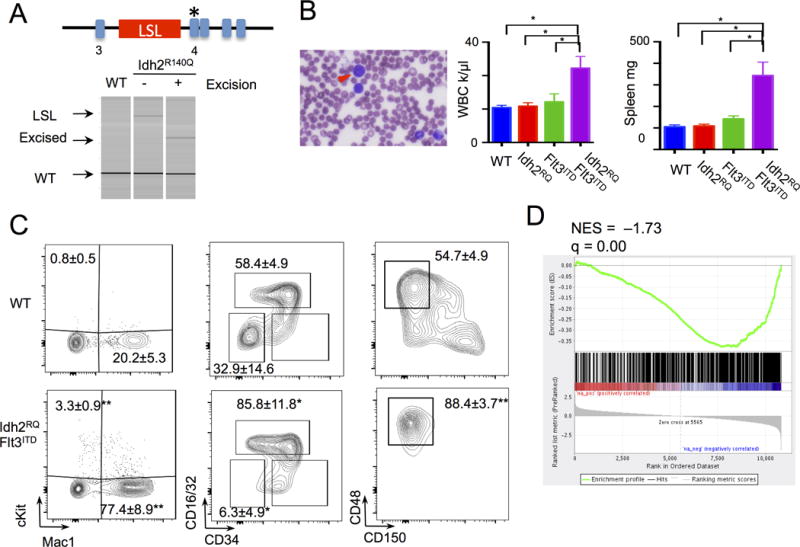
A, Targeting of endogenous Idh2 locus with Idh2R140Q mutation and the lox-Stop-lox (LSL) cassette. Genotyping PCR demonstrates Idh2WT, LSL Idh2R140Q, and excised alleles. B, Idh2R140Q Flt3ITD peripheral blood (PB) histology with blast cells (arrowhead), WBC (n=6 to 9) at 4–5 months, and spleen weight (n=4 to 6). Graph of mean ± sem. C, Immunophenotyping of PB Mac1 cKit (n=9 WT and Idh2R140QFlt3ITD), and bone marrow Myeloid progenitors (gated on lin−Sca1−cKit+) and stem cells (gated on lin−Sca1+cKit+) populations (n=3 WT, n=5 Idh2R140QFlt3ITD). Numbers indicate mean ± sem cKit+, Mac1+, GMP (CD34+CD16/32+), MEP (CD34−CD16/32−), and MPP (CD150−CD48+) percentages. Statistical comparisons to WT. D, GSEA analysis of differential expressed genes from RNA-seq of Idh2R140QFlt3ITD LSK (n=3) compared to WT LSK (n=3) against a human IDH2-mutant AML hypermethylated gene signature. **p<.01, *p<.05 by t-test.
We next used this murine model of IDH2-mutant AML to test the impact of small molecule IDH2 inhibition. IDH inhibition dose-dependently inhibited the ability of Idh2R140QFlt3ITD mutant AML cells to serially replate in vitro at doses that correlated with reductions in cellular 2HG levels and was dependent on the presence of the IDH2 mutation (Supplementary Fig. S2F–H). We next engrafted CD45.2+ Idh2R140QFlt3ITD mutant AML cells and CD45.1+ support marrow into CD45.1+ recipient mice and assessed the in vivo efficacy of AG-221, the first small molecule inhibitor of IDH2 in AML clinical trials (32). AG-221 therapy lowered serum 2HG levels in vivo, consistent with on-target inhibition of neomorphic IDH2 mutant enzyme function (Fig. 3A). It reduced the proportion of cKit+ cells in the peripheral blood, increased the proportion of Granulocyte Macrophage Progenitors (GMPs), but had variable effects on blood counts and the bone marrow (Fig. 3A and Supplementary Fig. S3A–E). Treatment induced a trend towards a decrease in CD48+CD150− leukemic stem cells (Supplementary Fig. S3F). Consistent with the flow cytometric data, we saw evidence of maturing cells and partial restoration of splenic germinal centers with AG-221 therapy (Fig. 3B).
Figure 3. Response of Idh2R140Q Flt3ITD leukemia model to AG-221 therapy.
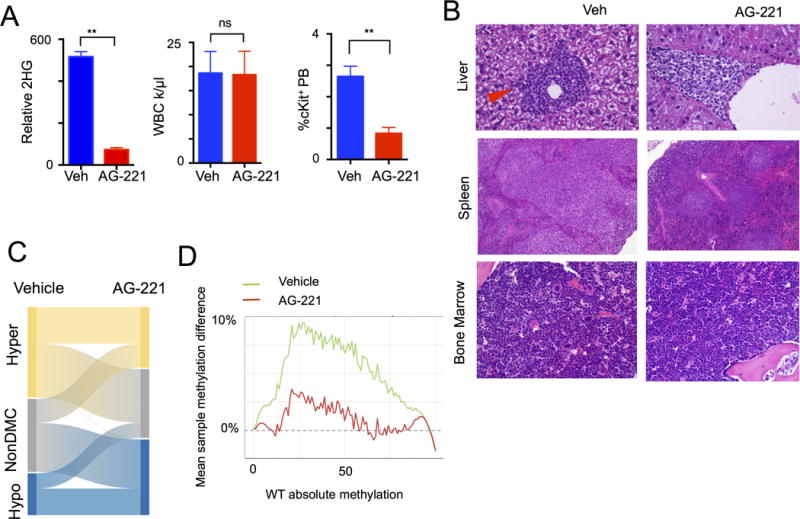
A, Idh2R140Q Flt3ITD transplant leukemia model following in vivo treatment trial with vehicle (n=7) or AG-221 (n=6) and serum 2-hydroxyglutarate (2HG) levels, WBC and peripheral blood (PB) cKit+ frequencies. Graphs of mean ± sem B, Representative histology of liver, spleen, and bone marrow after AG-221 treatment. (arrowhead indicates infiltrating leukemic cells). C, Graph of ERRBS analysis comparing vehicle (n=4) and AG-221 treatment (n=4), indicating proportion of hypo-, unchanged, and hyper-methylated differentiated methylated cytosines (DMCs). D, Graph of overall genomic methylation proportion comparing WT stem-progenitor cells lineage− Sca+ cKit+ (LSKs) to Idh2R140Q Flt3ITD vehicle (green line) and AG-221 (red line) treated LSKs. **p<.01, *p<.05 by t-test.
We next assessed the impact of small molecule IDH2 inhibition on DNA methylation in vivo by performing ERRBS on purified CD45.2+ Idh2R140QFlt3ITD LSK cells treated with 6 weeks of AG-221 or with vehicle. AG-221 therapy induced demethylation of a subset of aberrantly hypermethylated CpGs in leukemia derived cells (Fig. 3C and Supplementary Fig. S4A–C), resulting in reversal of hypermethylation towards wild-type stem cell levels (Fig 3D). AG-221 also induced additional regions of hypomethylation and even a small degree of hypermethylation, suggesting that additional epigenetic reprogramming effects may accompany the suppression of mutant IDH2 activity (Fig. 3C–D).
5-Aza and AG-221 induce differentiation of AML cells in vivo
Both 5-Aza and AG-221 therapy significantly reduced the proportion of lineage negative immature cells in the bone marrow of Tet2- or Idh2- mutant AML cells, respectively, consistent with a reduction in the proportion of stem-progenitor cells (Fig. 4A). In line with the impact of these two therapies on AML differentiation, we gated on the BM leukemic cell portion (CD45.2+) and observed that chronic 5-Aza or AG-221 therapy normalized terminal myeloid maturation, with reversal of aberrant monocytic (Mac+Gr1−) skewing associated with TET2 mutation (28, 33) and restoration towards normal neutrophil populations (Mac+Gr1+) (Fig. 4B). The data from these two therapeutic studies suggest that epigenetic therapies can restore differentiation and that therapeutic efficacy is associated with prolonged, reversible differentiation of AML cells as also observed in the in vitro setting (31).
Figure 4. Differentiation response to therapy to 5-Aza and AG-221.
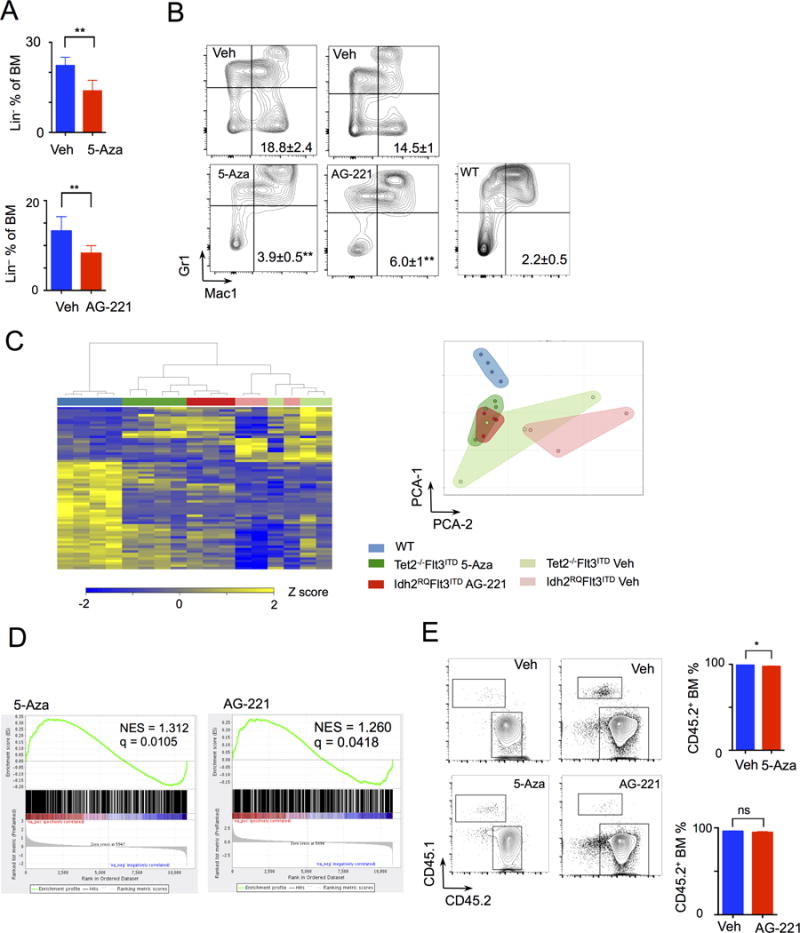
A, Bone marrow (BM) lineage negative fraction in vehicle (n=5) or 5-Aza (n=5) treated Tet2−/− Flt3ITD leukemic model and vehicle or AG-221 (n=6) treated Idh2R140Q Flt3ITD leukemic model. Graphs of mean ± sd B, Leukemic fraction (CD45.2+) gated, immunophenotypic Mac1 Gr1 expression analysis of bone marrow cells from Tet2−/− Flt3ITD and Idh2R140Q Flt3ITD leukemia models in response to treatment and from WT mice (n=6). Numbers of Mac1+Gr1− percentage mean ± sem. Statistical comparisons between vehicle and treated group. C, Heat map of RNA-seq analysis of LSK CD45.2+ leukemia cells from Tet2−/− Flt3ITD (TF) mice treated with vehicle (n=3) or 5-Aza (n=4) and Idh2R140Q Flt3ITD (IF) mice treated with vehicle (n=3) or AG-221 (n=3) therapy, and also of wild-type (WT) (n=4) LSK cells. And graph of principle component analysis (PCA) of RNA seq with clustering of samples. D, GSEA analysis of RNA differential expression between vehicle and 5-Aza treated, and between vehicle and AG-221 treated LSK cells compared to a human IDH2-mutant AML methylated genes signature. E, BM CD45.2+ (leukemic) and CD45.1+ (WT) flow cytometry gating and quantitation from Tet2−/− Flt3ITD and of Idh2R140Q Flt3ITD leukemia models in response to treatment with 5-Aza and AG-221, respectively. Graphs of mean ± sem. **p<.01, *p<.05 by t-test.
We next performed RNA-seq of LSK cells from Idh2R140QFlt3ITD mice treated with AG-221 or vehicle and from Tet2−/−Flt3ITD mice treated with 5-Aza or vehicle compared to control wild-type LSKs. We interrogated the effects of these two therapies on the expression of a set of genes we previously demonstrated are differentially expressed in Flt3ITD;Tet2-mutant AML (Supplementary Table S1). AG-221 therapy in the Idh2R140QFlt3ITD model, and 5-Aza therapy in the Tet2−/−;Flt3ITD AML model significantly attenuated, but did not fully reverse this gene expression signature (Fig. 4C). Unsupervised clustering analysis using all genes with variance in expression (sd >50 percentile) showed that in both cases drug treatment shifted gene expression patterns towards normal stem-progenitor cells, with convergence of the Idh2- and Tet2-mutant transcriptional profiles as illustrated by the almost fully overlapping fields (Fig. 4C). However, the normal stem-progenitor transcriptional pattern is only partially restored by these epigenetic therapies (Fig. 4C). Consistent with these data, we found that therapy with 5-Aza or AG-221 increased the expression of a significant subset of genes that are aberrantly hypermethylated in IDH1 or IDH2 mutant human AML (Fig. 4D). GSEA analysis of gene expression changes revealed significant enrichment for methylation, hematopoietic stem cell, and leukemic target gene signatures with AG-221 and 5-Aza therapy, thus impacting genes important for differentiation (Supplementary Fig. S5A).
We next assessed whether these epigenetic therapies were able to specifically reduce mutant allele burden. Although both therapies were able to reduce the proportion of morphologic leukemic blasts and restore normal hematopoietic differentiation, neither therapy could reduced a major proportion of CD45.2+ cells (Fig. 4E). This is consistent with persistence of the mutant AML clone (as differentiated cells) at the time of therapeutic response and a predominant in vivo differentiation effect. In addition, analysis of Tet2−/−Flt3ITD mice treated with 5-Aza and Idh2R140QFlt3ITD mice treated with AG-221 showed near complete excision of the Tet2 or Idh2 allele without expansion of the residual non-excised clone (Supplementary Fig. S5B–C). We also analyzed mice treated with 5-Aza and AG-221, by CD45.2+ leukemic vs. CD45.1+ wild-type cells, and found that leukemic cells are responsible for changes in differentiation rather than simply wild-type cells expanding to restore normal hematopoiesis (Supplementary Fig. S5D). There was also no change in the expression of wild-type FLT3 with treatment that would account for the change in the leukemic phenotype (Supplementary Fig. S5E) (34).
Combined signaling and epigenetic therapies increases therapeutic response and abrogates the competitive advantage of TET2- or IDH2- mutant AML cells
Given that neither epigenetic therapy alone was sufficient to suppress the malignant clone, we explored whether targeting the FLT3ITD cooperating oncogene might increase therapeutic efficacy. Combination therapy with 5-Aza and the FLT3 inhibitor AC220 in Flt3ITD;Tet2-mutant AML showed significant responses compared to vehicle or monotherapy, with reductions in spleen weight, leukocytosis, BM monocytosis, and immature or blast-like cells (Fig. 5A and Supplementary Fig. S6A–C). We next assessed the impact of combination therapy with AG-221 and AC220 in Idh2R140QFlt3ITD mutant AML. Combined therapy with AG-221 and AC220 was more effective in reducing leukocytosis compared to AG-221 monotherapy (Fig. 5B). Importantly, combined AG-221 and AC220 therapy resulted in a further reduction in leukemic blasts in the liver, spleen, and bone marrow compared to either therapy alone (Fig. 5C and Supplementary Fig. 7A). Moreover, combined therapy with AG-221 and AC220 improved platelet counts, increased the proportion of CD71+Ter119+ erythroid progenitors, and reduced peripheral blood immature cKit+ cells that neither monotherapy could not completely achieve independently (Supplementary Fig. S7B–D). Most importantly, combined 5-Aza+AC220 or AG-221+AC220 therapy altered hematopoietic differentiation and reduced the proportion of CD48+CD150− MPPs compared to monotherapy, consistent with a cooperative effect of combined therapy at reducing the proportion of leukemia stem cells in vivo (Fig. 5D–E and Supplementary Fig. S7E–J).
Figure 5. Combination signaling and epigenetic therapy increases efficacy.
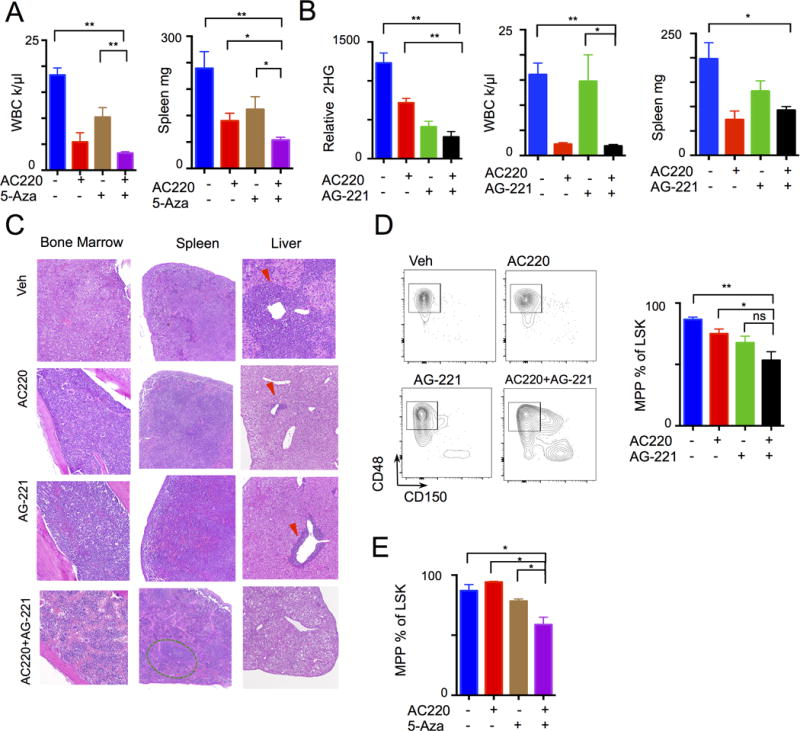
A, Tet2−/− Flt3ITD leukemia treated with Vehicle, AC220, 5-Aza, or combination (n= 5 per group) with resulting WBC and spleen weight. B–C, Idh2R140Q Flt3ITD leukemia treated with Vehicle, AC220, AG-221, or combination (n=5 to 6 per group) with resulting (B) 2HG levels, WBC, spleen weight, and (C) histology of liver, spleen, and bone marrow (arrowhead indicate leukemic infiltration, green oval – splenic follicle). D–E, Differentiation of stem cell compartment (gated on LSK lin−Sca1+cKit+) from (D) Idh2R140Q Flt3ITD and (E)Tet2−/− Flt3ITD treated leukemia models. Graphs of mean ± sem. **p<.01, *p<.05 by t-test.
We next investigated the impact of combined signaling and epigenetic therapy on DNA methylation in AML stem-progenitor cells. Combined therapy with AG-221 and AC220 resulted in more profound reversal of aberrant hypermethylation compared to AG-221 or AC220 monotherapy (Fig. 6A and Supplementary Table S2A). This includes a subset of target loci for which reversal of hypermethylation was only achieved by targeting both IDH2 and FLT3 (Fig. 6B). Similar effects were observed with 5-Aza + AC220 treatment of Tet2−/− Flt3ITD mice (Fig. 6A and Supplementary Fig. S8A). Combined treatment yielded proportionally greater hypomethylation and more significant reversal of aberrantly hypermethylated genes than either drug alone (Supplementary Table S2B). We previously showed that hypermethylation of the Gata2 locus is induced by concurrent mutations in Tet2 and Flt3ITD, and we have shown that Gata2 silencing is required for AML maintenance in Flt3ITD;Tet2 mutant AML in vivo (18). Combination therapy with AG-221+AC220 or 5-Aza+AC220 potently reversed aberrant DNA hypermethylation at the Gata2 locus in Tet2- and Idh-mutant AML stem-progenitor cells (Fig. 6C and Supplementary Fig. S8B), demonstrating the ability of combination therapy to restore more normal epigenetic regulation at key target loci in Tet2- or Idh2-mutant AML. Demethylation was also associated with a trend in increased Gata2 expression (Supplementary Fig. S8C–D). To assess the correlation of methylation with expression more globally, we annotated differentially methylated regions (DMRs) to genes and correlated methylation status with differential RNA expression compared to wild-type. Genes that contain hyper- or hypo-methylated regions are statistically different in RNA expression and as expected, hypermethylation resulted in a relative decrease in gene expression compared to wild-type (Supplementary Fig. S8E). We also ranked the genes by their log-fold change values and then checked to see if genes that contain hyper or hypo DMRs are enriched in the positive or negative leading edge of this rank list according to GSEA. We found that hypermethylation was associated a gene set demonstrating decreased expression (Supplementary Fig. S8F).
Figure 6. Enhanced methylation and leukemic response to combination signaling and epigenetic therapy.
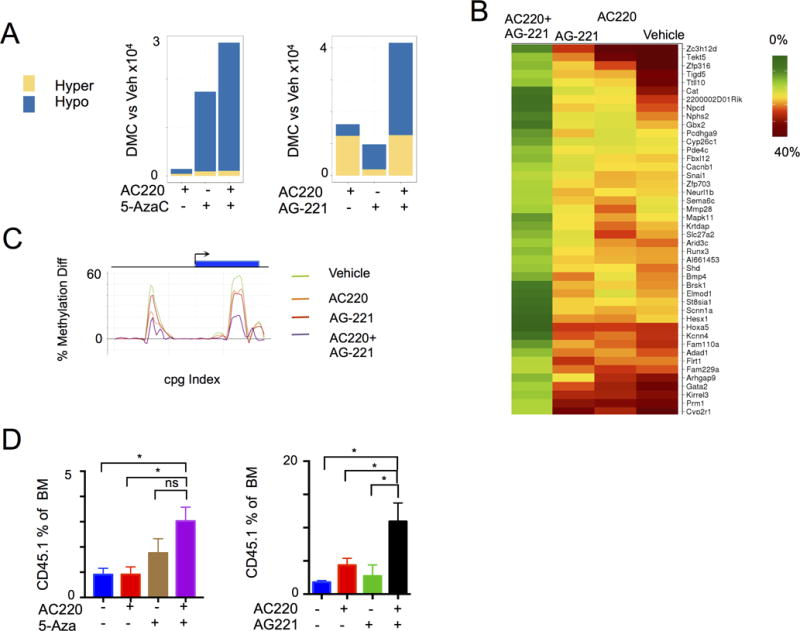
A, Graph of ERRBS analysis demonstrating differential hyper- and hypo- methylated cytosines (DMC) following treatment compared to vehicle in Tet2−/− Flt3ITD and Idh2R140Q Flt3ITD leukemia models (n=3). B, Heat map of genes demonstrating cooperative effects of AC220 and AG-221 therapy on methylation in the coding sequence in Idh2R140Q Flt3ITD leukemia model. C, Gata2 locus methylation status following treatment in Idh2R140Q Flt3ITD leukemia model. CpG indexed relative to Gata2 locus. D, Bone marrow CD45.1+ (normal) fraction following treatment in Tet2−/− Flt3ITD (n=5) and Idh2R140Q Flt3ITD (n=5–6) leukemia model. Graphs of mean ± sem. **p<.01, *p<.05 by t-test.
We next assessed the impact of combination therapy on the relative fitness of AML cells and wild-type hematopoietic cells in vivo. Combination therapy with AG-221+AC220 or 5-Aza+AC220 resulted in a reduction in CD45.2+ leukemic cells and an increase in CD45.1+ normal hematopoietic cells in the BM and spleen to an extent not observed with either agent given as monotherapy (Fig. 6D and Supplementary Fig. S9A–C). In the case of AG-221+AC220 there was also a small expansion of the un-excised Idh2-mutant allele clone in bone marrow progenitor cells further supporting loss of fitness of Idh2;Flt3-mutation clone with combination therapy, but not with AG221 or AC220 monotherapy (the minute fraction of un-excised Tet2 clone was too small to consistently measure) (Supplementary Fig. S9D–E). These data demonstrate that combination epigenetic and signaling therapy can specifically target the mutant clone in vivo, and abrogate the competitive advantage of AML cells and promote the expansion of normal hematopoietic cells.
Discussion
The identification of neomorphic, gain-of-function mutations in IDH1, IDH2, and EZH2 has led to the first clinical trials of molecularly targeted epigenetic therapies for human cancers. As these agents enter the clinic, it is critical to better elucidate their mechanisms of action in different malignant contexts, and to use insights from preclinical and clinical trials to understand how best to use these agents. Our studies show that epigenetic therapies, specifically HMAs in TET2-mutant AML and AG-221 in IDH2-mutant AML, achieve therapeutic efficacy through induction of differentiation and reversal of aberrant methylation patterns in AML stem-progenitor cells, and that this results in progressive differentiation of mutant AML cells at the time of clinical response. Changes with methylation were associated with gene expression changes in stem-progenitor cells, although other studies also suggest the increased importance of this methylation change in programing progeny cells, setting the potential for more dramatic changes in expression in later differentiated cell types (35).
The recent report of effectiveness of the HMA decitabine in AML with its increased potency towards affecting methylation underscores the potential for combination therapies employing epigenetic agents with other anti-leukemic therapies in AML (36, 37). The nature, kinetic, and mechanisms of therapeutic response to these agents are distinct from that observed with cytotoxic therapies and kinase inhibitors; as such it will require thoughtful use of specific criteria to measure and characterize response in the clinical context.
These data suggest the mechanism of action of these agents is to achieve therapeutic response through ongoing, chronic differentiation of mutant leukemia stem-progenitor cells. Previous studies and modeling approaches in patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors (TKI) have shown that mutant stem-progenitor cells can persist for years in the setting of clinically effective targeted therapy (38), of which only a subset of patients achieve sufficient responses to maintain remissions after cessation of TKI (39). Moreover, recent studies have shown that clonal hematopoiesis, driven by mutations in TET2, DNMT3A, IDH2, and other leukemia disease alleles, can persist for years as a pre-leukemic clonal state or in patients with prolonged clinical remission after cytotoxic chemotherapy (16, 17, 40–42). Detailed studies in clinical trial cohorts are needed to delineate the impact of HMAs in TET2-mutant AML and AG-221 in IDH2-mutant AML on mutational burden and clonal structure, and to determine if mutant cells can persist in a clonal, non-leukemic state in the setting of epigenetic therapies which induce differentiation of leukemia cells. Furthermore, although we observe a change in methylation with 5-azacytidine, its effect on RNA biology may also play a role in response (43, 44). Similarly, IDH2 inhibitors may affect other α-ketoglutarate dependent enzymes beyond TET2 and its effects on DNA methylation that may alter treatment response (45). Although these two leukemia models with parallel and replicate experiments show similar responses, these models do not capture the full complexity of human AML and thus not all human TET2 and IDH2 mutant leukemias may respond similarly through a differentiation effect or to combination therapy. Also given the limitations of our model, correlations with survival were not determined, and human trials will be needed to determine whether these treatments can translate to a meaningful survival advantage in patients with de novo or relapsed, refractory AML.
Our data suggest that these agents have the potential for significant clinical activity without significant toxicity and their effects can be potentiated through mechanism-based combinations with other effective anti-cancer therapies. We show that combined, targeted inhibition of oncogenic kinase signaling and of mutations which drive aberrant DNA methylation can increase therapeutic efficacy, and suggest the need for next-generation trials based on rational combination therapeutic approaches. Most importantly, the ability of epigenetic therapies to reversibly induce differentiation of cancer cells may serve as a platform by which these agents can increase the efficacy of different classes of anticancer agents, including small molecule kinase inhibitors, cell surface targeting therapies, and immunotherapy.
Methods
Mice
The conditional Vav-cre+ Tet2fl/fl Flt3ITD mice were previously described. The Idh2R140Q mutation was targeted by a codon change from CGA to CAA in exon 4 (see supplement for details). The generated knock-in mouse was then crossed to the Mx1-cre and Flt3ITD mice (Jackson Labs). Cre expression was induced by 5 intra-peritoneal injections of poly(I:C) (Invivogen) every other day at a dose of 20μg/g starting 4–5 weeks after birth. All animal procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees (IACUC) at MSKCC.
Bone marrow transplantation and cell harvesting
Dissected femurs and tibias were isolated. Bone marrow was flushed with a syringe into RPMI 10%FCS media or centrifuged at 8000rpm for 1 minute. Spleens were isolated and single cell suspensions made by mechanical disruption using glass slides. Cells were passed through a 70 μm strainer. RBCs were lysed in ammonium chloride-potassium bicarbonate lysis buffer for 10 minutes on ice. 1 × 106 total cells were transplanted via tail vein injection into lethally irradiated (2× 550 Rad) C57BL/6 or CD45.1 host mice (Jackson Labs).
In vivo treatment studies
Mice were treated daily using oral gavage with AC220 at 10 mg/kg, suspended in 5% 2-hydroxypropyl-β-cyclodextrin. 5-Azacytidine (Sigma) was administered at 5mg/kg dissolved in PBS using IP injection. Treatment was daily for 5 days every 21 days, for four cycles. AG-221 (provided by Agios) was administered twice daily using oral gavage at 40mg/kg to 100mg/kg, suspended in 0.5% methyl cellulose and 0.2% Tween80, for 4 to 6 weeks (no differences were observed between these dose levels). Pathology was obtained after fixation in 4% PFA and slides stained with H&E. Blood counts were obtained on an IDEXX ProCyte machine. Each treatment experiment (single or combination therapy) was performed with different donor disease mice (with more than 3 different donor disease mice used for each leukemic genotype) and repeated with representative experiment shown.
RNA sequencing and Analysis
Cell populations were sorted using BD FACSAria and RNA isolated using TrizolLS and RNeasy/AllPrep (Qiagen). RNA was prepared using RiboMinus from LifeTechnologies. The library was sequenced using Illumina HiSeq. Aligned RNA was analyzed for fold change. The raw output BAM files from the sequencer were first converted to FASTQ format using PICARD SamToFastq. Adapters were removed using cutadapt and any read shorter than 35bp was discarded. The trimmed reads were then mapped to the Mouse genome (mm9) using TopHat (ver 2) with a transcriptome index using the Ensembl GTF (ver 37.67) and specifying a strand specific library. Reads that were unmapped by TopHat were then re-mapped using BWA bwasw (ver 0.5.9) again to the mouse genome. The mapped reads from the two passes were merged and then quantitated with HTSeq-count with stranded and strict-intersection options.
ERRBS and Differential Methylation Analysis
ERRBS was performed using a protocol previously described (29). Briefly, genomic DNA were digested with MspI. DNA fragments were end-repaired, adenylated and ligated with Illumina kits. Library fragments of 150–250 bp and 250–400 bp were isolated. Bisulfite treatment was performed using the EZ DNA Methylation Kit (Zymo Research). Libraries were amplified and sequenced on an Illumina HiSeq. Differential methylation analysis was performed on the resulting ERRBS data using methylKit (46) for finding single nucleotide differences (q-value < 0.01 and methylation percentage difference of at least 25%) and eDMR (47) for finding differentially methylated regions. Gene annotations were carried out by overlapping the CpG sites and regions with the exons, introns, UTR5, UTR3 and 5kb upstream regions of refseq genes for genome assembly mm9. Synergy was determined using a generalized linear model, focusing on loci that that show differential methylation, using an adjusted p-value of < 0.01 and a synergy level of at least 20% methylation (absolute methylation difference of 20% above the sum of methylation differences of both single drugs). The data for these analyses are deposited at GEO under: GSE78690, GSE78691, and GSE86952.
Flow cytometry and FACS
Antibody staining and FACS analysis were performed as previously described (15). Cells were lysed in ACK lysis buffer except for CD71 Ter119 staining. Stem cell enrichment was performed using the Progenitor Cell Enrichment Kit (Stem Cell Technologies). See supplement for details.
2HG Measurement
2HG was measured as previously described (31). Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed using an AB Sciex 4000 (Framingham, MA) operating in negative electrospray mode. Multiple reaction monitoring (MRM) data were acquired for each compound, using the following transitions: 2HG (146.9/128.8 amu), 13C5-2HG (151.9/133.8 amu), and 3HMG (160.9/98.9 amu). Chromatographic separation was performed using an ion-exchange column (Fast Acid analysis, 9 μm, 7.8 × 100 mm; BioRad, Waltham, MA).
Supplementary Material
Significance.
Acute Myeloid Leukemias with mutations in TET2 and IDH2 are sensitive to epigenetic therapy through inhibition of DNA methyltransferase activity by 5-Azacytidine or inhibition of mutant IDH2 through AG-221. These inhibitors induce a differentiation response and can be used to inform mechanism-based combination therapy.
Acknowledgments
We thank the MSK IGO for assistance with sequencing and mutation analysis, MSK Flow Cytometry core, and Caroline Sheridan of the Weill Cornell Medicine Epigenomics Core.
Grant Support
This work was supported by a Gabrielle’s Angel Fund grant to RLL and AM, a Leukemia Lymphoma Society (LLS) Translational Research to RLL, grant CA172636-01 to RLL and AM, grant CA-1U54OD020355-01 to RLL, a supplement grant to P30 CA008748 to RLL and CBT, and by the Samuel Waxman Cancer Research Center. CEM is supported by R01HG006798, R01NS076465; MSKCC cores are supported by P30 CA008748. AM is a Burroughs Wellcome Clinical Translational Scholar and supported by the Sackler Center for Biomedical and Physical Sciences. AHS is supported by the Conquer Cancer Foundation, LLS, and NCI K08CA181507. FGB is supported by NCI K08CA169055 and the American Society of Hematology (ASHAMFDP-20121) under the ASH-AMFDP partnership with The Robert Wood Johnson Foundation. RLL is a Scholar of the Leukemia and Lymphoma Society
Footnotes
Conflicts of Interest: Jeremy Travis, Kim Straley, Camelia Gliser, Katharine Yen, and Craig B Thompson hold financial interests in Agios Pharmaceuticals.
References
- 1.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nature reviews Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 2.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Annals of oncology : official journal of the European Society for Medical Oncology. 2016;27:599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 3.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA Hydroxymethylation Profiling Reveals that WT1 Mutations Result in Loss of TET2 Function in Acute Myeloid Leukemia. Cell Rep. 2014;9:1841–55. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57:662–73. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.CGARN CGARN. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118:5593–603. doi: 10.1182/blood-2011-03-343988. [DOI] [PubMed] [Google Scholar]
- 14.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–81. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra18. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27:502–15. doi: 10.1016/j.ccell.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes & development. 2013;27:1974–85. doi: 10.1101/gad.226613.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-Oncogenic Role of Mutant IDH2 in Leukemia Initiation and Maintenance. Cell Stem Cell. 2014;14:329–41. doi: 10.1016/j.stem.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. 2016;22:1488–95. doi: 10.1038/nm.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nomdedeu J, Hoyos M, Carricondo M, Esteve J, Bussaglia E, Estivill C, et al. Adverse impact of IDH1 and IDH2 mutations in primary AML: experience of the Spanish CETLAM group. Leuk Res. 2012;36:990–7. doi: 10.1016/j.leukres.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 23.Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–10. doi: 10.1182/blood-2011-02-339747. [DOI] [PubMed] [Google Scholar]
- 24.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–81. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–12. doi: 10.1182/blood-2014-06-582809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–52. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 27.Madzo J, Liu H, Rodriguez A, Vasanthakumar A, Sundaravel S, Caces DB, et al. Hydroxymethylation at Gene Regulatory Regions Directs Stem/Early Progenitor Cell Commitment during Erythropoiesis. Cell Rep. 2014;6:231–44. doi: 10.1016/j.celrep.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–5. doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–6. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 31.Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood. 2015;125:296–303. doi: 10.1182/blood-2013-10-533604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein EM. IDH2 inhibition in AML: Finally progress? Best Pract Res Clin Haematol. 2015;28:112–5. doi: 10.1016/j.beha.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 33.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L, Bailey E, Greenblatt S, Huso D, Small D. Loss of the wild-type allele contributes to myeloid expansion and disease aggressiveness in FLT3/ITD knockin mice. Blood. 2011;118:4935–45. doi: 10.1182/blood-2011-01-328096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12:413–25. doi: 10.1016/j.stem.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010;5:e9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016;375:2023–36. doi: 10.1056/NEJMoa1605949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–70. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 39.Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–35. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 40.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine. 2014;371:2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aimiuwu J, Wang H, Chen P, Xie Z, Wang J, Liu S, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012;119:5229–38. doi: 10.1182/blood-2011-11-382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhuvanagiri M, Lewis J, Putzker K, Becker JP, Leicht S, Krijgsveld J, et al. 5-azacytidine inhibits nonsense-mediated decay in a MYC-dependent fashion. EMBO molecular medicine. 2014;6:1593–609. doi: 10.15252/emmm.201404461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chemical Society reviews. 2011;40:4364–97. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 46.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li S, Garrett-Bakelman FE, Akalin A, Zumbo P, Levine R, To BL, et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 2013;14(Suppl 5):S10. doi: 10.1186/1471-2105-14-S5-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.