

Abstract
Leigh syndrome (LS), or subacute necrotizing encephalomyelopathy, is a genetically heterogeneous, relentlessly progressive, devastating neurodegenerative disorder that usually presents in infancy or early childhood. A diagnosis of Leigh-like syndrome may be considered in individuals who do not fulfil the stringent diagnostic criteria but have features resembling Leigh syndrome.
We describe a unique presentation of Leigh-like syndrome in a 3-year-old boy with elevated 3-hydroxyisovalerylcarnitine (C5-OH) on newborn screening (NBS). Subsequent persistent plasma elevations of C5-OH and propionylcarnitine (C3) as well as fluctuating urinary markers were suggestive of multiple carboxylase deficiency (MCD). Normal enzymology and mutational analysis of genes encoding holocarboxylase synthetase (HLCS) and biotinidase (BTD) excluded MCD. Biotin uptake studies were normal excluding biotin transporter deficiency. His clinical features at 13 months of age comprised psychomotor delay, central hypotonia, myopathy, failure to thrive, hypocitrullinemia, recurrent episodes of decompensation with metabolic keto-lactic acidosis and an episode of hyperammonemia. Biotin treatment from 13 months of age was associated with increased patient activity, alertness, and attainment of new developmental milestones, despite lack of biochemical improvements. Whole exome sequencing (WES) analysis failed to identify any other variants which could likely contribute to the observed phenotype, apart from the homoplasmic (100%) m.8993T>G variant initially detected by mitochondrial DNA (mtDNA) sequencing.
Hypocitrullinemia has been reported in patients with the m.8993T>G variant and other mitochondrial disorders. However, persistent plasma elevations of C3 and C5-OH have previously only been reported in one other patient with this homoplasmic mutation. We suggest considering the m.8993T>G variant early in the diagnostic evaluation of MCD-like biochemical disturbances, particularly when associated with hypocitrullinemia on NBS and subsequent confirmatory tests. An oral biotin trial is also warranted.
Keywords: Acylcarnitines, Citrulline, Leigh syndrome, mtDNA mutation, Newborn screening (NBS)
Introduction
Leigh syndrome (LS) is the most common paediatric presentation of mitochondrial disease with an estimated pre-school incidence of 1 per 34,000 births (Darin et al. 2001). Higher incidences have however been reported in specific populations in the Faroe Islands, 1:1700 (Ostergaard et al. 2007) and Saguenay Lac-Saint-Jean region of Quebec, Canada, 1:2000 (Morin et al. 1993) which have been attributable to founder mutations.
Since its initial description (Leigh 1951), LS has evolved from a distinct neuropathological disorder defined by post-mortem histopathological findings to a clinical entity characterized by progressive neurodegenerative disease with symptoms and signs of brainstem and/or basal ganglia disease, raised lactate levels in blood and/or cerebrospinal fluid (CSF), and typical neuroimaging and/or neuropathological abnormalities (Rahman et al. 1996). More recently, Baertling et al. (2014) refined the diagnostic criteria to include the three most commonly described features: (1) neurodegenerative disease with variable symptoms; (2) bilateral neuroimaging or CNS lesions and (3) a variety of nuclear or mitochondrially encoded genetic causes of deficient mitochondrial energy metabolism. The term “Leigh-like syndrome” was proposed when these diagnostic criteria are only partially met but highly suggestive for LS (Rahman et al. 1996; Baertling et al. 2014).
The clinical presentation of Leigh syndrome can be highly variable with disease onset ranging from the neonatal period through adulthood. Typically, onset occurs between age three and 12 months, often triggered by an acute infection. Prenatal expression of mitochondrial disease has been described with oligohydramnios, intrauterine growth restriction and abnormal brain neuroimaging (Kumakura et al. 2009; Sofou et al. 2014). Late-onset Leigh syndrome has been associated with predominant extrapyramidal features, slow progression, acute deterioration, usually after decompensation with illness, and atypical presentations including features of Guillain–Barré syndrome, hypertrophic cardiomyopathy, anaemia and leukopenia (Huntsman et al. 2005).
LS is a genetically heterogeneous mitochondrial disorder. Causative variants have been identified in up to 75 genes involved in energy metabolism, encoded by either the nuclear or mitochondrial genomes, including each of the five OXPHOS complexes, electron carrier coenzyme Q10 (CoQ10) and components of the pyruvate dehydrogenase complex (Lake et al. 2015). Mitochondrial DNA (mtDNA) mutations underlie approximately 10–20% of LS cases (Rahman et al. 1996; Sofou et al. 2014). Approximately 10% of individuals have either the MT-ATP6 (mitochondrially encoded ATP synthase 6) m.8993T>G or m.8993T>C variants which represent the only established genetic cause of a Complex V-mediated LS (Santorelli et al. 1993; Rahman et al. 1996; Thorburn and Rahman 2014). The MT-ATP6 gene encodes a subunit of the 550 kKDa multi-subunit Complex V (ATP synthase or F1Fo ATPase), which is one of the key enzymes involved in the aerobic generation of energy, synthesizing ATP from ADP using the proton gradient generated across the mitochondrial inner membrane (Kucharczyk et al. 2009). Unlike many pathogenic mtDNA variants, the m.8993T>G and m.8993T>C variants display a strong genotype–phenotype correlation, with a lack of tissue or age-dependent variation in mutant load (White et al. 1999a) which enables accurate prediction of the probability of severe outcome and empirical recurrence risks (White et al. 1999b). Overall, LS develops whenever the m.8993T>G mutant load exceeds 90%, while the milder NARP (neurogenic muscle weakness, ataxia and retinitis pigmentosa) phenotype occurs with moderate levels of approximately 70–90% mutant loads (Thorburn and Rahman 2014). Other properties of m.8993T>G variant include de novo occurrences with rapid segregation toward homoplasmy, often within a single generation, observed in approximately 20% of families (White et al. 1999a). Likely explanations for these sporadic cases include spontaneous variants arising during oogenesis (Degoul et al. 1997); or presence of a mitochondrial genetic “bottleneck”, in which only a small subpopulation of mtDNA molecules are preferentially amplified to “repopulate” the oocyte (Blok et al. 1997; White et al. 1999b).
Here, we describe a male patient with increased C5-OH on NBS, presenting with psychomotor delay, central hypotonia, failure to thrive, episodes of decompensation with keto-lactic acidosis and persistent elevations in C3, C5-OH, with urinary markers suggestive of multiple carboxylase deficiency (MCD). Hypocitrullinemia was an additional feature which was also detected on NBS. Leigh-like disease was subsequently diagnosed secondary to a de novo homoplasmic m.8993T>G variant.
Materials and Methods
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. The family reported in this study has agreed to the publication and provided written consent as part of the informed consent process.
Case Report
The patient, a Caucasian male, was born at term with a birth weight of 3.2 kg (25th percentile) to non-consanguineous healthy parents. The patient had increased C5-OH (2.0 μmol/L, ref <0.6) on newborn screen and a similar bloodspot result at 3 weeks of age. His bloodspot citrulline on NBS was 7 μmol/L (1% in this age group on screening is 7 μmol/L; the 50% (median) is 14 μmol/L). Low citrulline is not used as a marker in newborn screening (NBS) in Western Australia. Plasma acylcarnitine analysis revealed increased C5-OH (2.1 μmol/L, ref <0.9) and C3 (1.1 μmol/L, ref <0.2). Plasma amino acid analysis at 3 weeks of age showed increased alanine (696 μmol/L, ref 130–460) and decreased citrulline (2 μmol/L ref 10–45). Venous blood gas was normal, apart from increased lactate (3.8 mmol/L, ref <1.5). Plasma ammonia and urine organic acid analysis were normal. Bloodspot biotinidase enzyme assays performed twice were normal.
For the first week of life, the patient was exclusively breastfed and developed feeding intolerance with vomiting and loose stools. He was diagnosed with breast milk protein-induced enterocolitis syndrome (FPIES) and feeding was switched to a hypoallergenic, elemental formula (Neocate Infant, Nutricia); coincident with the feeding change, the feeding intolerance resolved. However, weight gain remained poor over the first 6 months of life; this was attributed to recurrent viral infections. He failed to achieve age-related developmental milestones by 6 months of age; Griffith’s mental developmental assessment at 12 months confirmed global developmental delay with an overall mental age of 7 months and motor developmental age of 6 months. He was presented to the metabolic clinic at 13 months of age with failure to thrive, lethargy and central hypotonia. Physical examination at that time revealed brisk deep tendon reflexes, equivocal plantar reflexes and generalized muscle weakness. Laboratory investigations revealed mildly increased lactate (3.9 mmol/L, 0.5–2.0), with normal plasma ammonia, creatine kinase, liver enzyme tests and venous blood gas results. Plasma acylcarnitines showed increased C3 (3.1 μmol/L, ref <0.7) and C5-OH (0.97 μmol/L, ref <0.31); plasma amino acids revealed increased alanine (572 μmol/L, ref 143–439) and decreased citrulline (4 μmol/L, ref 10–45). Urine organic acid analysis showed mild to moderate increases in 3-hydroxyisovalerate, 3-hydroxypropionate, 3-methylcrotonylglycine, methylcitrate and 2-methyl-3-hydroxybutyrate. A biotin related inborn error was suspected, and oral biotin at 10 mg daily was initiated and subsequently increased to his current dose of 100 mg twice a day. Further investigations were carried out to exclude MCD including holocarboxylase synthetase (HLCS) gene sequencing, with deletion/duplication analysis; biotinidase (BTD) gene sequencing and fibroblast enzymology of propionyl-CoA carboxylase, methylcrotonyl-CoA carboxylase and pyruvate carboxylase using a low biotin culture medium (6 nmol/L). All these studies were normal.
Clinical improvements with increased patient activity levels, alertness and attainment of new developmental milestones were temporally associated with oral biotin treatment despite lack of significant correlation with biochemical improvements. Trio whole exome sequencing (WES) was carried out to exclude any novel genetic defects in the biotin pathway, and included analyses for any mutations in the mitochondrial carbonic anhydrase VA (CA5A) gene which presents with MCD phenotype (Karnebeek et al. 2014) but all tests were non-contributory.
At 15 months, he decompensated rapidly during an intercurrent pneumonia with human metapneumonia virus. He was ventilated in the ICU and had metabolic keto-lacticacidosis with blood pH 7.27, bicarbonate 12 mmol/L, base excess −14, lactate 8.8 mmol/L, plasma beta-hydroxybutyrate (6.0 mmol/L, ref <0.3) and hyperammonemia (119 μmol/L, ref <50). CSF lactate (1.7 mmol/L, ref 0.7–1.8) and pyruvate (0.08 mmol/L, ref 0.03–0.1) were normal. Apart from a small lactate doublet on spectroscopy, brain MRI was normal. He was discharged home after a week with temporary regression of milestones, particularly gross motor which returned to pre-morbid state a few weeks later.
A repeat developmental assessment performed at 17 months showed steady progress with a 6 months gain over the preceding 6 months. Persistent plasma increases of C5-OH (1.1 μmol/L, ref <0.2), C3 (3.0 μmol/L, ref <0.6) and fluctuating urinary markers (3-methylcrotonylglycine, 3-hydroxyisovalerate, 3-hydroxypropionate, methylcitrate and 2-methyl-3-hydroxybutyrate) were observed, despite significant clinical progress. Fundoscopy and slit-lamp examination were normal. Quadriceps muscle biopsy showed well-orientated muscle fibres with variable fibre diameter ranges of 10–25 mm (age appropriate range 16–18 mm) and small subsarcolemmal aggregates of mitochondria in some fibres with modified Gömöri trichrome stain. Electron microscopy displayed atrophic fibres, mild mitochondrial pleomorphism with lipid vacuoles and crowding of the cristae in rare fibres. Complex IV activity on snap frozen skeletal muscle homogenate was borderline low at 25% when expressed relative to citrate synthase, but in the lower end of the normal range 2.93 /min/mg (reference range 3.3–9.1) relative to protein. Complex V was not measured due to technical limitations. Identification of de novo homoplasmic (100%) m.8993T>G variant in muscle, blood and urine by next-generation sequencing of the mitochondrial genome led to the diagnosis of Leigh-like syndrome. Analysis of maternal blood and urine DNA tested negative for the m.8993T>G variant identified in her son. Therapy with riboflavin, thiamine, vitamin C, carnitine and coenzyme Q10 was started initially; oral supplementation of citrulline, creatine and alpha-lipoic acid was commenced at 24 months, 28 and 36 months, respectively. Repeat brain MRI at 31 months was again reported to be normal, apart from the presence of small lactate doublet. Early initiation of aggressive metabolic dietary support during unwell episodes has significantly decreased metabolic decompensations. Early institution of nasogastric feeding during intercurrent illness when oral feeding has been difficult has substantially reduced the frequency of hospital admissions and need to seek emergency care (from a 4 to 6 weekly frequency to approximately once in 6 months). The unwell dietary management plan includes increased caloric intake at 100 kcal/kg/day (fat 44%, protein 26% and carbohydrates 30%).
At 3 years 3 months of age, he has exhibited significant improvements in his overall developmental progress; particularly with gross motor milestones. He now has a healthy 2-month old sister. Prenatal testing through chorionic villus sampling for the sibling revealed no detectable m.8993T>G variant. She was born healthy with normal birth parameters, NBS and urine organic acids. She has shown normal weight gain and appears to be developing appropriately at 3 months of age.
Whole Exome Sequencing (WES)
After enrolment of the family within the TIDEX gene discovery project (UBC IRB approval H12-00067), WES was performed for the proband and his unaffected mother using the Agilent SureSelect kit and Illumina HiSeq 2000 (PerkinElmer, USA). The sequencing reads were mapped to the hg19 human reference genome. After multiple filtering steps (excluding sequencing errors, variants with MAF >0.01 etc.) and screening under multiple inheritance models, we uncovered eight genes harbouring homozygous recessive variants [one autosomal (NTN5) and seven X-linked (SYTL5, CLCN5, BEND2, CNGA2, RBBP7, HEPH and TCEAL4)], six compound heterozygous (TNRC6A, NAV3, CLCN2, ALKBH2, UNC5CL and BSG), and no nuclear de novo variants. None were deemed compatible with the patient’s phenotype. Each of these variants were further analyzed for their impact on gene function using multiple bioinformatic tools (including, SIFT, Polyphen and CADD scores), as well as published literature on their function to ascertain a potential correlation with the proband’s phenotype. The only variants of potential interest were the ones affecting CLCN5 and CLCN2; however, neither of these was decided to be explanatory for the observed MCD phenotype, and thus ATP6 was deemed the best fit.
Biotin Concentrations in Physiologic Fluids
All samples were initially assayed for biotin using a sequential solid phase avidin-binding assay as previously described (Mock 1997) that measures Total Avidin-Binding Substances (TABS) referred to hereafter as biotin unless noted otherwise.
Biotin Uptake Studies
Radioactive 3H-biotin (specific activity: 60 Ci/mmol; radiochemical purity >97%) was purchased from American Radiolabeled Chemicals (ARC) (St. Louis, MO). Other reagents and chemicals used in these studies were purchased from commercial vendors; all were of either analytical or molecular biology grade. Specific primers used for PCR amplifications were from Sigma Genosys (Woodlands, TX).
Biotin uptake was performed in lymphocytes from the proband and normal controls as previously reported (Said et al. 1998) using Krebs–Ringer (KR) buffer (in mM: 133 NaCl, 4.93 KCl, 1.23 MgSO4, 0.85 CaCl2, 5 glucose, 5 glutamine, 10 HEPES and 10 MES, pH 7.4). Briefly, lymphocytes were harvested by centrifugation and equal amount of cells were used for uptake studies for 5 min as described previously. The reaction was stopped by adding 2 ml of ice-cold KR buffer and cells were passed through PVDF membrane, rinsed twice with ice-cold buffer. The membrane was incubated further with scintillation fluid and radioactivity was measured in a scintillation counter (Beckman Coulter LS6500). Passive diffusion was quantified by performing uptake of 3H-biotin in the presence of excess unlabeled biotin (1 mM). Protein concentrations were measured by DC protein assay kit (Bio-Rad). Uptake was expressed as fmol/mg protein/5 min.
Real-Time PCR Analysis
Total RNA was isolated from lymphocytes using TRIzol reagent (Invitrogen). RNA samples were then treated with RNAse free DNAse I (Invitrogen) to remove contaminating DNA, and were reverse transcribed using iScript cDNA synthesis kit (Bio-Rad). Relative expression of the sodium dependent multivitamin transporter (SMVT; SLC5A6) transcript was quantified by real-time PCR (Bio-Rad CFX 96 real-time PCR system) using gene-specific primers for hSMVT (Forward 5′-TGTCTACCTTCTCCATCATGGA-3′ and Reverse 5′-TAGAGCCCAATGGCAAGAGA-3′). β-Actin was used as an internal control (Forward 5′-AAATGGGTTCTAGACCGCGGAGA-3′ and reverse 5′-CATGCTCGATGCGGTACTTCA-3′) and data were calculated following relative relationship method.
Western Blot Analysis
To determine the protein level of hSMVT, lymphocytes were lysed in RIPA buffer containing protease inhibitors, and the supernatant was collected by brief centrifugation. Equal amounts of protein (approximately 60 μg) were loaded into NuPAGE 4–12% Bis-Tris gradient mini gels (Invitrogen). The gel was electro-blotted onto PVDF membrane and incubated overnight with blocking buffer (LI-COR). The blot was then probed with specific human polyclonal antibodies to SMVT and β-actin (Santa Cruz Biotechnology), followed by probing with labelled secondary antibodies (anti-mouse IRDye 800 and anti-rabbit IRDye 680 in 1:25,000 dilutions) for 1 h. Fluorescent intensity of the specific band was quantified using the Odyssey Infrared imaging system (LI-COR Biosciences) and Odyssey application software (version 3.0).
Results
Biotin Studies:
In samples collected 10 days after initiating biotin therapy at 10 mg per day, the biotin level measured in plasma was ~1,500 times upper limit of reference range and the urine excretion rate was ~4,000 times upper limit of reference range (Table 1). These striking increases contrasted with the level in CSF, which was ~15 times upper limit of reference range. The discrepancies of biotin concentrations in the different fluids were of uncertain significance as the samples were collected post-biotin supplementation.
Table 1.
Biotin determinations of urine, CSF and plasma samples
| Sample | Proband | Normal range |
|---|---|---|
| Urine (pmol/mg creatinine) | 429,355 | 45–118 (n = 49) |
| CSF (pmol/ml) | 88.0 | 0.022–5.9 (n = 55) |
| Plasma (pmol/ml) | 1,398 | 0.55–1.1 (n = 80) |
Carrier-mediated biotin uptake was studied in EBV-transformed lymphocytes from the proband, and compared with similar studies performed in EBV-transformed lymphocytes from normal adult controls (ABC) and with those from cord blood controls (CBC). The results (Fig. 1) showed considerable biotin uptake that was slightly but significantly (p < 0.05) lower than uptake by EBV-transformed lymphocytes from control cord blood samples, yet somewhat higher than that of adult EBV-transformed lymphocytes. The higher uptake in cord blood controls compared to adult EBV-transformed lymphocytes could be attributed to developmental maturation. This has previously been described in a rodent model with higher intestinal biotin uptake observed during the suckling period compared to adulthood (Said and Redah 1988).
Fig. 1.
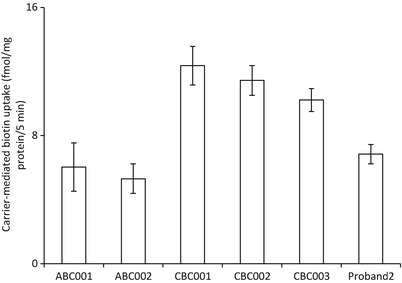
Carrier-mediated biotin uptake measured in EBV-transformed lymphocytes of proband compared to EBV-transformed lymphocytes from adults (ABC) and cord blood control (CBC). Data were expressed as fmol/mg protein/5 min and are mean ± SE of three independent experiments
SMVT Transcript and Protein Studies:
SMVT mRNA (Fig. 2a) and protein levels (Fig. 2b) in EBV-transformed lymphocytes from the proband were not different than those for EBV-transformed newborn (cord blood) and adult controls. In the light of these findings, we speculate that the relatively lower increases in CSF might represent saturation of a normal carrier-mediated transport across the blood–brain barrier. There is evidence for such a carrier in animal studies (Spector and Mock 1987, 1988).
Fig. 2.
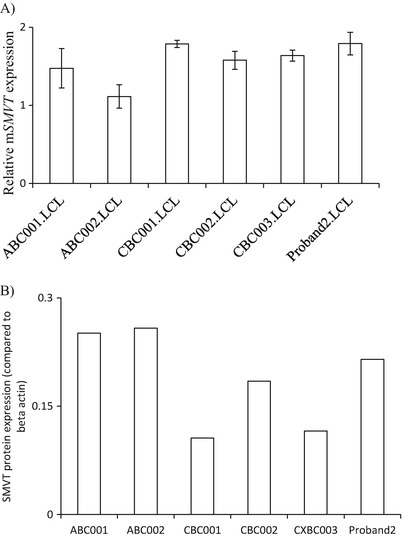
SMVT transcript and protein studies. (a) SMVT mRNA expression: real-time PCR was performed using total RNA isolated from lymphocytes. The level of hSMVT expression was normalized relative to β-actin as described in “Materials and Methods”. Data are mean ± SE of three independent experiments. (b) SMVT protein expression: total protein isolated from the lymphocytes was used for western blotting. The blot was probed with anti-SMVT antibody and expression was normalized relative to β-actin
Discussion
Persistent hypocitrullinemia has previously been reported in 13 patients with the m.8993T>G variant (Rabier et al. 1998; Parfait et al. 1999; Enns et al. 2006; Debray et al. 2010; Henriques et al. 2012; Mori et al. 2014). In an additional report, a female patient who was prenatally diagnosed with apparently mild ornithine transcarbamylase (OTC) deficiency succumbed during an acute febrile encephalopathic illness at 6 months of age, with neuroimaging suggestive of Leigh syndrome. Post-mortem genetic testing identified the m.8993T>G variant in the liver, muscle and blood with variant loads of 82–87% (Henriques et al. 2012). Persistent hypocitrullinemia has been described in other mitochondrial disorders including Pearson syndrome (Ribes et al. 1993), Leigh syndrome caused by isolated complex I deficiency (Debray et al. 2010), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS syndrome) (Naini et al. 2005), mitochondrial neurogastrointestinal encephalopathy (MNGIE) disease (Bindoff 2006), and with secondary mitochondrial respiratory chain dysfunction caused by organic acidemias (Atkuri et al. 2009) and deficiency of mitochondrial pyrroline-5-carboxylate synthetase (P5CS) (Baumgartner et al. 2000). In a small series of 16 Leigh syndrome patients, sensitivity and specificity of hypocitrullinemia as a marker for the m.8993T>G variant were reported to be 66% and 85%, respectively (Debray et al. 2010). Hypocitrullinemia was also found to be a characteristic unique to patients with the m.8993T>G mutation, seen in 90% of patients, as opposed to less than 20% for other respiratory chain deficiencies (Rabier et al. 1998). These findings are sufficiently convincing to consider hypocitrullinemia as a useful biochemical marker for the m.8993T>G mutation and, more generally, as a marker of impaired oxidative phosphorylation in the enterocyte (Rabier et al. 1998; Parfait et al. 1999; Debray et al. 2010; Henriques et al. 2012; Mori et al. 2014).
Citrulline is a non-protein amino acid that is not normally incorporated into proteins during protein synthesis. However, citrullinated proteins comprise citrulline residues that result from post-translational modification of arginine; these include keratinization-related proteins (Ishigami et al. 2002) and basic myelin protein, which makes up to 35% of the protein component of the central nervous system (Ishiyama et al. 2001). It is a non-essential amino acid, synthesized almost exclusively in the mitochondrial matrix of enterocytes, catalyzed by pathways involving P5CS, carbamoyl phosphate synthase 1 (CPS1) and OTC (Fig. 3). CPS I and P5CS activities are both tightly related to ATP concentration, while ADP acts as an inhibitor (Elliot and Tipton 1974). Citrulline metabolism within the urea cycle in the liver is strictly compartmentalized; involving ammonia detoxification and arginine synthesis.
Fig. 3.
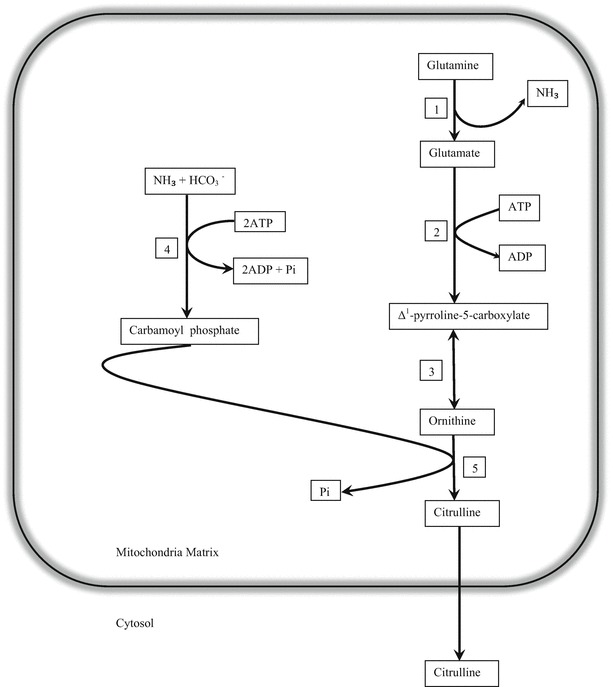
Citrulline synthesis in mitochondrial enterocyte: 1 glutaminase, 2 Δ1-pyrroline-5-carboxylate synthetase (P5CS), 3 ornithine δ-aminotransferase, 4 CPS I and 5 OTC
Reduced oxidative phosphorylation can lead to deficient activities of both P5CS and CPS1 in the enterocyte with subsequent enterocyte dysfunction and impairment of the intestinal citrulline biosynthetic pathway (Rabier et al. 1998). Studies have demonstrated citrulline as an efficient marker of the active small bowel mass (Crenn et al. 2000), as well as in a wide range of pathologies; villous atrophy-associated small bowel disease (Crenn et al. 2003); radiation-induced intestinal epithelial damage (Lutgens et al. 2003) or other myeloablative therapy (Blijlevens et al. 2004). The utility of citrulline as an effective marker of enterocyte mitochondrial dysfunction may correlate with the heteroplasmy in gut mucosa. A 97% heteroplasmy in gut mucosa was demonstrated in a patient with hypocitrullinemia (Rabier et al. 1998).
Abnormal acylcarnitine concentrations, particularly persistent elevations of C3 and C5-OH, have only been described in one other patient with the m.8993T>G variant (Hauser 2014). Our patient presented biochemically with profiles suggestive of MCD; however, extensive enzymatic and genetic analyses excluded that group of disorders. Biotin uptake and transport studies failed to identify any contributory cause. We speculate that defective oxidative phosphorylation not only affected citrulline biosynthesis, resulting in hypocitrullinemia, but also compromised ATP dependent carboxylases, causing the unusual biochemistry exhibited by this patient. Interestingly, this abnormal acylcarnitine profile has been associated with other MT-ATP6 variants, specifically m.8959G>A and m.9155A>T (unreported data, personal communication, A Mattman), hence raising the possibility this may be a feature specific to defects of the ATPase 6 subunit of Complex V. While citrulline is among the metabolites measured by expanded newborn screening in Western Australia, low citrulline values are not officially reported for further work-up due to low positive predictive value.
Larger studies are needed to ascertain the true prevalence of these abnormal biochemical parameters which may be detected as early as in NBS, and to determine the pathogenesis connecting the m.8993T>G mutation as well as other MT-ATP6 mutations and these metabolic abnormalities. We suggest expanding the differential diagnosis for persistent elevations in C3 and C5-OH detected on NBS, particularly when associated with hypocitrullinemia as potential NBS markers for mitochondrial disorders due to m.8993T>G associated Leigh-like syndrome and other MT-ATP6 mutations. This may facilitate rapid diagnosis through targeted mutational analysis and minimize invasive tissue biopsies for respiratory chain studies. Presymptomatic diagnosis of mitochondrial disorders may be of therapeutic benefit, particularly with this biochemical phenotype when oral biotin therapy is warranted along with close monitoring of relevant biochemical and neurodevelopmental parameters.
Acknowledgements
We are grateful to the patient and family for participation in this study; Dr Janice Fletcher (SA Pathology, Adelaide, South Australia) for advice on the diagnostic work-up; Mrs M Higginson for DNA extraction, sample handling and technical data; Dr W Wassermann for supervision of WES bio-informatics analyses; Dr CJ. Ross and Mrs X Han for Sanger sequencing (University of British Columbia, Vancouver, CA).
Compliance with Ethics Guidelines
Conflict of Interest
All authors declare that they have no conflicts of interest.
Details of the Contributions of Individual Authors
SB was the physician in charge of the family. BL, DMM, HMS, MTG, AM, CDvK, DRT and RJR performed/supervised/interpreted laboratory investigations. JC advised on the overall diagnostic work-up and management of the proband. SB and JC drafted the original manuscript. All authors have read/critically revised the manuscript.
Sources of Support
NIH grants R37 DK 36823 (DMM), R37 DK36823-26S1 (DMM), RO1 DK79890, DK79890-01S1 (DMM); the UAMS CTSA Award UL1 TR000039. B.C. Children’s Hospital Foundation (www.tidebc.org) (CVK); Canadian Institutes of Health Research grant #301221 (CDvK); British Columbia Clinical Genomics Network grant BCCGN00031 (CDvK). CDvK is recipient of the Michael Smith Foundation for Health Research Scholar Award.
Footnotes
The original version of this chapter was revised. An erratum to this chapter can be found at DOI 10.1007/8904_2017_588.
Contributor Information
Shanti Balasubramaniam, Email: saras329@hotmail.com.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Atkuri KR, Cowan TM, Kwan T, Ng A, Herzenberg LA, Enns GM. Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc Natl Acad Sci U S A. 2009;106(10):3941–3945. doi: 10.1073/pnas.0813409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertling F, Rodenburg RJ, Schaper J, et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014;85:257–265. doi: 10.1136/jnnp-2012-304426. [DOI] [PubMed] [Google Scholar]
- Baumgartner MR, Hu CA, Almashanu S, et al. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta (1)-pyrroline-5-carboxylate synthase. Hum Mol Genet. 2000;9(19):2853–2858. doi: 10.1093/hmg/9.19.2853. [DOI] [PubMed] [Google Scholar]
- Bindoff L. Mitochondrial gastroenterology. In: DiMauro S, Hirano M, Schon EA, editors. Mitochondrial medicine. Abingdon: Informa Healthcare; 2006. pp. 143–159. [Google Scholar]
- Blijlevens NMA, Lutgens LCHW, Schattenberg AVMB, Donnelly JP. Citrulline: a potentially simple quantitative marker of intestinal epithelial damage following myeloablative therapy. Bone Marrow Transplant. 2004;34:193–196. doi: 10.1038/sj.bmt.1704563. [DOI] [PubMed] [Google Scholar]
- Blok RB, Gook DA, Thorburn DR, Dahl H-HM. Skewed segregation of the mtDNA nt 8993 (T→G) mutation in human oocytes. Am J Hum Genet. 1997;60:1495–1501. doi: 10.1086/515453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crenn P, Coudray-Lucas C, Thuillier F, Cynober L, Messing B. Postabsorptive plasma citrulline concentration is a marker of absorptive enterocyte mass and intestinal failure in humans. Gastroenterology. 2000;119:1495–1505. doi: 10.1053/gast.2000.20227. [DOI] [PubMed] [Google Scholar]
- Crenn P, Vahedi K, Lavergne-Slove A, Cynober L, Matuchansky C, Messing B. Plasma citrulline: a marker of enterocyte mass in villous atrophy-associated small bowel disease. Gastroenterology. 2003;124:1210–1219. doi: 10.1016/S0016-5085(03)00170-7. [DOI] [PubMed] [Google Scholar]
- Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol. 2001;49(3):377–383. doi: 10.1002/ana.75. [DOI] [PubMed] [Google Scholar]
- Debray FG, Lambert M, Allard P, Mitchell GA. Low citrulline in Leigh disease: still a biomarker of maternally inherited Leigh syndrome. J Child Neurol. 2010;25(8):1000–1002. doi: 10.1177/0883073809351983. [DOI] [PubMed] [Google Scholar]
- Degoul F, Francois D, Diry M, et al. A near homoplasmic T8993G mtDNA mutation in a patient with atypic Leigh syndrome not present in the mother’s tissues. J Inherit Metab Dis. 1997;20(1):49–53. doi: 10.1023/A:1005357506614. [DOI] [PubMed] [Google Scholar]
- Elliot KRF, Tipton KF. Product inhibition studies on bovine liver carbamoyl phosphate synthetase. Biochem J. 1974;141:817–824. doi: 10.1042/bj1410817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns GM, Bai RK, Beck AE, Wong LJ. Molecular-clinical correlations in a family with variable tissue mitochondrial DNA T8993G mutant load. Mol Genet Metab. 2006;88(4):364–371. doi: 10.1016/j.ymgme.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Hauser N (2014) Persistent elevations in C3 and C5OH, heralding signs for an underlying mitochondrial defect: neurogenic weakness with ataxia and retinitis pigmentosa (NARP). 64th Annual meeting of the American Society of Human Genetics, San Diego
- Henriques M, Diogo L, Garcia P, Pratas J, Simoes M, Grazina M. Mitochondrial DNA 8993T>G mutation in a child with ornithine transcarbamylase deficiency and Leigh syndrome: an unexpected association. J Child Neurol. 2012;27(8):1059–1061. doi: 10.1177/0883073811431015. [DOI] [PubMed] [Google Scholar]
- Huntsman RJ, Sinclair DB, Bhargava R, Chan A. Atypical presentations of Leigh syndrome: a case series and review. Pediatr Neurol. 2005;32(5):334–340. doi: 10.1016/j.pediatrneurol.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Ishigami A, Ohsawab T, Asagab H, Akiyama K, Kuramotoa M, Maruyamaa N. Human peptidylarginine deiminase type II: molecular cloning, gene organization, and expression in human skin. Arch Biochem Biophys. 2002;407(1):25–31. doi: 10.1016/S0003-9861(02)00516-7. [DOI] [PubMed] [Google Scholar]
- Ishiyama N, Bates IR, Hill CM, et al. The effects of deimination of myelin basic protein on structures formed by its interaction with phosphoinositide-containing lipid monolayers. J Struct Biol. 2001;136(1):30–45. doi: 10.1006/jsbi.2001.4421. [DOI] [PubMed] [Google Scholar]
- Kucharczyk R, Zick M, Bietenhader M, et al. Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches. Biochim Biophys Acta. 2009;1793(1):186–199. doi: 10.1016/j.bbamcr.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Kumakura A, Asada J, Okumura R, Fujisawa I, Hata D. Diffusion-weighted imaging in preclinical Leigh syndrome. Pediatr Neurol. 2009;41(4):309–311. doi: 10.1016/j.pediatrneurol.2009.04.028. [DOI] [PubMed] [Google Scholar]
- Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2015 doi: 10.1002/ana.24551. [DOI] [PubMed] [Google Scholar]
- Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutgens LCHW, Deutz NEP, Gueulette J, et al. Citrulline: a physiologic marker enabling quantitation and monitoring of epithelial radiation-induced small bowel damage. Int J Radiat Oncol Biol Phys. 2003;57:1067–1074. doi: 10.1016/S0360-3016(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Mock DM. Determinations of biotin in biological fluids. In: McCormick DB, Suttie JW, Wagner C, editors. Methods in enzymology. New York: Academic; 1997. pp. 265–275. [DOI] [PubMed] [Google Scholar]
- Mori M, Mytingerm JR, Martin LC, Bartholomew D, Hickey S. 8993T>G-Associated Leigh syndrome with hypocitrullinemia on newborn screening. JIMD Rep. 2014;17:47–51. doi: 10.1007/8904_2014_332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin C, Mitchell G, Larochelle J, et al. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am J Hum Genet. 1993;53:488–496. [PMC free article] [PubMed] [Google Scholar]
- Naini A, Kaufmann P, Shanske S, Engelstad K, De Vivo DC, Schon EA. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox”. J Neurol Sci. 2005;229–230:187–193. doi: 10.1016/j.jns.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- Parfait P, de Lonlay P, von Kleist-Retzow JC, et al. The neurogenic weakness, ataxia and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia. Eur J Pediatr. 1999;158:55–58. doi: 10.1007/s004310051009. [DOI] [PubMed] [Google Scholar]
- Rabier D, Diry C, Rotig A, et al. Persistent hypocitrullinaemia as a marker for mtDNA NARP T8993G mutation? J Inherit Metab Dis. 1998;21:216–219. doi: 10.1023/A:1005391300203. [DOI] [PubMed] [Google Scholar]
- Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- Ribes A, Riudor E, Valcarel R, et al. Pearson syndrome: altered tricarboxylic acid and urea-cycle metabolites, adrenal insufficiency and corneal opacities. J Inherit Metab Dis. 1993;16(3):537–540. doi: 10.1007/BF00711675. [DOI] [PubMed] [Google Scholar]
- Said HM, Redah R. Ontogenesis of the intestinal transport of biotin in the rat. Gastroenterology. 1988;94(1):68–72. doi: 10.1016/0016-5085(88)90611-7. [DOI] [PubMed] [Google Scholar]
- Said HM, Ortiz A, McCloud E, Dyer D, Moyer MP, Rubin S. Biotin uptake by human colonic epithelial NCM460 cells: a carrier-mediated process shared with pantothenic acid. Am J Physiol. 1998;275(5):C1365–C1371. doi: 10.1152/ajpcell.1998.275.5.C1365. [DOI] [PubMed] [Google Scholar]
- Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol. 1993;34:827–834. doi: 10.1002/ana.410340612. [DOI] [PubMed] [Google Scholar]
- Sofou K, De Coo IF, Isohanni P, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9:52. doi: 10.1186/1750-1172-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector R, Mock DM. Biotin transport through the blood–brain barrier. J Neurochem. 1987;48:400–404. doi: 10.1111/j.1471-4159.1987.tb04107.x. [DOI] [PubMed] [Google Scholar]
- Spector R, Mock DM. Biotin transport and metabolism in the central nervous system. Neurochem Res. 1988;13:213–219. doi: 10.1007/BF00971535. [DOI] [PubMed] [Google Scholar]
- Thorburn DR, Rahman S (2014) Mitochondrial DNA-associated Leigh syndrome and NARP. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) GeneReviews™[Internet]. University of Washington, Seattle, 1993–2013; (updated 2014 Apr 17, http://www.ncbi.nlm.nih.gov/books/NBK1173/)
- van Karnebeek CDM, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet. 2014;94:453–461. doi: 10.1016/j.ajhg.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SL, Shanske S, McGill JJ, et al. Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue or age-related variation. J Inherit Metab Dis. 1999;22:899–914. doi: 10.1023/A:1005639407166. [DOI] [PubMed] [Google Scholar]
- White SL, Collins VR, Wolfe R, et al. Genetic counselling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. 1999;65:474–482. doi: 10.1086/302488. [DOI] [PMC free article] [PubMed] [Google Scholar]