

Abstract
The type, amount, and location of DNA methylation within a gene provides pivotal information on the enzymatic pathway by which it was achieved and its functional consequences. In plants (angiosperms specifically), gene body methylation (gbM) refers to genes with an enrichment of CG DNA methylation within the transcribed regions and depletion at the transcriptional start and termination sites. GbM genes often compose the bulk of methylated genes within angiosperm genomes and are enriched for housekeeping functions. Contrary to the transcriptionally repressive effects of other chromatin modifications within gene bodies, gbM genes are constitutively expressed. GbM has intrigued researchers since its discovery, and much effort has been placed on identifying its functional role. Here, we highlight the recent findings on the evolutionary origin and molecular mechanism of gbM and synthesize studies describing the possible roles for this enigmatic epigenetic phenotype.
Graphical Abstract
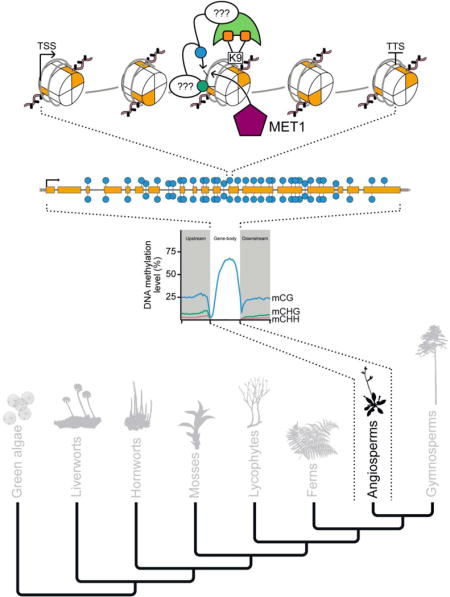
INTRODUCTION
Cytosine DNA methylation is an evolutionarily conserved chromatin modification that contributes to gene regulation and genome structure and integrity [1–4]. In plants, DNA methylation is categorized into distinct site classes based on the sequence context for which the methylated C (mC) is accompanying. These include CG, CHG (H = A|C|T) and CHH site classes, which are established and maintained by separate enzymatic pathways [5]. Methylated CG (mCG) is the most abundant form of mC in plant genomes [6–8]. It is catalyzed and maintained by METHYLTRANSFERASE 1 (MET1), which is the plant homolog of DNA methyltransferase 1 (DNMT1) in animals [9]. Methylated CHG (mCHG) is maintained by the plant-specific CHROMOMETHYLASE 3 (CMT3) through a reinforcing loop with histone H3 lysine 9 di-methylation (H3K9me2) catalyzed by the KRYPTONITE (KYP)/SU(VAR)3–9 HOMOLOG 4 (SUVH4), SUVH5 and SUVH6 lysine methyltransferases [9–12]. Methylation at CHH sites (mCHH) occurs through DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) in conjunction with the RNA-directed DNA methylation (RdDM) pathway. DRM2 to is recruited to target sites through short-interfering RNAs (siRNAs) produced by the RdDM pathway. However, in some cases alternative mechanisms preferentially methylate CHH and to some extent CHG [13,14]. For example, CHROMOMETHYLASE 2 (CMT2) – a homolog of CMT3 – preferentially methylates CHH sites of transposon bodies within the pericentromere [13].
The functional consequences of DNA methylation are an interplay between genomic location, underlying DNA sequence, and site class. Within genes, the type, amount, and distribution of mCG, mCHG and mCHH provide information on the enzymatic pathway by which it was achieved, gene ontology, expression, length, and rate of nucleotide substitutions [6,15–17]. Within angiosperms (flowering plants) there are at least five distinct classes of methylated genes that are classified based on enrichment tests for each site class (Fig. 1): (a) Un-methylated (UM): DNA methylation is absent at all three site classes throughout the entire transcribed region of the gene body; (b) Gene body methylated (gbM): enrichment of mCG within the transcribed regions, and depletion at the transcriptional start (TSS) and termination sites (TTS). GbM genes make up the bulk of DNA methylated genes, are often housekeeping, constitutively expressed, long (bp) relative to UM genes, and slowly evolving in terms of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site (dN/dS) compared to UM genes [7,15–18]. Furthermore, gbM is evolutionary conserved; orthologous proteins between species are typically gbM. (c) TSS: mCG is also enriched in this class of genes, however the distribution is limited to the TSS, which represses gene expression [7]; (d) CG/CHG gene: enrichment of mCHG and depletion of mCHH within the transcribed region. mCG may or may not be enriched. Unlike gbM, these genes are typically expressed at lower levels than all genes and gbM genes across a diverse set of angiosperms [7]; and (e) CHH/RdDM gene: enrichment of mCHH with possible enrichment of mCG and mCHG within the transcribed region. RdDM genes in Arabidopsis thaliana are mostly silenced throughout the plant with the exception of pollen and developing seeds [19]. Unlike gbM genes, DNA methylation levels of CG/CHG and CHH/RdDM genes are not conserved [7]. Additionally, non-gbM genes do not share similar and other conserved features as gbM genes when compared to UM genes [7]. Even though DNA methylation is commonly found in genes, the type and pattern of DNA methylation is often reflective of its expression status.
FIGURE 1. Stylized patterns and types of genic DNA methylation in land plants.
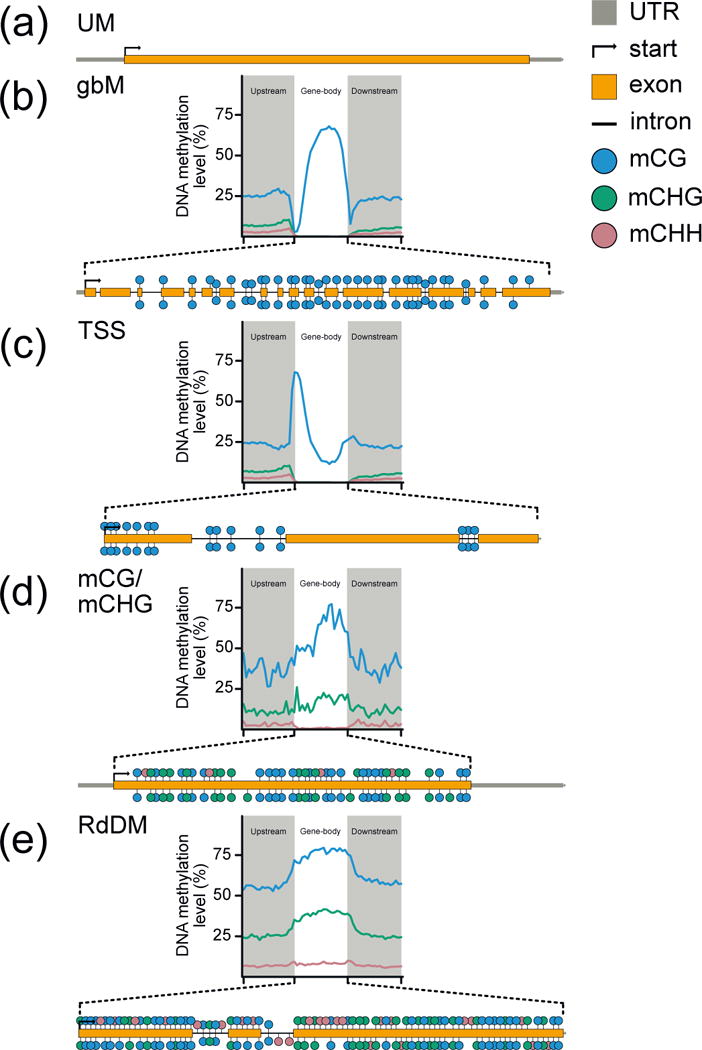
Genes can be categorized into different groups based on enrichment of DNA methylation in exonic sequence, which is deposited by different mechanisms. (a) UM: no enrichment of mCG, mCHG, mCHH; (b) gbM: enrichment of mCG only; (c) TSS: enrichment of mCG only at the TSS; (d) CG/CHG: enrichment for mCG and mCHG; and (e) RdDM: enrichment of mCHH. Additionally, the distribution of DNA methylation across the gene-body is indicative of mechanism and molecular consequences.
Although many of these patterns are observed across land plants (embryophyta), most attention has been placed on understanding the evolutionary origins and consequences of, and mechanism for establishing and maintaining gbM [8,17]. The function of gbM is unknown, but studies have provided evidence for possible functions, including gene regulation [20,21]. Furthermore, variation of DNA methylation (including gbM) between plant accessions has been hypothesized to be adaptive [22]. Conversely others have provided evidence for a lack of function. This review synthesizes the evolutionary and molecular studies on gbM, bringing to light mechanisms for establishment and maintenance, and possible function(s).
ON THE ORIGINS OF GBM
GbM was first described in the model plant species A. thaliana (Brassicaceae/mustard family) [23]. Since its discovery, studies have systematically branched out to include investigations of closely to more distantly related plant species, which has painted a picture of the evolution and conservation of gbM [7,16,18,21,24]. More recently, large, comparative studies incorporating species sister to flowering plants have helped to resolve this picture [4,6,8]. MCG can be found within gene bodies of angiosperms, gymnosperms (represented by Pinus taeda), ferns, lycopods (represented by Selaginella moellendorffii), liverworts (represented by Marchantia polymorpha), moss (represented by Physcomitrella patens), and green algae (represented by Chlorella sp. NC64A) [3,6,8]. Despite the presence of mCG in gene bodies of gymnosperms, ferns, lycopods, liverworts, mosses, and green algae, the levels and distributions have diverged [3,6]. In gymnosperms and ferns, levels and distribution of mCG in protein-coding genes are similar to angiosperms [6]. However, levels of mCG track that of mCHG [6], and thus most likely H3K9me2, a histone modification typically associated with a repressive chromatin state. Hence, H3K9me2 does not repress gene expression in species sister to angiosperms, or mCHG is maintained by an alternative mechanism in gymnosperms and ferns. In P. taeda the distribution of mCG flanking and within gene-bodies shares some resemblance to patterns in the basal-most angiosperm Amborella trichopoda [6]. However, there is also similarity to Chlorella sp. NC64A [3,6]. S. moellendorffii has low and constant levels of mCG flanking and within gene-bodies [3,6]. In M. polymorpha there is a spike of DNA methylation at all three sequence contexts around the TSS and TTS, with DNA methylation being depleted immediately at the TSS and TTS [6]. Furthermore, mCG does not increase across the gene body, but rather is depleted towards the center [6]. When all genes are considered in P. patens there is an apparent lack of mCG within gene bodies [3,6]. This is in contrast to Chlorella sp. NC64A, which has extremely high levels of mCG relative to embryophyta [3,6]. Additionally, mCG levels are reduced at the TSS and TTS, but not to the same extent as what is observed in angiosperms [3].
Enrichment tests are used to identify genes with statistically significant levels of mCG and depletion of non-mCG. The exclusivity of mCG along with the distribution across the entire gene body is used to describe gbM genes [7,8]. These tests have been heavily applied in angiosperms (e.g., [7]), but the application of these tests in species sister to angiosperms is sparse. Recent work by [6] has identified mCG-enriched genes in P. taeda, S. moellendorffii, and M. polymorpha. However, the distributions of the mCG in these genes are not typical of gbM genes identified in angiosperms with the exception being P. taeda [6]. Hence, it is conceivable then that gbM loci might exist in species sister to angiosperms. However do these mCG-enriched genes follow other patterns and consequences of gbM genes as in angiosperms? A question that remains outstanding at this time. Overall, current evidence based on distribution of DNA methylation across all genes suggests gbM is exclusive to angiosperms. However, deeper taxonomic sampling and higher coverage whole genome bisulfite sequencing (WGBS) might reveal the evolution of gbM to be earlier in plants. Changes in function of DNA methyltransferases and/or histone demethylases in the ancestor of angiosperms need to have occurred for gbM to arise. These observations suggest possible differences in underlying pathways and function between angiosperms and other viridiplantae (land plants and green algae).
The timing for the evolution of gbM in angiosperms corresponds with the evolution of CMT3 and orthologous proteins (ZMET2 and ZMET5) through a duplication event shared by all angiosperms [6]. A second duplication event at the base of eudicots gave rise to CMT1 and CMT3 [6]. Hence, ZMETs are co-orthologous to CMT1 and CMT3 [6]. In eudicots and monocots, chromomethylase proteins function in catalyzing and maintaining mCHG [11,12]. Although an unlikely candidate to contribute to a mechanism of gbM, independently derived naturally occurring mutants for CMT3 (Conringia planisiliqua and Eutrema salsugineum) have lost gbM from their genomes, yet retain mCG within repetitive DNA [17]. Based on monophyly of CMT3 and ZMETs, the latter might similarly contribute to a mechanism of gbM in monocots. However, a naturally occurring zmet species has yet to be confidently identified [6]. It is noteworthy that gbM is not immediately lost in A. thaliana cmt3 mutants most likely due to the maintenance of mCG by MET1 [17,25]. This suggests that the loss of gbM in mutants or species with defects in CMT3 is gradual over evolutionary time. Loss of gbM occurring over evolutionary time is also supported by species relationships of C. planisiliqua, E. salsugineum and other Brassicaceae species [26]. The most recent common ancestor for both species for which gbM is observed is ~24 and ~41 million years ago (MYA), respectively [26], suggesting the loss can take millions of years. However, the presence/absence of CMT3 and gbM phenotype are unknown for more closely related Brassicaceae species including other Conringia spp. and Eutrema spp. Additionally, CMT3 alleles and gbM phenotypes may have segregated differently between populations, thus gbM could be polymorphic within a species suggesting a quicker loss of gbM.
The accumulation of gbM also appears to occur slowly over time. In met1 plants, mCG and gbM is lost genome-wide, however, even after reintroducing wild-type MET1 alleles gbM does not immediately return whereas mCG at many repeats and transposons does [17,25]. A recent analysis of DNA methylomes of A. thaliana that have had wild-type alleles of MET1 for eight generations, after met1 was originally mutated, revealed that in rare cases gbM begins to accumulate. Why does it accumulate at certain loci and why does it accumulate slowly remain open questions, although potential models have been proposed [17,27] (Fig. 2). Possible hints come from studies of the histone lysine demethylase, INCREASED IN BONSAI METHYLATION 1 (IBM1), that when mutated leads to the accumulation of H3K9me2 and hypermethylation of CHG and CHH sites mostly at gbM loci. This indicates that in wild-type cells, IBM1 functions to remove H3K9me2 from transcribed loci. Therefore, rare events where IBM1 fails to remove H3K9me2 leads to recruitment of CMT3 and methylation of CHG sites (Fig. 2). How the presence of CMT3 and mCHG at transcribed genes leads to methylation at CG sites is unclear. However, the presence of methylated DNA serves as a substrate for continual recruitment of SRA-containing proteins such as KYP/SUVH4, SUVH5 and SUVH6. These enzymes produce H3K9me2 at DNA methylated loci, which leads to a constant tug-of-war between IBM1 and KYP/SUVH4, SUVH5 and SUVH6. Although IBM1 functions to remove H3K9me2 from transcribed loci, the rare failure to do so results in footprints in the form of mCG that accumulate over evolutionary time due to maintenance activity of MET1.
FIGURE 2. GbM is dependent on a chromomethylase protein.
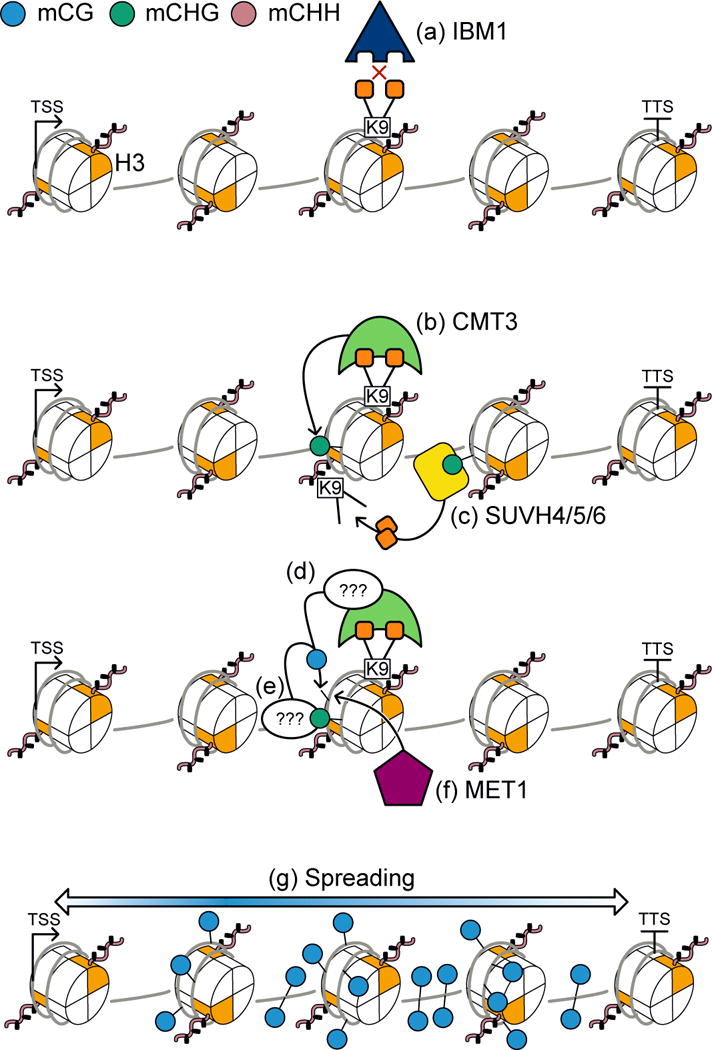
In the model proposed by [17], (a) rare events where IBM1 fails to remove H3K9me2 leads to (b) recruitment of CMT3 and methylation of CHG sites. (c) Methylation of CHG sites recruits KYP/SUVH4/5/6, di-methylating H3K9, and initiating a reinforcing loop with CMT3 and gbM. (d) and (e) CG sites are methylated by a yet undiscovered mechanism. (f) Subsequently, any CG site methylated will be maintained by MET1. (g) Over time, mCG spreads throughout the gene body.
GBM FUNCTION REMAINS ENIGMATIC
Genes underlie many phenotypic differences within and between species. Hence, by nature gbM is interesting due to its occupancy within coding sequence, and its potential contributions to phenotypes. This has led to several hypotheses for its possible function. GbM has been hypothesized to regulate splicing and gene expression [20,21]. GbM possibly enhances splicing accuracy through exon definition [28–30], and by excluding non-CG DNA methylation at acceptor splice sites that have been shown to reduce splicing efficiency [21]. Furthermore, mCG might inhibit DNA polymerase II (Pol II) and transcriptional initiation, which provides an explanation for depletion at the TSS and low expression levels of genes with DNA methylation at this region [1,7,28]. However, DNA methylation within the gene body has limited to no effect on transcriptional elongation [1]. Together these observations suggest gbM and transcription are associated at some level, but further details remain at large.
H2A.Z is a conserved eukaryotic histone variant, and is preferentially enriched at the beginnings of genes where it contributes to promoter competence [30–33]. Hence, DNA methylation and H2A.Z are anticorrelated, and DNA methylation may prevent H2A.Z from encroaching into gene bodies and aberrant transcription [20]. Alternatively, H2A.Z may prevent DNA methylation from encroaching into promoters and disrupting gene expression [20,34]. However, in light of identification of species without gbM and A. thaliana mutant lines, these hypotheses seem to go dark as none of the predicted changes for splicing, gene expression, H2A.Z occupancy, and other molecular phenotypes are observed [17,25]. Specifically, no differences between gbM genes and evolutionary or synthetically removed gbM genes was observed for splicing, gene expression and H2A.Z occupancy [25]. Thus, gbM is dispensable in some species.
Several lines of evidence suggest gbM may serve a yet undiscovered function: (i) gbM is found in 47 of 49 angiosperms investigated to date, which spans angiosperm diversification and evolutionary time [7]. More losses would be expected if gbM is functionally obsolete in every angiosperm; (ii) Across eudicots, the gbM-dependent protein CMT3 evolved under purifying selection with potentially deleterious allelic variation being selectively removed [6]. When purifying selection is weakened – as in certain clades within the Brassicaceae – null alleles are introduced, which negatively affect the mechanism of gbM and are associated with lower numbers of gbM loci. However, (i) and (ii) are confounded by the role of CMT3 in maintaining mCHG at transposons and other regions of the genome [25]. (iii) The addition of a methyl group to a cytosine requires the transcription and translation of methyltransferases and protein complexes, and is a biochemical reaction with energetic and metabolic costs [35]. With costs, benefits most likely ensue. A potential function of gbM, whether universally shared among all angiosperms, clades of closely related species, or species-specific, is unknown.
One possibility is that gbM represents standing epigenomic variation that could be advantageous under novel selection pressures. Epigenomic variation, including epialleles – heritable, genetically identical alleles that differ in transcriptional level [36] – are observable across >1000 A. thaliana accessions [19,22,37]. For example, the same gene can be polymorphic for DNA methylation (or a “poly-epiallele”), where it can be found as un-methylated, gbM methylated or possess transposable element-like DNA methylation (teM; i.e., RdDM) across accessions [37] (Fig. 3). Additionally, the presence of gbM genes are correlated with latitude of origin [22]. A. thaliana accessions found in colder environments have higher levels of gbM for a significant fraction of the genome, which might be adaptive [22]. How selection or neutral processes act on epigenetic variation is unknown. Understanding how epigenetic variation arises and segregates within and between populations can shed light on selection for epigenetic variation, and elucidate possible adaptive epiallelic variation and ultimately function. Although the selection pressure to maintain epigenomic variation, including gbM, for adaptive purposes will most likely be population-specific, and thus rare. Alternatively, epigenomic variation is effectively neutral or the background result of other epigenetic processes.
FIGURE 3. Poly-epialleles between A. thaliana accessions might provide the substrate for adaptive evolution.
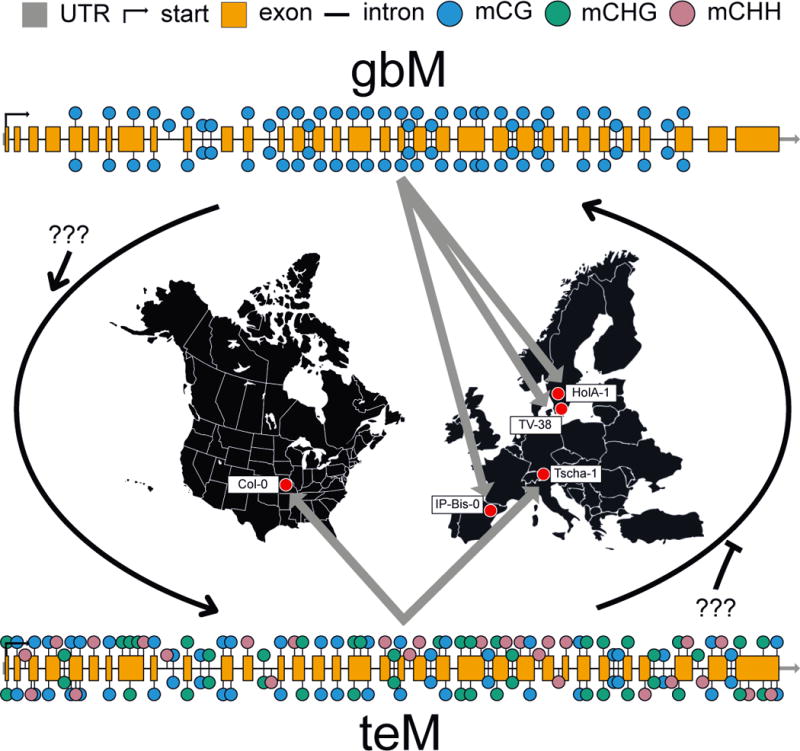
A gene might “flip” DNA methylation state due to a change in targeting by a DNA methylation pathway. Changes to DNA methylation might be stable for many generations or represent more plastic changes occupying shorter periods of time. This variation could be adaptive, for example, in the face of environmental changes. The figure represents one example of a poly-epiallele (AT2G07680) identified by [37].
GBM POSSIBLY EVOLVED AS A BYPRODUCT OF DNA METHYLATION SILENCING OF REPEATS
The evolution of gbM may represent an inconsequential byproduct of DNA methylation silencing of transposable elements (TEs) and other repetitive DNA sequences. Insertion of TEs or other repetitive elements within or in close proximity of genes can disrupt gene expression, which can produce negative phenotypic and fitness consequences [38]. Mechanisms have evolved to silence TEs and other repetitive elements as to maintain proper stoichiometry of gene expression [39,40]. An evolutionarly conserved mechanism is through the RdDM pathway [41], which uses siRNAs produced from double-stranded RNA (dsRNA) generated by sense–antisense pairing or hairpin structures of inverted repeats [42]. SiRNAs are also generated by RNA polymerase IV (Pol IV) by producing long single-stranded RNA transcripts. The RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) then converts these transcripts to dsRNA, which are further processed into the appropriate size and loaded into protein complexes that associate with DNA and the de novo methyltransferase DRM2 [43,44]. Non-coding transcripts generated by RNA polymerase V (Pol V) additionally recruit these complexes and DRM2 [45]. Although this mechanism is typically reserved for repetitive DNA (and a small subset of genes), other modifications affect both transcribed and repetitive DNA, but the regulation of these has evolved to ensure expression and silencing, respectively. For example, H3K9me2 is required for establishment of transcriptional silencing of TEs and repetitive DNA [46], but is also transiently found within genes, being removed by IBM1 [47]. These modifications and accompanying DNA element-specific mechanisms provide a potential link between DNA methylation silencing of repeats and the evolution of gbM. Furthermore, the deposition of DNA methylation may be more tolerable within genes as compared to TEs due to differences in transcriptional regulation [27]. Overall, divergent regulatory mechanisms of shared modifications between genes and TEs evolved to ensure expression and silencing of these DNA elements, respectively. However, as a byproduct, DNA methylation may have been introduced into gene-bodies and subsequently maintained over evolutionary time.
CONCLUSIONS
GbM is characterized by enrichment of mCG within the transcribed region, and depletion at the TSS and TTS. Maintenance of gbM is dependent on a CHG methyltransferase belonging to the chromomethylase gene family, which may have diverged in function since the split of angiosperms from gymnosperms. Phylogenetic relationships of chromomethylase genes and patterns of DNA methylation within gene bodies corroborate differences between angiosperms and earlier diverged plants. Recent identification of naturally occurring gbM mutants suggests dispensability of this evolutionary conserved and genomically abundant type of DNA methylation. Thus, the function of gbM remains elusive. Overall, a simple scenario for the evolution of gbM is not at hand, and several steps likely occurred to remove repressive marks from genes, but be maintained in TEs and vice versa. Consequently, the origins of gbM likely share deep roots with transcriptional silencing of repetitive DNA elements.
HIGHLIGHTS.
GbM is typified by mCG within coding regions, and depletion at the TSS and TTS.
Species without gbM revealed an unexpected role for CMT3 in the evolution of gbM.
Loss of gbM does not result in expected changes, which suggests dispensability.
GbM might have evolved as a byproduct of transcriptional silencing of transposons.
The function of gbM is enigmatic, but natural variation will aid in a discovery.
Acknowledgments
We thank Xiaoyu Zhang for comments. This work was supported by the by the National Science Foundation (MCB – 1339194), the National Institutes of Health (R00GM100000) and by the Office of the Vice President of Research at UGA to R.J.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1•.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2006;39:61–69. doi: 10.1038/ng1929. One of the earlier studies investigating DNA methylation from a genomic perspective and perturbations to DNA methylation caused by met1. [DOI] [PubMed] [Google Scholar]
- 2•.Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. A comparative study illustrating the similarities and differences of DNA methylation between eight diverse plant and animal, and the prevalence of DNA methylation within genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. DNA methylation analysis of 17 eukaryotic genomes. This study substantially expanded taxonomic sampling regarding DNA methylomes, and provided evidence for conservation of DNA methylation within gene bodies and H2A.Z. [DOI] [PubMed] [Google Scholar]
- 4.Huff JT, Zilberman D. Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell. 2014;156:1286–1297. doi: 10.1016/j.cell.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bewick AJ, Niederhuth CE, Rohr NA, Griffin PT, Jim L-M, Schmitz RJ. The evolution of CHROMOMETHYLASES and gene body DNA methylation in plants. biorxiv. 2016 doi: 10.1101/054924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Niederhuth CE, Bewick AJ, Lexiang J, Magdy A, Do Kim K, Page JT, Li Q, Rohr NA, Rambani A, Burke JM, et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016;17:194. doi: 10.1186/s13059-016-1059-0. An in-depth study of DNA methylation across 34 angiosperms utilizing and incorporating statistical tests and biological discoveries from nearly a decade of plant DNA methylation research. Additionally, browsers for each species are publically available providing a comprehensive resource of plant DNA methylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8•.Takuno S, Ran J-H, Gaut BS. Evolutionary patterns of genic DNA methylation vary across land plants. Nat Plants. 2016;2:15222. doi: 10.1038/nplants.2015.222. Using a combination of molecular phylogenetics and WGBS this study provides insights into evolutionary processes that contribute and shape DNA methylation within the genome and specifically within gene bodies. [DOI] [PubMed] [Google Scholar]
- 9.Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16:519–532. doi: 10.1038/nrm4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan SW-L, Henderson IR, Xiaoyu Z, Govind S, Chien JS-C, Jacobsen SE. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in Arabidopsis. PLoS Genet. 2006;2:e83. doi: 10.1371/journal.pgen.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du J, Zhong X, Bernatavichute YV, Stroud H, Feng S, Caro E, Vashisht AA, Terragni J, Chin HG, Tu A, et al. Dual Binding of Chromomethylase Domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell. 2012;151:167–180. doi: 10.1016/j.cell.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du J, Johnson LM, Groth M, Feng S, Hale CJ, Li S, Vashisht AA, Gallego-Bartolome J, Wohlschlegel JA, Patel DJ, Jacobsen SE. Mechanism of DNA methylation-directed histone methylation by KRYPTONITE. Mol Cell. 2014;55:495–504. doi: 10.1016/j.molcel.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takuno S, Gaut BS. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 2012;29:219–227. doi: 10.1093/molbev/msr188. [DOI] [PubMed] [Google Scholar]
- 16.Takuno S, Gaut BS. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc Natl Acad Sci USA. 2013;110:1797–1802. doi: 10.1073/pnas.1215380110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17••.Bewick AJ, Ji L, Niederhuth CE, Willing EM, Hofmeister BT, Shi X, Wang L, Lu Z, Rohr NA, Hartwig B, et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc Natl Acad Sci USA. 2016;113:9111–9116. doi: 10.1073/pnas.1604666113. Exploring natural variation in DNA methylation and underlying proteins lead to the discovery of two cmt3 species. The consequence of this genotype in relation to DNA methylation identified a link between CMT3 and gbM, and provided evidence for the inconsequential effects of lacking gbM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seymour DK, Koenig D, Hagmann J, Becker C, Weigel D. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PLoS Genet. 2014;10:e1004785. doi: 10.1371/journal.pgen.1004785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, et al. Patterns of population epigenomic diversity. Nature. 2013;495:193–198. doi: 10.1038/nature11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456:125–129. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regulski M, Lu Z, Kendall J, Donoghue MT, Reinders J, Llaca V, Deschamps S, Smith A, Levy D, McCombie WR, et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013;23:1651–1662. doi: 10.1101/gr.153510.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubin MJ, Zhang P, Meng D, Remigereau MS, Osborne EJ, Paolo Casale F, Drewe P, Kahles A, Jean G, Vilhjálmsson B, et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife. 2014;4:p.e0525. doi: 10.7554/eLife.05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran RK, Henikoff JG, Zilberman D, Ditt RF, Jacobsen SE, Henikoff S. DNA methylation profiling identifies CG methylation clusters in Arabidopsis genes. Curr Biol. 2005;15:154–159. doi: 10.1016/j.cub.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Beyene G, Zhai J, Feng S, Fahlgren N, Taylor NJ, Bart R, Carrington JC, Jacobsen SE, Ausin I. CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc Natl Acad Sci USA. 2015;112:13729–13734. doi: 10.1073/pnas.1519067112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Stroud H, Hume S, Greenberg MVC, Suhua F, Bernatavichute YV, Jacobsen SE. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 2013;152:352–364. doi: 10.1016/j.cell.2012.10.054. A systematic approach to studying the function of chromatin modifying genes through WGBS in 86 silencing mutants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arias T, Beilstein MA, Tang M, McKain MR, Pires JC. Diversification times among Brassica (Brassicaceae) crops suggest hybrid formation after 20 million years of divergence. Am J Bot. 2014;101:86–91. doi: 10.3732/ajb.1300312. [DOI] [PubMed] [Google Scholar]
- 27••.Inagaki S, Kakutani T. What triggers differential DNA methylation of genes and TEs: contribution of body methylation? Cold Spring Harb Symp Quant Biol. 2012;77:155–160. doi: 10.1101/sqb.2013.77.016212. A logical thought process proposing hypotheses for the link between DNA methylation and transcriptional regulation. Differences between gene and TE transcriptional regulation govern the consequences of DNA methylation within the transcriptional body. [DOI] [PubMed] [Google Scholar]
- 28.Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 29.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112:725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- 32.Brickner DG, Cajigas I, Fondufe-Mittendorf Y, Ahmed S, Lee PC, Widom J, Brickner JH. H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007;5:e81. doi: 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deal RB, Topp CN, McKinney EC, Meagher RB. Repression of flowering in Arabidopsis requires activation of FLOWERING LOCUS C expression by the histone variant H2A.Z. Plant Cell. 2007;19:74–83. doi: 10.1105/tpc.106.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coleman-Derr D, Devin C-D, Daniel Z. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 2012;8:e1002988. doi: 10.1371/journal.pgen.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groth M, Moissiard G, Wirtz M, Wang H, Garcia-Salinas C, Ramos-Parra PA, Bischof S, Feng S, Cokus SJ, John A, et al. MTHFD1 controls DNA methylation in Arabidopsis. Nat Commun. 2016;7:11640. doi: 10.1038/ncomms11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eichten SR, Schmitz RJ, Springer NM. Epigenetics: Beyond chromatin modifications and complex genetic regulation. Plant Physiol. 2014;165:933–947. doi: 10.1104/pp.113.234211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Kawakatsu T, Huang SS, Jupe F, Sasaki E, Schmitz RJ, Urich MA, Castanon R, Nery JR, Barragan C, He Y, et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell. 2016;166:492–505. doi: 10.1016/j.cell.2016.06.044. The largest simultaneous sampling of methylomes and transcriptomes from a single species to date. This study reveals how DNA methylation is shaped by natural genomic variation and by the environment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kidwell MG, Lisch D. Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci USA. 1997;94:7704–7711. doi: 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hollister JD, Smith LM, Guo YL, Ott F, Weigel D, Gaut BS. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci USA. 2011;108:2322–2327. doi: 10.1073/pnas.1018222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu C, Chen J, Zhang Y, Hu Q, Su W, Kuang H. Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. Mol Biol Evol. 2012;29:1005–1017. doi: 10.1093/molbev/msr282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coruh C, Cho SH, Shahid S, Liu Q, Wierzbicki A, Axtell MJ. Comprehensive annotation of Physcomitrella patens small RNA loci reveals that the heterochromatic short interfering RNA pathway is largely conserved in land plants. Plant Cell. 2015;27:2148–2162. doi: 10.1105/tpc.15.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. 2014;15:394–408. doi: 10.1038/nrg3683. [DOI] [PubMed] [Google Scholar]
- 43.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 44.Matzke M, Kanno T, Daxinger L, Huettel B, Matzke A. RNA-mediated chromatin-based silencing in plants. Curr Opin Cell Biol. 2009;21:367–376. doi: 10.1016/j.ceb.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 45.Haag JR, Pikaard CS. Multisubunit RNA polymerases IV and V: purveyors of non-coding RNA for plant gene silencing. Nat Rev Mol Cell Biol. 2011;12:483–492. doi: 10.1038/nrm3152. [DOI] [PubMed] [Google Scholar]
- 46.Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 47.Saze H, Shiraishi A, Miura A, Kakutani T. Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science. 2008;319:462–465. doi: 10.1126/science.1150987. [DOI] [PubMed] [Google Scholar]