

Abstract
Background
The imprinted small nucleolar RNA (snoRNA) Snord116 is implicated in the aetiology of Prader-Willi syndrome (PWS), a disease associated with hyperphagia and obesity. Germline deletion of Snord116 in mice has been found to lead to increased food intake but not to the development of obesity. To determine the role of Snord116 independent of potential compensatory developmental factors, we investigated the effects of conditional adult-onset deletion of Snord116 in mice.
Methods
Deletion of Snord116 was induced at 8 weeks of age by oral administration of tamoxifen to male Snordlox/lox; ROSAcre/+ mice, with vehicle-treated mice used as controls. Body weight (BW) was monitored weekly and body composition was measured by dual-energy X-ray absorptiometry and tissue dissection. Non-fasted and fasting-induced food intake was determined, and glucose and insulin tolerance tests were performed. Twenty-four-hour energy expenditure and physical activity were assessed by indirect calorimetry.
Results
Adult-onset deletion of Snord116 led to reduced food intake and increased adiposity, albeit with no concomitant change in BW or lean mass compared to controls. Adult onset Snord116 deletion was also associated with worsened glucose tolerance and insulin sensitivity.
Conclusions
This study identified a key role for Snord116 in feeding behaviour and growth. Further, it is likely that the effects of this gene are modulated by developmental stage, as mice with adult-onset deletion showed an opposite phenotype, with respect to food intake and body composition, to previously published data on mice with germline deletion.
Keywords: Mouse, neurodevelopmental disorders, Prader-Willi syndrome (PWS), Snord116, small nucleolar RNA (snoRNA)
Introduction
Prader-Willi syndrome (PWS), a genomic imprinting disorder, is the leading genetic cause of obesity, characterized by failure-to-thrive and muscular hypotonia in infancy followed by hyperphagia and development of obesity in childhood and adulthood (1). The genetic locus which is affected in PWS is a large imprinted region on chromosome 15 encompassing a number of coding and non-coding genes (2). The PWS locus is conserved between species, with only minor alterations between humans and mice (3).
The degree of conservation between the human and mouse PWS loci offers a valuable means of studying the contribution of individual genes to the syndrome—genetic knockout (KO) mouse models. In recent years, a number of single- and multi-gene deletion models, demonstrating some phenotypic features of PWS, have been developed (4,5). As the critical PWS region has been narrowed down and refined over time through human genetic analyses, so have PWS mouse models concentrated more and more on specific genes within that region using sophisticated gene targeting methods.
A number of patients with micro-deletions in the PWS critical region have been identified. Of these, the smallest deletions that give rise to classical PWS phenotypic features are in a region that encompasses the small nucleolar RNA (snoRNA) SNORD116 (6-10). The analogue of this gene in mice, Snord116, is present in the corresponding murine PWS critical region, conserved on chromosome 7 (11,12). Snord116 is the first snoRNA to be investigated via KO mouse models. Thus far two independently engineered models have been created which share some common phenotypic features, as outlined below.
Skryabin et al. reported that their Snord116 deficient mice had a slower growth pattern from postnatal day 5 to adulthood compared to wildtype (WT) (13). KO animals did not become obese and fertility was conserved. Food intake, body composition and metabolic parameters were not investigated in that study.
A more in-depth study assessed the metabolic and behavioural effects of an independently generated Snord116 deletion mouse line (14), which reported stunted early growth beginning from postnatal day 2. Mice survived to adulthood but remained smaller than WT and, while normal fertility was observed, there was a delay in sexual maturation. Behavioural tests showed that these Snord116-deficient mice had increased anxiety and a defect in motor learning, although motor function was normal. This mouse model was the first in PWS research to display hyperphagia. Strikingly, food intake was not different to that of controls at 7 weeks of age but hyperphagia developed with age in Snord116-deficient mice, with males exhibiting increased food intake (corrected for BW) at 3 months and females at 6 months of age. This hyperphagia was accompanied by elevated circulating levels of the gut-derived anorexigenic hormone ghrelin, but did not result in obesity. To partially explain this apparently discrepant finding of hyperphagia in the absence of obesity, the authors investigated parameters related to energy expenditure and reported increased oxygen consumption and a stronger preference for carbohydrate oxidation in Snord116-deficient mice versus controls. This finding is in contrast to human PWS, in which reduced resting energy expenditure in line with lower lean mass has been observed (15-18).
The finding that germline deletion of Snord116 in mice leads to hyperphagia is the first proof in an animal model that this molecule is critical for the development of PWS. Indeed, the fact that hyperphagia is only observed after an initial period of failure to thrive aligns closely with the clinical progression of PWS in humans. The elevated ghrelin levels and increased anxiety observed (14) also mimic features of the syndrome.
It has been suggested that postnatal failure to thrive could itself shape the later hyperphagic stage of PWS (14). In this scenario, hypotonia and poor suck in neonates causes malnutrition and stunted growth at a period when significant development is taking place in appetite-regulatory neuronal pathways. It is hypothesized that the brain is then rewired, in a compensatory manner, in favour of a strong drive to eat and accumulate adipose tissue, leading to hyperphagia and obesity in the second stage of the disease. Although it is not possible to test this hypothesis in humans, recently developed gene manipulation techniques allow the generation of inducible KO mouse models, in which gene deletion can be induced at any stage of development.
In the current study, we aimed to determine the role of Snord116 in the regulation of growth and energy homeostasis independent of any compensatory developmental influences. In order to do this, we assessed the metabolic phenotype of a mouse model in which Snord116 was globally deleted in adult mice.
Methods
Generation of mouse lines
Mice with loxP sites flanking the Snord116 locus (14) were obtained from Jackson laboratory. Conditional deletion mice (mice in which deletion has been targeted to a specific time point) were generated by crossing these Snord116lox/lox mice with mice with a copy of the Cre-recombinase gene fused to the estrogen receptor under the control of the ubiquitin promoter (Snordlox/lox; ROSAcre/+). Deletion of Snord116 was induced at 8 weeks of age by the oral administration of 2.5 mg/kg tamoxifen (tamoxifen dissolved in 100% ethanol at a dilution of 1 mg/µL and mixed into 0.5 g of peanut butter; n=7). Vehicle-treated Snordlox/lox;ROSAcre/+ littermates were used as controls (n=8).
Animal experiments
All animal experiments for this study were approved by the Garvan Institute/St Vincent’s Hospital Animal Ethics Committee and conducted in accordance with relevant regulations and guidelines. Mice were housed in groups of 2–5 under controlled temperature conditions (22 °C), with a 12-hour light, 12-hour dark cycle (lights on at 7:00 am) and standardized environmental enrichment. Mice had ad libitum access to water and a normal chow diet (8% of energy from fat, 21% from protein and 71% from carbohydrate, 10.9 kJ/g; Gordon’s Specialty Stock Feeds, Yanderra, NSW, Australia).
Body weight (BW) and composition
BW was measured at weekly intervals, always at the same time of day, from 8 weeks of age onwards. To assess body composition (whole body lean mass and fat mass) at 8 weeks of age (prior to tamoxifen administration) and at 14 weeks of age, dual-energy X-ray absorptiometry (DXA; Lunar PIXImus2 mouse densitometer, GE Medical Systems Lunar, Madison, WI, USA) was performed on isoflurane-anaesthetized mice, ventral side down. The heads of the mice were excluded from analysis, which was performed using software provided by the manufacturer.
Food intake
Food intake was measured at 11 weeks of age, both under non-fasted conditions (spontaneous food intake) and in response to an overnight fast (fasting-induced food intake). To acclimatize to experimental conditions, mice were individually housed for 3 days before the experiment and fed powdered chow. They were then transferred to individual specialized Nalgene metabolic chambers (Medtex, Notting Hill, VIC) and given ad libitum access to powdered chow and water. Food spillage, water spillage, faeces and urine were collected into discrete compartments. BW, hopper weight and faecal output were recorded over three consecutive days, and the amount of food consumed (i.e., food given minus food remaining) was calculated on a daily basis.
To determine the effect of fasting on food intake, animals’ food was removed at 5:00 pm on the day before the experiment. Food was given at 9:00 am the following day and food intake was measured as described above at 1, 2, 4, 8, 24, 48 and 72 hours after reintroduction of food. BW was monitored daily.
Indirect calorimetry
Energy expenditure and substrate utilization were measured at 13 weeks of age by indirect calorimetry using an eight-chamber open-circuit calorimeter (Oxymax Series; Columbus Instruments, OH, USA). Locomotor activity was also determined in these chambers using an OPTO-M3 sensor system (Columbus Instruments). An airflow rate of 0.6 L/min was maintained, and mice were acclimatized to chambers for 24 hours before the beginning of recording. Oxygen consumption (VO2) and carbon dioxide production (VCO2) were measured and respiratory exchange ratio (RER) was calculated as VCO2/VO2. Energy expenditure (in kCal) was calculated as calorific value (CV) × VO2, where CV =3.815+1.232× RER. Where specified, energy expenditure was normalized to lean body mass as determined by DXA as described below. Physical activity was assessed as ambulatory counts, consecutive beam breaks in the X and Y horizontal dimensions. Data collected in the 24-hour measurement period were expressed as hourly averages, and physical activity data were presented as hourly sums.
Glucose tolerance tests
Glucose tolerance tests were performed when mice were 12 weeks of age. The day prior to the procedure, food was removed from the hopper at 5:00 pm and mice were transferred to clean cages with fresh bedding. Animals were fasted overnight for 20 hours, with the glucose tolerance test performed at 1:00 pm the next day. A 10% glucose solution for injection was prepared by diluting a sterile solution of 50% w/v glucose (Pharmalab, Lane Cove, NSW, Australia) with sterile physiological saline. Glucose was injected intraperitoneally at a dose of 1 mg/kg BW in a volume of 10 µL/g BW. Blood was taken from the tip of the tail immediately prior to injection (0 minutes) and at 15, 30, 60 and 90 minutes post-injection, and blood glucose concentrations were measured with an Accu-chek® Go glucometer (Roche, Dee Why, NSW, Australia).
Insulin tolerance tests
Four days after the glucose tolerance test, animals of 12 weeks of age were fasted from 9:00 am on the day of the procedure, by placing them in clean cages as described above. The insulin tolerance test was performed at 2:00 pm after 5 hours’ fast. A 0.1 ìU/ìL insulin solution was prepared by diluting 20 µL of hospital-grade insulin (Actrapid, Novo Nordisk, Baulkham Hills, NSW, Australia) with 20 mL of sterile physiological saline. Insulin was administered by intraperitoneal (IP) injection at a dose of 0.75 µU/g BW. Whole blood glucose was measured in blood collected from the tip of the tail immediately prior to injection (0 minutes), as well as at 15, 30, 60 and 90 minutes post-injection.
Tissue collection
At 14 weeks of age, mice were sacrificed by cervical dislocation and decapitation between 1:00 and 5:00 pm. White adipose tissue (WAT) depots [right (R) inguinal, R epididymal, R retroperitoneal and mesenteric], intrascapular brown adipose tissue, pancreas, liver, spleen, R kidney, heart, R testis and seminal vesicle were excised and weighed. Brains were collected, frozen on dry ice and stored at −80 °C for subsequent sectioning and in situ hybridization as described below.
Brain sectioning
Frozen brains were embedded in OCT medium (Tissue-Tek OCT compound, Sakura Finetek USA, Torrance, CA, USA). Coronal brain sections of 25 µm thickness were cut with a cryostat (CM3050S, Leica Microsystems, Wetzlar, Germany) and thaw-mounted on superfrost glass slides (SuperFrost Plus®, Menzel-Gläser, Braunschweig, Germany). Slides were stored at −20 °C until subsequent and in situ hybridization.
In situ hybridization for determination of Snord116 mRNA expression
In order to establish the effectiveness of our adult-onset Snord116 deletion strategy, in situ hybridization was performed to assess hypothalamic Snord116 mRNA expression. This was done according to the method of Young (19), with minor modifications. Unless specified otherwise, all steps were carried out at room temperature. Mounted sections were fixed in 2% paraformaldehyde (PFA) for 10 minutes on ice, then washed with phosphate-buffered saline (PBS). After acetylation with acetic anhydride in TEA, sections were dehydrated by successive incubation in an increasing ethanol (EtOH) concentration gradient (70–100%), chloroform, 100% EtOH and 90% EtOH.
Hybridization was carried out overnight at 42 °C in a humidity chamber (Hybaid Omniside Thermal Cycler, Thermo Scientific, Milford, MA, USA). Sections were incubated with radio-labelled DNA oligonucleotides complementary to murine Snord116 mRNA ('5-GTTCAGCTTTTCCAAGGAATGTTTGACTGGGAATCATCATAGATCC-3'; Sigma-Aldrich Inc., St Louis, MO, USA).
The following day, sections were washed in 5× SSC buffer with 1M dithiothreitol (DTT), then incubated at 40 °C in 2× SSC/50% formamid first with and then without 1M DTT. After cooling to room temperature, sections were washed in 1× SSC, distilled water, then 70% EtOH. Finally, sections were air-dried for three hours and exposed to Kodak Biomax film (Kodak, Rochester, NY, USA) for 3 hours in complete darkness.
Expression levels of Snord116 mRNA were quantified by densitometry with ImageJ software (US National Institutes of Health).
Statistical analysis
Analyses were performed using GraphPad Prism software (version 6.0, GraphPad software, San Diego, CA). Differences in individual parameters between groups were assessed using Student’s t-test. Time course or multiple-parameter analyses were performed using two-way ANOVA for repeated measures and, where appropriate, calculation of area under the curve (AUC). Significance, unless specified as a genotype/time interaction, indicates a genotype effect. Data are expressed as mean ± standard error of the mean. A P value <0.05 was considered statistically significant.
Results
The Snordlox/lox;ROSAcre/+ mice in which the KO of Snord116 was induced by oral tamoxifen treatment was designated as the experimental group. Vehicle-treated littermate control mice were designated as the control group.
As shown in Figure 1, hypothalamic expression of Snord116 mRNA was significantly reduced by approximately 16% in the experimental group compared to controls. One reason for the low efficiency of Cre-mediated deletion could be the rather large size of the loxP flanked region of DNA (>150 KB) that needs to be removed. However, while this is not a 100% reduction in expression as is typically seen in germline KO mice, considering the extremely high levels of Snord116 mRNA expression within the brain (20) it still represents significant downregulation, potentially sufficient to cause relevant physiological changes.
Figure 1.
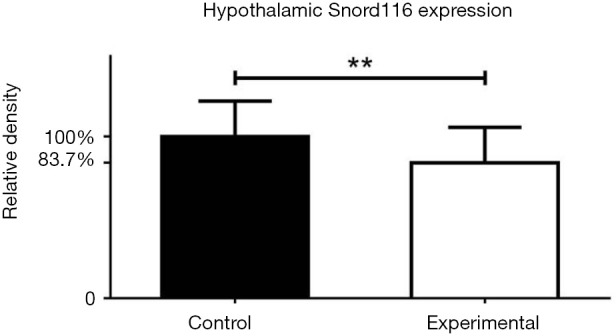
Conditional knockdown of Snord116. Expression levels of hypothalamic Snord116 mRNA in experimental and control mice, detected by in situ hybridisation, demonstrating partial deletion of Snord116. **, P<0.01; data expressed as mean ± SE.
Interestingly, in contrast to global germline deletion of Snord116, the conditional deletion of Snord116 in adult mice had no significant effect on BW or growth rate (Figure 2A). Analysis of body composition, assessed by DXA at 13 weeks of age did not show any significant differences in fat or lean mass between experimental and control mice (Figure 2B). When adipose tissue was analysed by the more precise measurement of masses of individually dissected adipose tissue depots, the experimental group exhibited significantly increased total WAT mass compared to controls (Figure 2C). There was no effect of Snord116 deletion on the weight of any of the organs dissected (Figure 2D).
Figure 2.

Growth and body composition. BW curve and body composition in experimental and control mice. (A) Weekly BW from 8 to 14 weeks of age; (B) total lean and fat mass as a percentage of BW at 13 weeks of age; (C) mass of dissected adipose tissue depots (% BW) at 14 weeks of age; (D) mass of dissected organs (% BW) at 14 weeks of age. Control group n=8; experimental group n=7. *, P<0.05; data expressed as mean ± SE. BW, body weight; WAT, white adipose tissue; BAT, brown adipose tissue; SV, seminal vesicle.
As hyperphagia is the most prominent feature of people with PWS and of germline Snord116 KO mice, we investigated whether adult-onset Snord116 knockdown also leads to changes in food intake by measuring spontaneous and fasting-induced food intake at 11 weeks of age. Unexpectedly, the experimental group exhibited decreased spontaneous food intake as a percentage of BW compared to controls, as well as reduced fasting-induced food intake measured over a 48-hour period as a percentage of BW (Figure 3A,B).
Figure 3.

Food intake at 11 weeks of age in experimental and control mice. (A) Three-day average basal food intake as a percentage of BW; (B) food intake 1, 2, 4, 8, 24 and 48 hours post-refeeding after overnight fast as a percentage of BW. Control group n=8; experimental group n=7. **, P<0.01; ***, P<0.005; data expressed as mean ± SE. BW, body weight.
In order to understand why decreased food intake was not accompanied by reduced BW or fat mass in the experimental group, energy metabolism and physical activity were measured at 13 weeks of age. Both experimental and control mice displayed classical 24-hour energy expenditure profiles, with energy expenditure higher in the dark, active phase than in the light, somnolent phase (Figure 4A). There were no differences between groups, either in absolute values or when normalized to lean body mass. However, there was a shift in the timing of energy expenditure changes between groups, as indicated by a significant time-genotype interaction (Figure 4A). RER values were similar between the experimental group and controls, in both the light and dark phases (Figure 4B). Similar to that observed in energy expenditure measurements, there was a shift in the timing of RER changes between groups, significant when analysed as a time-genotype interaction. The RER of the experimental group appeared to lag behind controls slightly in the transition between phases. Mice in the experimental group displayed a similar 24-hour physical activity pattern to controls, with no significant differences in the light or dark phase and no significant time-genotype interaction (Figure 4C).
Figure 4.

Indirect calorimetry and physical activity. Twenty-four-hour indirect calorimetry and physical activity assessment at 13 weeks of age in experimental and control mice (white area represents light phase, grey area represents dark phase). (A) Absolute energy expenditure; (B) RER; (C) ambulatory physical activity. Control group n=8; experimental group n=7. **, P<0.01; ***, P<0.005 (genotype/time interaction); data expressed as mean ± SE. RER, respiratory exchange ratio.
To investigate glucose homeostasis, glucose and insulin tolerance were measured at 12 weeks of age. Five-hour and 20-hour fasting glucose levels were not different between the experimental group and controls, as seen from the pre-injection (0 minute) time points on Figures 5A and 5C. The experimental group showed impaired glucose tolerance compared to controls, with elevated blood glucose levels seen in the 90 minutes after glucose injection and in the AUC (Figure 5A,B), and decreased insulin sensitivity compared to controls (Figure 5C,D).
Figure 5.

Glucose and insulin tolerance. Glucose and insulin tolerance tests at 12 weeks of age in experimental and control mice. (A) Ninety minutes blood glucose response to IP glucose administration; (B) blood glucose AUC during 90 minutes post glucose administration; (C) 90 minute blood glucose response to IP insulin administration; (D) blood glucose AUC during 90 minutes post insulin administration. Control group n=8; experimental group n=7. *, P<0.05; **, P<0.01; data expressed as mean ± SE. AUC, area under the curve; IP, intraperitoneal.
Discussion
To investigate the contribution of the imprinted snoRNA gene cluster Snord116 to PWS without developmental influences, we determined the effects of the global deletion of Snord116 when conferred in adult mice. This model showed some similarity with the features of human PWS but, interestingly, different effects were observed between it and a previously published germline Snord116 model (14). This suggests that the function of Snord116 is indeed modulated by developmental factors. Germline Snord116 deletion mice exhibited reduced BW (although growth rate from 12 weeks onwards was normal), hyperphagia, decreased physical activity and altered substrate oxidation (14). The present study, on the other hand, showed that mice with adult-onset Snord116 knockdown displayed normal growth, reduced food intake and increased adiposity without changes in BW. Both models exhibited impaired glucose tolerance.
It is striking that growth patterns of adult mice—whether they were germline or conditional Snord116-deficient—were the same as their respective controls. Once germline KOs overcame initial failure to thrive, they grew at a normal weight-adjusted rate (14), although they did not exhibit the “catch-up” growth seen in other models with postnatal underweight (21,22). The experimental group did not differ from controls in post-deletion BW or growth rate. This indicates that the growth-detrimental effects of Snord116 may only be conferred in the early stages of development.
The most arresting part of the phenotype of the experimental group in this study was that—in contrast to germline Snord116 KOs, which showed adult-onset hyperphagia—they showed reduced food intake compared to controls. The food intake changes observed in this study indicate that SNORD116 is indeed a likely candidate gene whose absence is involved in the appetite dysregulation in PWS, but the discrepancy between the present adult-onset conditional deletion and the previously-published germline deletion model (14) suggests that the mechanism behind this dysregulation is complex and influenced by extra-genetic factors, such as the stage of development at which the deletion is induced.
The growth/food intake phenotype seen in germline Snord116 models—i.e., initial underweight accompanied by hyperphagia in adulthood—accords with the theory that PWS hyperphagia may be induced by a hypercorrection of postnatal malnutrition (14). The phenotype resulting from adult-onset knockdown of Snord116 outlined in this study supports the corollary of this hypothesis: that when Snord116 deletion does not stunt growth, no compensatory hyperphagia ensues. In this model, early development was normal because the deletion of Snord116 had not yet been induced; upon the induction of the deletion in adulthood, hyperphagia was not observed. Thus this study suggests that, although Snord116 plays a role in the regulation of feeding behaviour, it is only associated with the development of hyperphagia when it is absent from an early age or for a longer period of time. Further, Snord116’s effects on food intake may act in compensation for initial failure to thrive.
Not only were mice in the experimental group not hyperphagic, they actually had decreased food intake compared to controls, which may parallel with failure to thrive in neonates. Despite this reduction in caloric intake, we saw increased adiposity in the experimental group. Miller et al. found that in infants with PWS, adiposity began to increase before the onset of hyperphagia, which followed soon after (23). It may be that mice with conditional knockdown of Snord116 follow a similar pattern. A long-term study would be required to assess whether these mice eventually move into a second phase of hyperphagia and obesity.
Energy expenditure, when expressed as a percentage of lean mass, was not different between the experimental group and controls. It is well known that lean tissue (in particular muscle and liver mass) is the main predictor of energy expenditure (24-26). The fact that correction for lean mass equalized 24-hour energy expenditure between groups suggests that muscle was metabolically equal between them. These results suggest that Snord116 does not play a major role in the regulation of cellular energy metabolism.
In conclusion, this is the first study to compare the effects of the adult-onset knockdown of Snord116. While the cre-mediated deletion of Snord116 was not complete, most likely due to the large size of the loxP flanked sequence and reduced efficiency under these circumstances, there was a significant reduction in Snord116 expression. Importantly, even this partial reduction of Snord116 expression in this model resulted in major phenotypic differences from germline Snord116 deletion mice, with the most marked of these being reduced food intake in the adult-onset deletion. Thus each model mimics a separate nutritional phase of PWS, suggesting a role for Snord116 in neurodevelopment. This study identified a role for Snord116 in the regulation of food intake, growth and fat accretion, and suggested that its effects are modulated by developmental stage. The absence of SNORD116 is likely to contribute to the feeding dysregulation seen in people with PWS, from the hypophagia of infancy to the hyperphagia of childhood and later life, thus making it a potential target for appetite therapies in the future.
Acknowledgements
Funding: We would like to thank Professor Arabella Smith for her input into the interpretation of this study’s findings. This work was sponsored by the National Health and Medical Research Council of Australia (grant number 1045166).
Ethical Statement: All animal experiments for this study were approved by the Garvan Institute/St Vincent’s Hospital Animal Ethics Committee and conducted in accordance with relevant regulations and guidelines.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med 2012;14:10-26. 10.1038/gim.0b013e31822bead0 [DOI] [PubMed] [Google Scholar]
- 2.Butler MG. Prader-Willi Syndrome: Obesity due to Genomic Imprinting. Curr Genomics 2011;12:204-15. 10.2174/138920211795677877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholls RD, Gottleib W, Avidano K, et al. Mouse chromosome mapping of clones from the PWS/AS genetic region. Mouse Genome 1991;89:254. [Google Scholar]
- 4.Bervini S, Herzog H. Mouse models of Prader-Willi Syndrome: a systematic review. Front Neuroendocrinol 2013;34:107-19. 10.1016/j.yfrne.2013.01.002 [DOI] [PubMed] [Google Scholar]
- 5.Relkovic D, Isles AR. Behavioural and cognitive profiles of mouse models for Prader-Willi syndrome. Brain Res Bull 2013;92:41-8. 10.1016/j.brainresbull.2011.09.009 [DOI] [PubMed] [Google Scholar]
- 6.Duker AL, Ballif BC, Bawle EV, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 2010;18:1196-201. 10.1038/ejhg.2010.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bieth E, Eddiry S, Gaston V, et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet 2015;23:252-5. 10.1038/ejhg.2014.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahoo T, del Gaudio D, German JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 2008;40:719-21. 10.1038/ng.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Smith AJ, Purmann C, Walters RG, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet 2009;18:3257-65. 10.1093/hmg/ddp263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SJ, Miller JL, Kuipers PJ, et al. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet 2012;20:283-90. 10.1038/ejhg.2011.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leff SE, Brannan CI, Reed ML, et al. Maternal imprinting of the mouse Snrpn gene and conserved linkage homology with the human Prader-Willi syndrome region. Nat Genet 1992;2:259-64. 10.1038/ng1292-259 [DOI] [PubMed] [Google Scholar]
- 12.Chaillet JR, Knoll JH, Horsthemke B, et al. The syntenic relationship between the critical deletion region for the Prader-Willi/Angelman syndromes and proximal mouse chromosome 7. Genomics 1991;11:773-6. 10.1016/0888-7543(91)90090-2 [DOI] [PubMed] [Google Scholar]
- 13.Skryabin BV, Gubar LV, Seeger B, et al. Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet 2007;3:e235. 10.1371/journal.pgen.0030235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding F, Li HH, Zhang S, et al. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS One 2008;3:e1709. 10.1371/journal.pone.0001709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purtell L, Viardot A, Sze L, et al. Postprandial metabolism in adults with Prader-Willi syndrome. Obesity (Silver Spring) 2015;23:1159-65. 10.1002/oby.21041 [DOI] [PubMed] [Google Scholar]
- 16.Schoeller DA, Levitsky LL, Bandini LG, et al. Energy expenditure and body composition in Prader-Willi syndrome. Metabolism 1988;37:115-20. 10.1016/S0026-0495(98)90003-8 [DOI] [PubMed] [Google Scholar]
- 17.van Mil EA, Westerterp KR, Gerver WJ, et al. Energy expenditure at rest and during sleep in children with Prader-Willi syndrome is explained by body composition. Am J Clin Nutr 2000;71:752-6. [DOI] [PubMed] [Google Scholar]
- 18.Butler MG, Theodoro MF, Bittel DC, et al. Energy expenditure and physical activity in Prader-Willi syndrome: comparison with obese subjects. Am J Med Genet A 2007;143A:449-59. 10.1002/ajmg.a.31507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young WS., 3rd In situ hybridization histochemical detection of neuropeptide mRNA using DNA and RNA probes. Methods Enzymol 1989;168:702-10. 10.1016/0076-6879(89)68051-2 [DOI] [PubMed] [Google Scholar]
- 20.Zhang Q, Bouma GJ, McClellan K, et al. Hypothalamic expression of snoRNA Snord116 is consistent with a link to the hyperphagia and obesity symptoms of Prader-Willi syndrome. Int J Dev Neurosci 2012;30:479-85. 10.1016/j.ijdevneu.2012.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozanne SE, Hales CN. Lifespan: catch-up growth and obesity in male mice. Nature 2004;427:411-2. 10.1038/427411b [DOI] [PubMed] [Google Scholar]
- 22.Bieswal F, Ahn MT, Reusens B, et al. The importance of catch-up growth after early malnutrition for the programming of obesity in male rat. Obesity (Silver Spring) 2006;14:1330-43. 10.1038/oby.2006.151 [DOI] [PubMed] [Google Scholar]
- 23.Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A 2011;155A:1040-9. 10.1002/ajmg.a.33951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunningham JJ. Body composition as a determinant of energy expenditure: a synthetic review and a proposed general prediction equation. Am J Clin Nutr 1991;54:963-9. [DOI] [PubMed] [Google Scholar]
- 25.Nelson KM, Weinsier RL, Long CL, et al. Prediction of resting energy expenditure from fat-free mass and fat mass. Am J Clin Nutr 1992;56:848-56. [DOI] [PubMed] [Google Scholar]
- 26.Illner K, Brinkmann G, Heller M, et al. Metabolically active components of fat free mass and resting energy expenditure in nonobese adults. Am J Physiol Endocrinol Metab 2000;278:E308-15. [DOI] [PubMed] [Google Scholar]