

Abstract
Troyer syndrome is an autosomal recessive form of hereditary spastic paraplegia (HSP) caused by deleterious mutations in the SPG20 gene. Although the disease is associated with a loss of function mechanism of spartin, the protein encoded by SPG20, the precise pathogenesis is yet to be elucidated. Recent data indicated an important role for spartin in both mitochondrial maintenance and function. Here we report a child presenting with progressive spastic paraparesis, generalized muscle weakness, dysarthria, impaired growth, and severe isolated decrease in muscle cytochrome c oxidase (COX) activity. Whole exome sequencing identified the homozygous c.988A>G variant in SPG20 gene (p.Met330Val) resulting in almost complete loss of spartin in skeletal muscle. Further analyses demonstrated significant tissue specific reduction of COX 4, a nuclear encoded subunit of COX, in muscle suggesting a role for spartin in proper mitochondrial respiratory chain function mediated by COX activity. Our findings need to be verified in other Troyer syndrome patients in order to classify it as a form of HSP caused by mitochondrial dysfunction.
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2016_580) contains supplementary material, which is available to authorized users.
Keywords: Cytochrome c oxidase, Hereditary spastic paraplegia, Mitochondria, Oxidative phosphorylation, SPG20, Troyer syndrome
Introduction
Hereditary spastic paraplegia (HSP) comprises a highly heterogeneous group of neurogenetic disorders with multiple identified causative genes involved in various cellular pathophysiologic pathways. HSP is further classified as pure or complicated depending on accompanying clinical (mainly neurologic) features. In general complicated HSP is mainly inherited as autosomal recessive and is associated with variable manifestations such as intellectual disability, peripheral neuropathy, optic atrophy, and others (Hensiek et al. 2015). Several HSP forms may be caused by mutations in genes involved in mitochondrial function. Few recent examples include FARS2 (Yang et al. 2016), C12ORF65 (Shimazaki et al. 2012), SPG7 (Shanmughapriya et al. 2015), HSPD1 (Bross et al. 2008), IBA57 (Lossos et al. 2015), and others.
Troyer syndrome (MIM # 275900) is an autosomal recessive complicated HSP characterized in addition by distal amyotrophy, dysarthria, cerebellar signs, developmental delay, and short stature caused by mutations in the SPG20 gene leading to loss of the encoded spartin protein (Patel et al. 2002). Since the initial identification of the founder c.1110delA mutation in the Amish population only two other mutations have been reported, both predicted deleterious and associated with the Troyer phenotype (Manzini et al. 2010; Tawamie et al. 2015; Alazami et al. 2015; Butler et al. 2016). Herein, we report a patient with a novel homozygous mutation in SPG20 gene with concomitantly reduced mitochondrial cytochrome c oxidase (COX) activity, suggesting a link between spartin and the mitochondrial respiratory chain.
Patients and Methods
Case Report
The patient is the first child of healthy Arab Muslim parents who are first degree cousins once removed. She has twin male siblings with confirmed glycogen storage disease type 1a. The patient was born vaginally at term with no further complications. Her pregnancy was unremarkable except for intrauterine growth retardation initially documented at the third trimester. Her birth weight was 2,270 g and the Apgar score was normal. Growth retardation was a problem since infancy as weight and height remained below the third centile. Development was mildly delayed as she started to walk at 1 year and 9 months and to gain her first words at 2 years. At the age of 5 years she was referred for neurological evaluation due to global developmental delay dominated by hypotonia and gross motor impairment. At that time she already displayed dysarthric speech. A year later she was diagnosed with attention deficit disorder and treatment with methylphenidate was started.
Disease course was progressive with gait disturbance being the major problem manifested by significant clumsiness while walking, recurrent falls, and instability associated with further worsening of dysarthria. At the age of 8 years intention tremor occurred in both hands. This was relatively mild at rest but exacerbated during exercise or anxiety. Physical examination at that time showed weight and height below the third centile, elongated face with drooling, and reduced facial movements compatible with pseudobulbar palsy. She had generalized muscle wasting reflected by pectus carinatum deformity and clear atrophy of her four limbs as well as distal amyotrophy. Her neurological examination revealed mildly delayed cognitive skills, positive Gower sign consistent with proximal muscle weakness, generalized tendon hyperreflexia predominantly in the lower limbs associated with bilateral ankle clonus and positive Babinski sign. In addition upper extremities dysmetria was also noted. Brain MR imaging at this age was normal.
The patient had an extensive metabolic investigation that included among others blood count, serum creatine phosphokinase, liver transaminases, renal functions, thyroid hormones, repeated lactate and ammonia in plasma, amino acids in plasma and urine, acylcarnitine profile, organic acids in urine, total plasma homocysteine, serum transferrin isoelectric focusing, and serum very long chain fatty acids all of which were normal. Serum fibroblast growth factor 21 (FGF21), a novel biomarker for mitochondrial diseases (Suomalainen et al. 2011), was mildly elevated to 455 pg/ml (normal range < 200 pg/ml). Chromosomal microarray assay did not reveal pathological copy number variations.
Methods
Biochemical Assay of OXPHOS
Enzymatic activities of the five respiratory chain complexes, rotenone sensitive NADH CoQ reductase (Complex I), succinate cytochrome c reductase (complex II + III), succinate dehydrogenase (complex II) COX (complex IV), and Mg2+ATPase (complex V) were determined in isolated muscle mitochondria from the patient. We used the same methodology as the one employed in a previous study (Saada et al. 2004). The activities were normalized to citrate synthase and compared to normal control means.
Whole Exome Sequencing
Exonic sequences from DNA sample of the patient were enriched with the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA, USA). Sequences (100-bp paired-end) were generated on a HiSeq2000 (Illumina, San Diego, CA, USA). Read alignment and variant calling were performed with DNAnexus (Palo Alto, CA, USA) using default parameters with the human genome assembly hg19 (GRCh37) as reference.
Western Blot Analysis of Muscle and Fibroblasts
Proteins from muscle and fibroblast homogenates were treated with sample solubilization buffer and separated by SDS-polyacrylamide gel electrophoresis on a 12% gel. Proteins were transferred to PVDF membranes and probed with the indicated antibodies using the Enhanced Chemiluminescence Western blotting method as according to the manufacturer’s instruction (Biological Industries, HaEmek, Israel). The primary antibodies used were mouse-anti-B-tubulin-loading control (Santa Cruz Biotechnology), mouse-anti-COX 1, 2, 4 subunits (Molecular Probes), and rabbit anti-SPG20 (Proteintech). The secondary antibodies were HRP-conjugated anti-mouse and anti-rabbit (Jackson).
Results
Muscle biopsy from the right quadriceps was obtained at the age of 9 years. Light microscopy, immunohistochemical staining including COX, and electron microscopy were largely normal (supplementary Fig. S1). Enzymatic activities of the five respiratory chain complexes determined in muscle and mitochondria assays disclosed significantly reduced COX (respiratory chain complex IV) activity in muscle mitochondria, 209 nmol/min/mg (controls 1,061 ± 47 nmol/min/mg) which is 25% of control mean when normalized to citrate synthase (CS). The activities of complexes I–III and V were within normal limits. However, COX activity was normal in fibroblast homogenate (Table 1). ATP production in fibroblasts was tested normal both on glucose and galactose.
Table 1.
Enzymatic activities of mitochondrial respiratory chain complexes
| Assaya | Muscle mitochondria | Fibroblast homogenate | ||
|---|---|---|---|---|
| Patient | Controls ± SD (range) n = 50 | Patient | Control mean n = 4 | |
| CI | 106 [98%] | 98 ± 46 (46–215) | nd | nd |
| CI + III | 159 [65%] | 305 ± 148 (104–717) | nd | nd |
| CII | 141 [116%] | 124 ± 57 (52–230) | nd | nd |
| CII + III | 75 [105%] | 80 ± 40 (41–172) | nd | nd |
| CIV | 209 [25%] | 1,061 ± 47 (265–2,896) | 61 [115%] | 72 ± 22 (62–135) |
| CS | 980 | 1,132 ± 438 (438–2,350) | 42 | 45 ± 12 (30–66) |
nd not determined
anmol/min/mg (%) percentage residual activity normalized to citrate synthase (CS) activity compared to controls
Molecular genetic investigations for the patient and her family were initiated after obtaining the relevant written informed consents and local ethical review board approval. Given the complicated HSP phenotype and the presumed nuclear encoded mitochondrial impairment we elected to proceed with whole exome sequencing (WES) under a rare allele, recessively inherited hypothesis. WES of the patient sample yielded 42.5 million mapped reads with a mean coverage of X56. Following alignment and variant calling, we performed a series of filtering steps. These included removing variants called less than X8, were off-target, synonymous, heterozygous, or had MAF > 0.5% at ExAC (Exome Aggregation Consortium, Cambridge, MA [URL: http://exac.broadinstitute.org]). We also removed variants with MAF > 4% at the Hadassah in-house database (~800 ethnic matched exome analyses) or variants which were predicted benign (Mutation Taster, http://mutationtaster.org/). Only three variants survived this filtering (supplementary Table S1). Of these three variants only SPG20 is associated with HSP phenotype whereas the other two variants are associated with completely unrelated phenotypes and non-recessive inheritance.
WES underscored a homozygous c.988A>G in the SPG20 gene, predicting the substitution of highly evolutionary conserved methionine at position 330 with valine (p.Met330Val) as the only relevant HSP-targeted gene. This previously unreported variant was predicted to be disease causing by Mutation Taster (http://mutationtaster.org), Polyphen 2 (http://genetics.bwh.harvard.edu/pph2), and SIFT (http://siftdna.org). Both parents were heterozygous for this variant and it segregated perfectly in the family.
In order to further assess the pathogenicity of this c.988A>G variant, Western blot analysis was performed on homogenate muscle tissue and fibroblasts from the patient using commercial monoclonal antibodies. There was an almost complete reduction in steady state SPG20 protein level in patient’s muscle and decrease but to a lesser degree in patient’s fibroblasts (Fig. 1a).
Fig. 1.
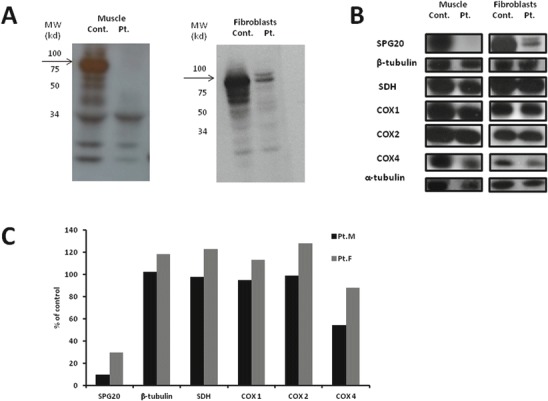
Western blot of spartin and COX subunits in muscle and fibroblasts. (a) Control and patient’s muscle (right) and fibroblasts (left) were exposed to anti-spartin antibodies and detected with Enhanced Chemiluminescence. The location of molecular weight markers is marked and the spartin protein (arrow) is located as expected at 85 kd. (b) Western blot in muscle (right) and fibroblasts (left) specific antibodies after exposure to Enhanced Chemiluminescence. (c) Quantification relative to control (Image J software). Pt. M = patient’s muscle, Pt. F = patient’s fibroblasts
Discussion
Our patient presented with clinical features consistent with Troyer syndrome. This phenotype is usually attributed to deleterious mutations in SPG20 gene leading to loss of expression of the encoded protein termed spartin (Bakowska et al. 2008). In contrast with previously reported mutations that were all caused by frameshift mutations resulting in early truncation and absent protein (Patel et al. 2002; Manzini et al. 2010; Alazami et al. 2015; Butler et al. 2016) the missense mutation identified in our patient was not predicted to disrupt the protein. This mutation was not located in either of the two conserved domains of the spartin, i.e., the microtubule interacting and trafficking motif and the plant-related senescence domain but it is located in an evolutionary highly conserved residue (supplementary Fig. S2) rendering an unstable protein as suggested by the absence of spartin demonstrated by Western blot analysis. In order to assess for absence of mRNA resulting in lack of protein, RNA was isolated from control and patient’s fibroblasts, reverse transcribed and the cDNA was subjected to PCR using the following primers: Forward: GCT CAG AGG GAT CAG CAT TT and Reverse: CCT CCT TTA CTT CCT TCG TCT. No difference in the length and abundance was noted (data not shown). Notably, spartin was almost completely depleted in muscle compared with some residual protein in fibroblasts thus inferring tissue variability (Fig. 1b, c).
Spartin is a multifunctional protein widely expressed in human tissues and is localized to several cellular organelles and compartments including endosomes, lipid droplets, and mitochondria (Bakowska et al. 2007; Eastman et al. 2009; Lu et al. 2006). The precise role of spartin in mitochondrial function is yet to be elucidated but elegant studies by Joshi et al. illustrated that spartin binds to mitochondrial cardiolipin via its plant-related senescence domain where it associates with mitochondrial outer membrane playing a crucial role in mitochondrial Ca+2 influx which is essential in the maintenance of mitochondrial membrane potential (Joshi and Bakowska 2011). Furthermore, recent evidence obtained from studies on C. elegans SPG20 orthologue showed a protective effect of spartin on mitochondrial oxidation stress thereby allowing more effective mitochondrial ATP production in wild type worms compared with animals harboring a null allele (Truong et al. 2015).
A major clinical feature observed in our patient was abnormal skeletal muscle function manifested by generalized muscle wasting, facial weakness, and Gower sign suggesting proximal muscle weakness as well as clear evidence of distal muscle weakness. These manifestations prompted us to perform a muscle biopsy which revealed severe isolated reduction in COX activity relative to CS. The seemingly unremarkable COX stain is due the qualitative rather than quantitative nature of the histochemical stain. Importantly, the decreased COX activity was in correlation with the almost undetectable spartin protein levels in muscle while intact COX activity in fibroblasts where spartin was clearly present albeit in lower amount. Moreover, the nuclear encoded subunit of COX, COX 4, was significantly reduced in muscle but only mildly decreased in fibroblasts (Fig. 1b, c). Given that spartin associates with cardiolipin located mainly in the mitochondrial outer membrane (Joshi and Bakowska 2011) its effect on the inner mitochondrial membrane COX enzyme is not intuitively explained. We therefore speculate a link between spartin and mitochondrial oxidative phosphorylation mediated by COX activity via COX4 although the exact mechanism remains to be elucidated. This may explain the tissue specificity seen in our patient where high energy producing tissues such as skeletal muscles are more severely affected following the lack of spartin expression. However, similar findings should be verified in another patient with Troyer syndrome in order to confirm pathogenesis caused by mitochondrial dysfunction. Of note, tissue specific COX assembly defects were previously reported with variable levels of the enzymatic subcomplexes (Stiburek et al. 2005) but the profile seen in our patient is yet uncommon. Noteworthy, another form of HSP caused by mutations in the SPG7 gene encoding paraplegin, a mitochondrial metallopeptidase result in a secondary assembly defect of mitochondrial complex I of the respiratory chain of affected patients (Atorino et al. 2003).
In conclusion Troyer syndrome is a complex disease with variable clinical features caused by lack of spartin in various tissues including skeletal muscle, central nervous system, growth plates of bones, and adipose tissue. Although the pathogenesis of the disease is not completely understood, growing evidence implies that mitochondrial dysfunction plays a major role. Our study supports this assumption and adds more evidence to disease pathogenesis that needs to be further investigated in future research.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Suspected variants identified by WES (DOCX 29 kb)
A. Light microscopy showing normal structure of skeletal muscle. H&E stain X200. B. Skeletal muscle immune-histochemistry showing normal COX staining (JPG 326 kb)
High evolutionary conservation of Met residue at position 330 is illustrated (the mutated residue is marked by black box) (JPG 161 kb)
Acknowledgements
Corinne Alban is acknowledged for technical assistance. This research was in part funded by the Hadassah Compensatory fund.
Authors’ Contributions
Dr. Spiegel leads the composition and evaluation of the manuscript; designed and conceptualized the study, and interpreted the data. Dr. Spiegel serves as guarantor for the article, accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Dr. Soiferman evaluated the manuscript for content and analyzed and interpreted the biochemical studies as well as Western blot analyses.
Dr. Shaag evaluated the manuscript for content and analyzed and interpreted the genetic data.
Prof. Shalev evaluated the manuscript for content and analyzed and interpreted the clinical data.
Prof. Elpeleg evaluated the manuscript for content, including medical writing for content, analyzed and interpreted the genetic data in particular the exome sequencing.
Prof. Saada leads the composition and evaluation of the manuscript, designed and conceptualized the study, and analyzed and interpreted the biochemical data as well as the Western blot analysis.
Conflict of Interests
Dr Spiegel, Dr. Soiferman, Dr. Shaag, Prof. Shalev, Prof. Elpeleg and Prof. Saada all declare that they have no conflict of interests.
Ethics and Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained accordingly as approved by the local ethical review board.
Contributor Information
Ronen Spiegel, Email: spiegelr@zahav.net.il, Email: spiegel_ro@clalit.org.il.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Atorino L, Silvestri L, Koppen M, et al. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol. 2003;163:777–787. doi: 10.1083/jcb.200304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowska JC, Jupille H, Fatheddin P, Puertollano R, Blackstone C. Troyer syndrome protein spartin is mono-ubiquitinated and functions in EGF receptor trafficking. Mol Biol Cell. 2007;18:1683–1692. doi: 10.1091/mbc.E06-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowska JC, Wang H, Xin B, Sumner CJ, Blackstone C. Lack of spartin protein in Troyer syndrome: a loss-of-function disease mechanism? Arch Neurol. 2008;65:520–524. doi: 10.1001/archneur.65.4.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bross P, Naundrup S, Hansen J, et al. The Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J Biol Chem. 2008;283:15694–15700. doi: 10.1074/jbc.M800548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler S, Helbig KL, Alcaraz W, Seaver LH, Hsieh DT, Rohena L. Three cases of Troyer syndrome in two families of Filipino descent. Am J Med Genet A. 2016;170(7):1780–1785. doi: 10.1002/ajmg.a.37658. [DOI] [PubMed] [Google Scholar]
- Eastman SW, Yassaee M, Bieniasz PD. A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J Cell Biol. 2009;184:881–894. doi: 10.1083/jcb.200808041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensiek A, Kirker S, Reid E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J Neurol. 2015;262:1601–1612. doi: 10.1007/s00415-014-7598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi DC, Bakowska JC. SPG20 protein spartin associates with cardiolipin via its plant-related senescence domain and regulates mitochondrial Ca2+ homeostasis. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0019290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossos A, Stümpfig C, Stevanin G, et al. Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology. 2015;84:659–667. doi: 10.1212/WNL.0000000000001270. [DOI] [PubMed] [Google Scholar]
- Lu J, Rashid F, Byrne PC. The hereditary spastic paraplegia protein spartin localises to mitochondria. J Neurochem. 2006;98:1908–1919. doi: 10.1111/j.1471-4159.2006.04008.x. [DOI] [PubMed] [Google Scholar]
- Manzini MC, Rajab A, Maynard TM, et al. Developmental and degenerative features in a complicated spastic paraplegia. Ann Neurol. 2010;67:516–525. doi: 10.1002/ana.21923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Cross H, Proukakis C, et al. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- Saada A, Bar-Meir M, Belaiche C, Miller C, Elpeleg O. Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal Biochem. 2004;335:66–72. doi: 10.1016/j.ab.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, et al. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell. 2015;60:47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki H, Takiyama Y, Ishiura H, et al. A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55) J Med Genet. 2012;49:777–784. doi: 10.1136/jmedgenet-2012-101212. [DOI] [PubMed] [Google Scholar]
- Stiburek L, Vesela K, Hansikova H, et al. Tissue-specific cytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1. Biochem J. 2005;392:625–632. doi: 10.1042/BJ20050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A, Elo JM, Pietiläinen KH, et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawamie H, Wohlleber E, Uebe S, Schmäl C, Nöthen MM, Abou Jamra R. Recurrent null mutation in SPG20 leads to Troyer syndrome. Mol Cell Probes. 2015;29:315–318. doi: 10.1016/j.mcp.2015.05.006. [DOI] [PubMed] [Google Scholar]
- Truong T, Karlinski ZA, O’Hara C, Cabe M, Kim H, Bakowska JC. Oxidative stress in Caenorhabditis elegans: protective effects of spartin. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0130455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu W, Fang Z, et al. A newly identified missense mutation in FARS 2 causes autosomal recessive spastic paraplegia. Hum Mutat. 2016;37:165–169. doi: 10.1002/humu.22930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suspected variants identified by WES (DOCX 29 kb)
A. Light microscopy showing normal structure of skeletal muscle. H&E stain X200. B. Skeletal muscle immune-histochemistry showing normal COX staining (JPG 326 kb)
High evolutionary conservation of Met residue at position 330 is illustrated (the mutated residue is marked by black box) (JPG 161 kb)