

Abstract
Mitochondria are important regulators of cell death and cell survival. Mitochondrial Ca2+ levels are critically involved in both of these processes. On the one hand, excessive mitochondrial Ca2+ leads to Ca2+-induced mitochondrial outer membrane permeabilization and thus apoptosis. On the other hand, mitochondria need Ca2+ in order to efficiently fuel the tricarboxylic acid cycle and maintain adequate mitochondrial bioenergetics. For obtaining this Ca2+, the mitochondria are largely dependent on close contact sites with the endoplasmic reticulum (ER), the so-called mitochondria-associated ER membranes. There, the inositol 1,4,5-trisphosphate receptors are responsible for the Ca2+ release from the ER. It comes as no surprise that this Ca2+ release from the ER and the subsequent Ca2+ uptake at the mitochondria are finely regulated. Cancer cells often modulate ER-Ca2+ transfer to the mitochondria in order to promote cell survival and to inhibit cell death. Important regulators of these Ca2+ signals and the onset of cancer are the B-cell lymphoma 2 (Bcl-2) family of proteins. An increasing number of reports highlight the ability of these Bcl-2-protein family members to finely regulate Ca2+ transfer from ER to mitochondria both in healthy cells and in cancer. In this review, we focus on recent insights into the dynamic regulation of ER–mitochondrial Ca2+ fluxes by Bcl-2-family members and how this impacts cell survival, cell death and mitochondrial energy production.
Keywords: endoplasmic reticulum–mitochondria contact sites, Ca2+-transport systems, apoptosis, autophagy, mitochondrial bio energetics, IP3 receptors, voltage-dependent anion channels, Bcl-2
Introduction
Ca2+ signaling plays important roles in a vast amount of cell physiological processes (1). In cancer cells, Ca2+ signaling is altered to promote mitochondrial bioenergetics, cell proliferation, migration, and survival while inhibiting cell death (2–6). The involvement of Ca2+ signaling in the development of cancer and consequently the potential of Ca2+ signaling as a target for treatment is becoming increasingly apparent (5–11). In cancer cells, proteins involved in Ca2+ signaling have been reported to have differential expression profiles compared to healthy cells (12–15). In addition, an increasing number of proto-oncogenes and tumor suppressors impact Ca2+-signaling pathways by directly modulating intracellular Ca2+-transport systems with critical functions in cell survival and cell death (16–19).
An important Ca2+-signaling pathway involved in both cell death and cell survival is the transfer of Ca2+ from the endoplasmic reticulum (ER) to the mitochondria (20). These Ca2+ transfers occur at the so-called mitochondria-associated ER membranes (MAMs), which are close contact sites between the ER and the mitochondria (21). A continuous small Ca2+ transfer to the mitochondria is necessary to maintain proper energy production (22). Ca2+ is required by several enzymes of the tricarboxylic acid (TCA) cycle (like pyruvate dehydrogenase, isocitrate dehydrogenase and α-ketoglutarate) to promote NADH and ATP production (23). Besides this, Ca2+ also modulates the ATP synthase complex V and the adenine nucleotide translocator (24). In addition to this mitochondrial pathway, pro-survival Ca2+ oscillations activate calcineurin, which in turn dephosphorylates the nuclear factor of activated T-cells (NFAT), conferring its translocation into the nucleus (25). Here, NFAT triggers the transcription of genes involved in cell proliferation. In contrast, large Ca2+ transfers from the ER to the mitochondria may result in both Ca2+-induced mitochondrial outer membrane permeabilization (MOMP) and opening of the mitochondrial permeability transition pore (mPTP), the latter formed by dimers of the F0F1 ATP synthase (4, 26, 27). In this process, Ca2+ overload in the mitochondria triggers cardiolipin oxidation, resulting in the disassembly of the respiratory chain complex 2 (also known as succinate dehydrogenase), subsequently leading to excessive reactive oxygen species (ROS) production (28). Mitochondrial produced ROS can open the mPTP, ultimately leading to MOMP. At the level of the ER, the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) (29) is an important intracellular Ca2+-release channel involved in these Ca2+ transfers, whereas at the mitochondria, the voltage-dependent anion channel (VDAC) (at the outer mitochondrial membrane) (30) and the mitochondrial Ca2+ uniporter (MCU) (at the inner mitochondrial membrane) (31, 32) are important for transporting Ca2+ into the mitochondrial matrix.
The B-cell lymphoma 2 (Bcl-2)-protein family, consisting of both anti- and pro-apoptotic members, is critically involved in regulating cell death and survival (33–36). Dysregulated expression and function of Bcl-2 proteins have been not only implicated in oncogenesis but also represent an “Achilles’ heel” in cancer cells that can be exploited by the use of Bcl-2 inhibitors (37–39). Anti-apoptotic Bcl-2 proteins (like Bcl-2, Bcl-XL and Mcl-1) have been extensively described to inhibit apoptosis by neutralizing the pro-apoptotic Bcl-2-family members (like Bax, Bak, Bim, Bid, etc.). The mechanism involves binding of the Bcl-2 homology (BH) 3 domains of the pro-apoptotic proteins to the hydrophobic cleft formed by the BH1, BH2 and BH3 domains of the anti-apoptotic members, thereby inhibiting cell death (40). A recently developed class of compounds, so-called BH3-mimetic drugs (40–42), is able to compete with pro-apoptotic Bcl-2-family members for the hydrophobic cleft of the anti-apoptotic Bcl-2-family members. Hence, BH3-mimetics alleviate the inhibition of Bax and Bak by the anti-apoptotic Bcl-2-family members, effectively killing cancer cells that are dependent on anti-apoptotic Bcl-2 proteins for their survival. In addition to this, the BH4 domain of Bcl-2 also contributes to the interaction with Bax via a site that is distinct from Bax’s BH3 domain (43). Moreover, the isolated BH4 domain, delivered as a stapled peptide, neutralized the pro-apoptotic activity of Bim-derived BH3 peptides by restricting Bax’s conformational change (44).
Anti-apoptotic Bcl-2 proteins are also known to regulate ER to mitochondrial Ca2+ signaling at both organelles, and several Bcl-2-family members, including Bcl-2 and Bcl-XL, are present in the MAMs (45, 46) (Figure 1). At the ER, anti-apoptotic Bcl-2, Bcl-XL and Mcl-1 promote pro-survival IP3R-mediated Ca2+ oscillations, enhancing cell proliferation and mitochondrial energy production (47–49). Bcl-2 (and Bcl-XL at high concentrations) also inhibits excessive pro-apoptotic IP3R-mediated Ca2+ release (50–53), thereby preventing Ca2+-induced MOMP. At the mitochondrial side of the MAMs, anti-apoptotic Bcl-2 and Bcl-XL proteins inhibit VDAC1-mediated Ca2+ uptake in the mitochondria (45, 54, 55). However, also stimulatory roles of Bcl-2-family members on VDAC1-mediated mitochondrial Ca2+ transfer have been described, thereby maintaining adequate mitochondrial Ca2+ levels that promote survival and mitochondrial bioenergetics (56, 57). Besides IP3Rs and VDAC, anti-apoptotic Bcl-2-family members also regulate other members of the Ca2+ toolkit at different locations in the cell [extensively reviewed in Ref. (33)]. Mcl-1, located at the inner mitochondrial membrane, was also shown to be crucial for normal mitochondrial bioenergetics by regulating the assembly of the F0F1 ATP synthase oligomers (58). Finally, the F0F1 ATP synthase emerged as a target for anti-apoptotic Bcl-XL, allowing the direct regulation of ATP production (59, 60). In this review, we will focus on recent insights into the dynamic regulation of ER–mitochondrial Ca2+ fluxes, the involvement of anti-apoptotic Bcl-2-family members and how this impacts cell survival, cell death, and mitochondrial energy production (Figure 2), three important aspects of cancer development.
Figure 1.
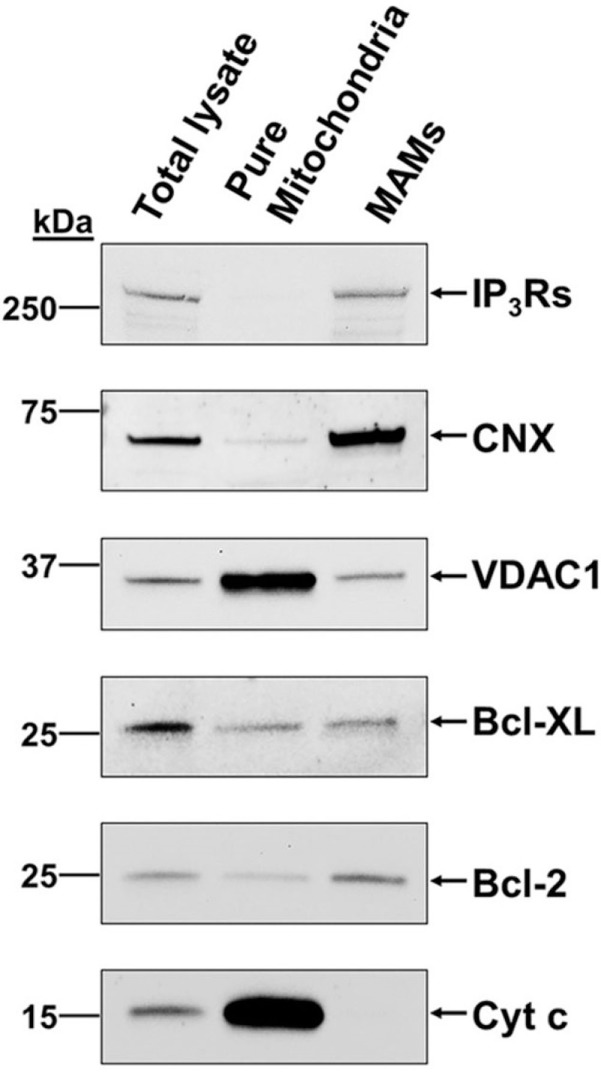
B-cell lymphoma (Bcl)-2 and Bcl-XL and their targets in Ca2+ signaling, inositol 1,4,5-trisphosphate receptor (IP3R) and voltage-dependent anion channel 1 (VDAC1), are present in the mitochondria-associated endoplasmic reticulum membranes (MAMs). Representative immunoblots showing the presence of VDAC1, IP3Rs, Bcl-2, and Bcl-XL in the MAMs of MEFs. Calnexin (CNX) and cytochrome c (Cyt c) served as specific MAMs and mitochondrial markers, respectively. These data were originally published in Journal of Biological Chemistry with following reference: Monaco et al. (45). © The American Society for Biochemistry and Molecular Biology. Authors of articles in Journal of Biological Chemistry have the rights to reuse their own material and are automatically granted a permission to reuse figures from their articles in future works. The original results have been produced by Dr. Alex van Vliet in the laboratory of Prof. Patrizia Agostinis (KU Leuven, Belgium).
Figure 2.

Modulation of endoplasmic reticulum (ER) to mitochondrial Ca2+ transfers by anti-apoptotic B-cell lymphoma (Bcl)-2 proteins. ER to mitochondrial Ca2+ transfers are critical for the regulation of cell death and cell survival decisions. In order to fuel the tricarboxylic acid (TCA) cycle, a continuous influx of Ca2+ into the mitochondria is required (green arrow), thereby promoting cell survival. Excessive mitochondrial Ca2+ uptake leads to Ca2+-induced mitochondrial outer membrane permeabilization (MOMP) and cell death (red arrow). The anti-apoptotic side of the Bcl-2-protein family regulates these Ca2+ transfers at both organelles. During pro-survival Ca2+ signaling at the ER, Bcl-2, Bcl-XL, and Mcl-1 modulate inositol 1,4,5-trisphosphate receptor (IP3R)-mediated Ca2+ release to generate Ca2+ oscillations. At the mitochondria, Bcl-XL and Mcl-1 can increase voltage-dependent anion channel 1 (VDAC1)-mediated Ca2+ uptake. Combining the effects at the two organelles results in an efficient and finely regulated Ca2+ uptake at the mitochondria, which increases mitochondrial bioenergetics and promotes cell survival. In addition, Mcl-1 and Bcl-XL target the F0F1 ATP synthase, thereby regulating ATP-production. During pro-death signaling, Bcl-2 and Bcl-XL can inhibit both pro-apoptotic Ca2+ release from the IP3R and the Ca2+ uptake into the mitochondria via VDAC. Finally, abolishing ER to mitochondrial Ca2+ transfers by either blocking IP3Rs or knocking down the mitochondrial Ca2+ uniporter (MCU) induces autophagy. When this is coupled to decreased cell proliferation (healthy cells), this increase in autophagy may rescue the cell. However, when proliferation is not halted (cancer cells) this results in cell death.
ER Side of the MAMs
ER Ca2+ release is an important determinant for cell survival by regulating mitochondrial bioenergetics and for cell death via promoting mPTP opening. In most cells, including cancer cells, the IP3R is an important intracellular Ca2+-release channel responsible for Ca2+ release from the ER. Cancer cells have developed several ways to modulate IP3R-mediated Ca2+ release, among which Bcl-2-dependent regulation.
IP3R
A continuous Ca2+ flux from the ER to the mitochondria is necessary in order to maintain normal energy production. At the ER, the IP3R is responsible for the Ca2+ release and is present at the MAMs (Figure 1). Inhibition of the IP3R and thus of the continuous Ca2+ transfer to the mitochondria was already shown to result in the induction of autophagy, thereby managing the decrease in mitochondrial energy production (22). New findings emerged, showing that cancer cells are addicted to constitutive IP3R-driven Ca2+ transfer to the mitochondria (61, 62). Similar to normal/non-tumorigenic cells, cancer cells increase their autophagic flux upon IP3R inhibition in order to cope with the loss of Ca2+ influx into the mitochondria and subsequent reduction in energy production. However, in normal cells, the increased autophagy is accompanied by a decrease in the proliferation rate at the G1/S checkpoint (63), addressing the decreased availability of mitochondrial substrates for biosynthetic pathways of nucleosides and other cellular building blocks. In this way, cells may survive until normal Ca2+ transfer to the mitochondria is restored. In cancer cells, this increase in autophagy is not accompanied by a reduction in cell proliferation, likely due to a loss of the link between the monitoring of the mitochondrial health and the G1/S checkpoint. As such, these malignant cells proceed through the cell cycle without the necessary pool of nucleosides, resulting in a mitotic catastrophe and necrotic cell death.
Anti-apoptotic Bcl-2-family members have been shown to regulate the IP3R. Both inhibitory (50–52) and stimulatory (47, 49) effects, largely dependent on the Bcl-2-family member involved (64, 65) and the strength of IP3R activation (25, 53), have been described. As such, it was reported that in T-cell models, Bcl-2 suppresses IP3R-mediated Ca2+ release generated by strong T-cell receptor stimulation, thereby preventing excessive Ca2+ transfer into the mitochondria. This interaction occurs via Bcl-2’s BH4 domain and a stretch of 20 amino acids in the central coupling domain of the IP3R (52, 64). Peptides derived from this amino acid stretch were able to disrupt IP3R/Bcl-2 complexes in several cell types and models, thereby augmenting cell death in response to apoptotic triggers that act through Ca2+ signaling (25, 66). The efficient IP3R inhibition by anti-apoptotic Bcl-2 critically depended on the presence of Bcl-2’s C-terminal transmembrane domain, which interacted with the C-terminal domain of the IP3R channel (67). Bcl-2 lacking its transmembrane domain failed to inhibit IP3R-mediated Ca2+ release and to suppress Ca2+-dependent apoptosis in an in cellulo context. In contrast, the hydrophobic cleft of Bcl-2, responsible for scaffolding pro-apoptotic family members, was dispensable for IP3R binding and inhibition.
Bcl-2, Bcl-XL and Mcl-1 were reported to sensitize the IP3R to low levels of IP3 in order to promote pro-survival Ca2+ oscillations, thereby feeding Ca2+ into the mitochondria to maintain adequate mitochondrial bioenergetics (47–49). The interaction of Bcl-2-family members with the C-terminus of the IP3R has been proposed as the underlying molecular mechanism for generating these Ca2+ oscillations (68). Recently, the mechanism underlying IP3R sensitization by Bcl-XL has been identified with a prominent role for its hydrophobic cleft (53). Two BH3-like domains were identified in the C-terminus of IP3Rs. Indeed, in contrast to Bcl-2, for which its hydrophobic cleft was shown to be dispensable for IP3R modulation, Bcl-XL via its hydrophobic cleft could target, with different affinities, both BH3-like domains present in the C-terminal region of the IP3R. At low concentrations, Bcl-XL increased the open probability of the IP3R in response to low levels of IP3 by simultaneous binding to both BH3-like domains. Similar to Bcl-2, high Bcl-XL concentrations were able to inhibit IP3R-mediated Ca2+ release in response to strong IP3R stimulation. The “dual” interaction with the BH3-like domain which conferred the highest affinity toward Bcl-XL as well as the region in the coupling domain of the IP3R targeted by Bcl-2’s BH4 domain, was important for IP3R inhibition by Bcl-XL. Binding of Bcl-XL to the coupling domain appeared with much lower affinity than the binding to the C-terminal tail, which is in line with our previous study that focused on the binding efficiency of Bcl-2 versus Bcl-XL for both IP3R domains (48). This may indicate that moderate levels of Bcl-XL will most likely operate in IP3R-sensitizing modus and thus will promote Ca2+ oscillations, whereas high levels of Bcl-XL will be needed to operate in IP3R-inhibiting modus. Finally, binding of Bcl-XL to both BH3-like domains is involved in maintaining cell viability and in protecting cells from stress inducers. These molecular results substantiate the previously observed sensitization of the IP3R by Bcl-XL (68), resulting in pro-survival Ca2+ oscillations, and underscore the importance of this interaction for cell viability.
The role of Bcl-XL in modulating IP3R-mediated Ca2+ release in order to promote mitochondrial bioenergetics was recently further highlighted (69). The authors showed that Bcl-XL interacts with IP3R3 at the MAMs, where it increased Ca2+ transfer into the mitochondria, thereby enhancing TCA cycling. Upon ER-stress induction, Bcl-XL translocated more to the MAMs, where the subsequent facilitation of Ca2+ transfer to the mitochondria and thus increased energy production helped the cells cope with the induced ER stress. This further highlights that Bcl-XL exerts its protective effects against stress inducers in large part via modulating Ca2+ signaling.
Mitochondrial Side of the MAMs
Cancer cells are highly dependent on the mitochondria for their energy production. For sustaining this energy production, adequate control of mitochondrial Ca2+ levels is important. Anti-apoptotic Bcl-2 proteins are known regulators of this mitochondrial Ca2+ influx, thereby regulating mitochondrial bioenergetics. In addition, the F0F1 ATP synthase has also been identified as a target for anti-apoptotic Bcl-2-family members, thereby directly linking them to the production of ATP (58–60).
VDAC
The large conductance channel VDAC, of which three isoforms are known to exist, is located at the outer mitochondrial membranes (30). At the MAMs, VDAC is physically linked to the IP3R via molecular tethers like the chaperone protein, glucose-regulated protein 75, allowing efficient Ca2+ transfer from the ER into the mitochondria (70). Close regulation of mitochondrial Ca2+ uptake via VDAC is critical for maintaining mitochondrial energy production. Anti-apoptotic Bcl-2-family members are known to modulate this mitochondrial Ca2+ transfer through interactions with VDAC. Both Bcl-2 and Bcl-XL have been reported to inhibit VDAC1-mediated Ca2+ uptake into the mitochondria, thereby protecting cells from Ca2+-induced MOMP (45, 54, 55, 71). The BH4 domain of Bcl-XL, but not the one of Bcl-2, was sufficient to bind to VDAC1 and to directly inhibit VDAC1 single-channel activity (45). Although different regions of Bcl-2 and Bcl-XL seem to be involved in this interaction, both anti-apoptotic proteins target the N-terminus of VDAC1. Introducing VDAC1’s N-terminal into cells was shown to inhibit both Bcl-2’s and Bcl-XL’s anti-apoptotic function, illustrating that VDAC1 could be a target for anti-cancer drugs (54, 71–73). However, at the level of the BH4 domains, the N-terminal peptide of VDAC1 could only counteract the inhibitory action of Bcl-XL’s, but not that of Bcl-2’s BH4 domain. The BH4 domain of Bcl-2 also suppressed agonist-induced mitochondrial Ca2+ uptake and staurosporine-induced cell death, but acted through inhibition of IP3Rs, since IP3R-derived peptides were able to alleviate the inhibitory effects of Bcl-2’s, but not those of Bcl-XL’s BH4 domain (45).
Although the interaction of Bcl-XL with VDAC1 is well established, the impact of Bcl-XL on VDAC1’s functional properties may be dichotomous. Besides inhibiting VDAC1 (45, 54), Bcl-XL has been reported to enhance VDAC1 activity. Bcl-XL knockout MEF cells displayed a reduced VDAC1-mediated Ca2+ uptake in the mitochondria compared with the wild-type MEF cells (56). Similarly, N-terminal peptides derived from VDAC1 that disrupt Bcl-XL binding to VDAC1 could also antagonize mitochondrial Ca2+ uptake in wild-type MEF cells, while these peptides lacked any effect in Bcl-XL-deficient MEF cells. While differences in experimental conditions may underlie the seemingly contrasting observations, these results indicate that Bcl-XL might have a dual impact on VDAC1’s Ca2+-flux properties dependent on VDAC1’s function as a pro-survival or pro-death protein. Hence, Bcl-XL could stimulate basal pro-survival and inhibit excessive pro-apoptotic VDAC1-mediated mitochondrial Ca2+ transfer, thereby fine-tuning mitochondrial Ca2+ handling according to cellular needs, with respect to cell fate decisions. The molecular basis for these opposite effects of Bcl-XL on VDAC1 remains poorly understood.
Mcl-1 has also been shown to positively regulate VDAC in non-small cell lung carcinoma cells (57). In these cancer cells, Mcl-1 interacted with VDAC, with a pronounced role for its N-terminus, thereby increasing mitochondrial Ca2+ uptake, resulting in increased ROS production and cell migration. Disrupting the Mcl-1/VDAC interaction utilizing N-terminal VDAC-derived peptides could inhibit ROS production and cell migration. The importance of Mcl-1 at the mitochondria was further underscored by a recent study concerning different Mcl-1 splice variants (74). In this study, the increased expression of the short pro-apoptotic Mcl-1 isoform resulted in increased mitochondrial fusion via a reduced Mcl-1-dependent recruitment of dynamin-related protein 1 to the mitochondria. This was accompanied by hyperpolarization of the mitochondrial potential and increased mitochondrial Ca2+ uptake, thereby increasing susceptibility to apoptotic stimuli. Whether this increase in mitochondrial Ca2+ uptake was also mediated through the interaction with VDAC was not evaluated. Nevertheless, it would be interesting to assess whether the short pro-apoptotic Mcl-1 isoform would shift VDAC-mediated mitochondrial Ca2+ uptake toward more pro-apoptotic levels in comparison to the long pro-survival Mcl-1 isoform.
F0F1 ATP Synthase
In cultured hippocampal neurons, Bcl-XL was shown to be present at the inner mitochondrial membranes, where it directly targets the β-subunit of the F0F1 ATP synthase (59, 60). The interaction stabilized the mitochondrial membrane potential via the closure of a membrane leak pathway. This increased the enzymatic activity of the F0F1 ATP synthase, thereby promoting ATP production during neural activity. In addition, the interaction seems to occur via Bcl-XL’s hydrophobic cleft, since ABT-737 could reverse the effects of Bcl-XL on the F0F1 ATP synthase. Recently, this process was further explored and was shown to be important for neuronal survival (75). In response to excitotoxic stimuli, cyclin B1 and cyclin-dependent kinase 1 (CdK1) accumulated in the mitochondria. There, the cyclin B1-Cdk1 complex phosphorylated Bcl-XL, leading to its dissociation from the ATP-synthase. This led to decreased ATP synthesis and production of ROS species, resulting in the inhibition of respiratory chain complex I, mitochondrial dysfunction, and potentially neuronal death.
Potential Therapeutic Opportunities
Promoting ER–Mitochondrial Ca2+ Transfer
Many chemotherapeutics trigger intracellular Ca2+ release from the ER, causing, or at least contributing to, mitochondrial Ca2+ overload. This Ca2+ release is often considered as a nonspecific side effect of the drug, but in many cases, it contributes to obtain maximal therapeutic effects (76). Moreover, recent studies have unraveled the molecular mechanisms underlying the impact of chemotherapeutics and photodynamic therapy on intracellular Ca2+ homeostasis (18, 77, 78). These anti-cancer regimens caused the accumulation of the tumor suppressor p53 at the ER membranes, where it enhanced sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 2b activity. The effects were independent of the transcriptional roles of p53. Recruitment of p53 at the ER augmented the Ca2+ filling state of the ER stores, increasing the susceptibility to apoptotic stimuli and the likelihood for mitochondrial Ca2+ overload. Cells deficient in p53 did not display this effect and were resistant to chemotherapy. This resistance could be overcome by SERCA and/or MCU overexpression.
Inhibiting ER–Mitochondrial Ca2+ Transfer
The therapeutic potential of dampening Ca2+ transfer from ER to mitochondria has recently been proposed as an anti-cancer strategy (9, 61). It was shown that dysregulation of the ER to mitochondrial Ca2+ transfer via inhibition of IP3Rs, results in the induction of autophagy in both cancer and normal cells. However, when this increase in autophagy is not accompanied by a halt in proliferation, the cancer cells will die mainly through necrosis. This could prove to be a very specific way of eliminating cancer cells by effectively turning the increased proliferative capacity of cancer cells against themselves, whereas healthy cells can cope with this loss of Ca2+ transfer to the mitochondria. A major challenge will be to selectively target the Ca2+ transfer into the mitochondria without affecting global Ca2+ signaling and to mainly limit the effect of IP3R-inhibiting drugs to the malignant cells.
Antagonizing Anti-apoptotic Bcl-2 Proteins
Major efforts have been dedicated towards the development of BH3-mimetic drugs, which target the hydrophobic cleft of anti-apoptotic Bcl-2-family members. The first generation of BH3 mimetics (ABT-737 and ABT-263) inhibited both Bcl-2 and Bcl-XL, resulting in severe side effects related to thrombocytopenia due to the dependence of thrombocytes on Bcl-XL for their survival (41, 79). More recently, a Bcl-2-selective BH3-mimetic inhibitor was developed, namely ABT-199/venetoclax, which is a very promising anti-cancer drug that has been approved for the treatment of chronic lymphocytic leukemia (80). Whether these BH3-mimetic drugs also influence the ability of anti-apoptotic Bcl-2-family members to modulate intracellular Ca2+ release is less well understood, although some recent studies aimed to address this. With the identification of the two BH3-like domains at the C-terminus of the IP3R, the ABT-737 compound was shown to disrupt the binding of Bcl-XL to the C-terminus of the IP3R, thereby abolishing both the stimulatory and inhibitory effects of Bcl-XL on IP3R-mediated Ca2+ release (53). However, the contribution of Ca2+ signaling to ABT-737-induced cell death requires further investigation, since ABT-737 could cause cell death in primary chronic lymphocytic leukemia cells without inducing elevations in intracellular [Ca2+] (81).
Besides a direct impact on IP3R/Bcl-XL complexes, ABT-737 has also been proposed to modulate the sensitivity of cancer cells to chemotherapy via a mechanism that involves remodeling of ER–mitochondrial contact sites (82). As such, cisplatin-resistant ovarian cancer cells could be re-sensitized to cisplatin by ABT-737. This drug increased ER–mitochondrial contact sites, thereby increasing cisplatin-induced elevations in mitochondrial Ca2+. When co-applied with cisplatin in cholangiocarcinoma cells, ABT-737 has been shown to induce mitochondrial fragmentation and mitophagy, resulting in cell death, whereas cisplatin alone induced mitochondrial hyperfusion, potentially underlying cell-death resistance (83). The combined ABT-737/cisplatin treatment led to a decreased Mcl-1 and an increased Bax expression. Interestingly, Mcl-1 has recently been shown to be implicated in controlling mitochondrial dynamics (74).
Consistent with the lack of contribution of Bcl-2’s hydrophobic cleft to the interaction with and regulation of IP3R, IP3R/Bcl-2-protein complexes and IP3R inhibition by Bcl-2 were resistant to ABT199/venetoclax treatment (67). Acute addition of ABT199/venetoclax to a variety of permeabilized and intact cell systems did neither trigger Ca2+ release by itself nor directly affected ER-located Ca2+-uptake and -release systems. Related to this, ABT199/venetoclax-induced apoptosis in Bcl-2-dependent cancer cells appeared to occur independently of intracellular Ca2+ overload (67, 84). However, the inhibition of Bcl-2 by BH3-mimetics has been reported to result in a rapid impairment of mitochondrial oxidative phosphorylation (85). This may underlie the increased sensitivity of Bcl-2-dependent cancer cells to ABT199/venetoclax in the presence of the intracellular Ca2+ buffer, BAPTA-AM (67, 84).
Over the years, it has become clear that Bcl-2 inhibition via targeting its BH4 domain has potential as an effective anti-cancer treatment (38, 86–88). Targeting Bcl-2/IP3R complex with Bcl-2/IP3 receptor disrupter-2 (BIRD-2), a stabilized TAT-linked peptide containing the 20 amino acids that represent the Bcl-2 interaction motif of IP3Rs, triggers intracellular Ca2+ overload and apoptotic cell death in a variety of cancer cell models, including chronic lymphocytic leukemia (81), diffuse large B-cell lymphoma (89), multiple myeloma, follicular lymphoma (90), and small-cell lung carcinoma (91). The cell death could be suppressed by buffering intracellular Ca2+ and by inhibiting IP3R activity (81, 89). Very recently, a small molecule (BDA-366) that targets the BH4 domain of Bcl-2 has been developed and shown to be effective in lung cancers and multiple myeloma (92, 93). The mechanism involved a conformational switch in Bcl-2 that turned it from a pro-survival to a pro-death protein by exposing its BH3 domain. A decrease in Bcl-2 phosphorylation may contribute to this pro-apoptotic switch induced by BDA-366. BDA-366 also impaired IP3R/Bcl-2 complex formation and raised cytosolic Ca2+ levels, although further work is needed to determine the contribution of Ca2+ signaling to BDA-366-induced cell death in cancer cells.
Mcl-1 gene amplifications are frequently found in many types of cancer (94). Very recently, an Mcl-1 inhibitor (S63845) targeting Mcl-1’s hydrophobic cleft has been developed (95). This compound was shown to be very specific for Mcl-1, well-tolerated by animal models and efficient at triggering cell death in Mcl-1-dependent tumor cells. As the regulation of VDAC1 by Mcl-1 also stimulates cancer cell migration (57), Mcl-1 inhibitors may not only be useful to eliminate Mcl-1-dependent cancers by provoking cell death but also by counteracting metastasis. However, at this point, it is not clear whether these Mcl-1 inhibitors can disrupt VDAC1/Mcl-1 complex formation.
Conclusions
Ca2+ transfer from ER to mitochondria is important for maintaining proper energy production and balance between cell survival and cell death. The anti-apoptotic Bcl-2-family members regulate these Ca2+ transfers at the level of the ER as well as of the mitochondria by directly targeting Ca2+-transport systems located at the ER and mitochondria. Moreover, the molecular determinants underlying the complex formation between the Bcl-2 proteins and these systems are emerging as a hot topic, which allows the development of strategies and tools to interfere with the Bcl-2 protein-mediated control of Ca2+-signaling. These mechanisms also appear to be exploited by cancer cells to promote survival and mitochondrial bioenergetics, to contribute to cell-death resistance and control metastasis. Thus, targeting the Ca2+-modulating abilities of Bcl-2 proteins may offer novel anti-cancer strategies. In addition to this, Ca2+ signaling might contribute to the cell-death properties of recently developed Bcl-2 inhibitors, including BH3-mimetics and BH4-domain antagonists.
Author Contributions
TV and GB drafted the manuscript. All authors critically read, amended, and/or corrected the manuscript. TV made the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all lab members for fruitful discussions and Alex van Vliet and Patrizia Agostinis (Laboratory of Cell Death Research and Therapy, Department of Cellular and Molecular Medicine, KU Leuven, Belgium) for the MAM analysis.
Funding
This work was supported by grants from the Research Foundation-Flanders (FWO grants 6.057.12, G.0819.13, G.0C91.14, and G.0A34.16), by the Research Council of the KU Leuven (OT grant 14/101), and by the Interuniversity Attraction Poles Program (Belgian Science Policy; IAP-P7/13). HI is a recipient of a PhD fellowship of the Research Foundation-Flanders. TV and GM are recipients of a post-doctoral fellowship of the Research Foundation-Flanders.
References
- 1.Berridge MJ. The versatility and complexity of calcium signalling. Novartis Found Symp (2001) 239:52–64; discussion 64–7, 150–9. 10.1002/0470846674.ch6 [DOI] [PubMed] [Google Scholar]
- 2.Cardenas C, Foskett JK. Mitochondrial Ca2+ signals in autophagy. Cell Calcium (2012) 52:44–51. 10.1016/j.ceca.2012.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harr MW, Distelhorst CW. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol (2010) 2:a005579. 10.1101/cshperspect.a005579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimm S. The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta (2012) 1823:327–34. 10.1016/j.bbamcr.2011.11.018 [DOI] [PubMed] [Google Scholar]
- 5.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer (2011) 11:609–18. 10.1038/nrc3105 [DOI] [PubMed] [Google Scholar]
- 6.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer (2008) 8:361–75. 10.1038/nrc2374 [DOI] [PubMed] [Google Scholar]
- 7.Dang D, Rao R. Calcium-ATPases: gene disorders and dysregulation in cancer. Biochim Biophys Acta (2016) 1863:1344–50. 10.1016/j.bbamcr.2015.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoth M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim Biophys Acta (2016) 1863:1408–17. 10.1016/j.bbamcr.2015.12.009 [DOI] [PubMed] [Google Scholar]
- 9.Lovy A, Foskett JK, Cardenas C. InsP3R, the calcium whisperer: maintaining mitochondrial function in cancer. Mol Cell Oncol (2016) 3:e1185563. 10.1080/23723556.2016.1185563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer (2007) 7:519–30. 10.1038/nrc2171 [DOI] [PubMed] [Google Scholar]
- 11.Padanyi R, Paszty K, Hegedus L, Varga K, Papp B, Penniston JT, et al. Multifaceted plasma membrane Ca2+ pumps: from structure to intracellular Ca2+ handling and cancer. Biochim Biophys Acta (2016) 1863:1351–63. 10.1016/j.bbamcr.2015.12.011 [DOI] [PubMed] [Google Scholar]
- 12.Dubois C, Vanden Abeele F, Lehen’kyi V, Gkika D, Guarmit B, Lepage G, et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell (2014) 26:19–32. 10.1016/j.ccr.2014.04.025 [DOI] [PubMed] [Google Scholar]
- 13.McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther (2011) 10:448–60. 10.1158/1535-7163.MCT-10-0923 [DOI] [PubMed] [Google Scholar]
- 14.Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem (2012) 287:31666–73. 10.1074/jbc.R112.343061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Liu Y, Song F, Zheng H, Hu L, Lu H, et al. Functional SNP in the microRNA-367 binding site in the 3’UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc Natl Acad Sci U S A (2011) 108:13653–8. 10.1073/pnas.1103360108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akl H, Bultynck G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta (2013) 1835:180–93. 10.1016/j.bbcan.2012.12.001 [DOI] [PubMed] [Google Scholar]
- 17.Bittremieux M, Parys JB, Pinton P, Bultynck G. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca2+ signaling. Biochim Biophys Acta (2016) 1863:1364–78. 10.1016/j.bbamcr.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 18.Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, et al. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget (2015) 6:1435–45. 10.18632/oncotarget.2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedgepeth SC, Garcia MI, Wagner LE, II, Rodriguez AM, Chintapalli SV, Snyder RR, et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J Biol Chem (2015) 290:7304–13. 10.1074/jbc.M114.611186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giorgi C, Wieckowski MR, Pandolfi PP, Pinton P. Mitochondria associated membranes (MAMs) as critical hubs for apoptosis. Commun Integr Biol (2011) 4:334–5. 10.4161/cib.4.3.15021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal (2015) 22:995–1019. 10.1089/ars.2014.6223 [DOI] [PubMed] [Google Scholar]
- 22.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. 10.1016/j.cell.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol (2012) 13:566–78. 10.1038/nrm3412 [DOI] [PubMed] [Google Scholar]
- 24.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry (2012) 51:2959–73. 10.1021/bi2018909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong F, Davis MC, McColl KS, Distelhorst CW. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol (2006) 172:127–37. 10.1083/jcb.200506189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A (2013) 110:5887–92. 10.1073/pnas.1217823110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle (2013) 12:674–83. 10.4161/cc.23599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang MS, Schwall CT, Pazarentzos E, Datler C, Alder NN, Grimm S. Mitochondrial Ca2+ influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death Differ (2014) 21:1733–45. 10.1038/cdd.2014.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parys JB, De Smedt H. Inositol 1,4,5-trisphosphate and its receptors. Adv Exp Med Biol (2012) 740:255–79. 10.1007/978-94-007-2888-2_11 [DOI] [PubMed] [Google Scholar]
- 30.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med (2010) 31:227–85. 10.1016/j.mam.2010.03.002 [DOI] [PubMed] [Google Scholar]
- 31.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene (2016) 35:5079–92. 10.1038/onc.2016.31 [DOI] [PubMed] [Google Scholar]
- 34.Distelhorst CW, Shore GC. Bcl-2 and calcium: controversy beneath the surface. Oncogene (2004) 23:2875–80. 10.1038/sj.onc.1207519 [DOI] [PubMed] [Google Scholar]
- 35.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell (2010) 37:299–310. 10.1016/j.molcel.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci (2009) 122:437–41. 10.1242/jcs.031682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev (2011) 21:12–20. 10.1016/j.gde.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vervloessem T, La Rovere R, Bultynck G. Antagonizing Bcl-2’s BH4 domain in cancer. Aging (Albany NY) (2015) 7:748–9. 10.18632/aging.100828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene (2008) 27:6398–406. 10.1038/onc.2008.307 [DOI] [PubMed] [Google Scholar]
- 40.Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol (2012) 30:3127–35. 10.1200/JCO.2011.37.0981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature (2005) 435:677–81. 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]
- 42.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med (2013) 19:202–8. 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- 43.Ding J, Zhang Z, Roberts GJ, Falcone M, Miao Y, Shao Y, et al. Bcl-2 and Bax interact via the BH1-3 groove-BH3 motif interface and a novel interface involving the BH4 motif. J Biol Chem (2010) 285:28749–63. 10.1074/jbc.M110.148361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, et al. Inhibition of pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell (2015) 57:873–86. 10.1016/j.molcel.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem (2015) 290:9150–61. 10.1074/jbc.M114.622514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewis A, Hayashi T, Su TP, Betenbaugh MJ. Bcl-2 family in inter-organelle modulation of calcium signaling; roles in bioenergetics and cell survival. J Bioenerg Biomembr (2014) 46:1–15. 10.1007/s10863-013-9527-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem (2010) 285:13678–84. 10.1074/jbc.M109.096040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monaco G, Beckers M, Ivanova H, Missiaen L, Parys JB, De Smedt H, et al. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem Biophys Res Commun (2012) 428:31–5. 10.1016/j.bbrc.2012.10.002 [DOI] [PubMed] [Google Scholar]
- 49.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat Cell Biol (2005) 7:1021–8. 10.1038/ncb1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol (2004) 166:193–203. 10.1083/jcb.200309146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanson CJ, Bootman MD, Distelhorst CW, Wojcikiewicz RJ, Roderick HL. Bcl-2 suppresses Ca2+ release through inositol 1,4,5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell Calcium (2008) 44:324–38. 10.1016/j.ceca.2008.01.003 [DOI] [PubMed] [Google Scholar]
- 52.Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A (2009) 106:14397–402. 10.1073/pnas.0907555106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J, Vais H, Gu W, Foskett JK. Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc Natl Acad Sci U S A (2016) 113:E1953–62. 10.1073/pnas.1517935113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J Biol Chem (2012) 287:23152–61. 10.1074/jbc.M112.345918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature (1999) 399:483–7. 10.1038/20959 [DOI] [PubMed] [Google Scholar]
- 56.Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem (2013) 288:19870–81. 10.1074/jbc.M112.448290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang H, Shah K, Bradbury NA, Li C, White C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis (2014) 5:e1482. 10.1038/cddis.2014.419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol (2012) 14:575–83. 10.1038/ncb2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol (2011) 13:1224–33. 10.1038/ncb2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol (2011) 195:263–76. 10.1083/jcb.201108059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 14:2313–24. 10.1016/j.celrep.2016.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bultynck G. Onco-IP3Rs feed cancerous cravings for mitochondrial Ca2+. Trends Biochem Sci (2016) 41:390–3. 10.1016/j.tibs.2016.03.006 [DOI] [PubMed] [Google Scholar]
- 63.Finkel T, Hwang PM. The Krebs cycle meets the cell cycle: mitochondria and the G1-S transition. Proc Natl Acad Sci U S A (2009) 106:11825–6. 10.1073/pnas.0906430106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ (2012) 19:295–309. 10.1038/cdd.2011.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Monaco G, Vervliet T, Akl H, Bultynck G. The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol Life Sci (2013) 70:1171–83. 10.1007/s00018-012-1118-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell (2008) 31:255–65. 10.1016/j.molcel.2008.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ivanova H, Ritaine A, Wagner L, Luyten T, Shapovalov G, Welkenhuyzen K, et al. The trans-membrane domain of Bcl-2alpha, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget (2016) 7:55704–20. 10.18632/oncotarget.11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foskett JK, Yang Y, Cheung KH, Vais H. Bcl-xL regulation of InsP3 receptor gating mediated by dual Ca2+ release channel BH3 domains. Biophys J (2009) 96:391a. 10.1016/j.bpj.2008.12.2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams A, Hayashi T, Wolozny D, Yin B, Su TC, Betenbaugh MJ, et al. The non-apoptotic action of Bcl-xL: regulating Ca2+ signaling and bioenergetics at the ER-mitochondrion interface. J Bioenerg Biomembr (2016) 48:211–25. 10.1007/s10863-016-9664-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175:901–11. 10.1083/jcb.200608073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem (2010) 285:6053–62. 10.1074/jbc.M109.082990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shoshan-Barmatz V, Ben-Hail D, Admoni L, Krelin Y, Tripathi SS. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim Biophys Acta (2015) 1848:2547–75. 10.1016/j.bbamem.2014.10.040 [DOI] [PubMed] [Google Scholar]
- 73.Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci (2009) 122:1906–16. 10.1242/jcs.040188 [DOI] [PubMed] [Google Scholar]
- 74.Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M, et al. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell (2016) 27:20–34. 10.1091/mbc.E15-01-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veas-Perez de Tudela M, Delgado-Esteban M, Maestre C, Bobo-Jimenez V, Jimenez-Blasco D, Vecino R, et al. Regulation of Bcl-xL-ATP synthase interaction by mitochondrial cyclin B1-cyclin-dependent kinase-1 determines neuronal survival. J Neurosci (2015) 35:9287–301. 10.1523/JNEUROSCI.4712-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bonora M, Giorgi C, Pinton P. Novel frontiers in calcium signaling: a possible target for chemotherapy. Pharmacol Res (2015) 99:82–5. 10.1016/j.phrs.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 77.Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112:1779–84. 10.1073/pnas.1410723112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bittremieux M, Bultynck G. p53 and Ca2+ signaling from the endoplasmic reticulum: partners in anti-cancer therapies. Oncoscience (2015) 2:233–8. 10.18632/oncoscience.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood (2011) 118:1663–74. 10.1182/blood-2011-04-347849 [DOI] [PubMed] [Google Scholar]
- 80.Green DR. A BH3 mimetic for killing cancer cells. Cell (2016) 165:1560. 10.1016/j.cell.2016.05.080 [DOI] [PubMed] [Google Scholar]
- 81.Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood (2011) 117:2924–34. 10.1182/blood-2010-09-307405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xie Q, Su J, Jiao B, Shen L, Ma L, Qu X, et al. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int J Oncol (2016) 49:2507–19. 10.3892/ijo.2016.3733 [DOI] [PubMed] [Google Scholar]
- 83.Fan Z, Yu H, Cui N, Kong X, Liu X, Chang Y, et al. ABT737 enhances cholangiocarcinoma sensitivity to cisplatin through regulation of mitochondrial dynamics. Exp Cell Res (2015) 335:68–81. 10.1016/j.yexcr.2015.04.016 [DOI] [PubMed] [Google Scholar]
- 84.Vervloessem T, Ivanova H, Luyten T, Parys JB, Bultynck G. The selective Bcl-2 inhibitor venetoclax, a BH3 mimetic, does not dysregulate intracellular Ca2+ signaling. Biochim Biophys Acta (2016). 10.1016/j.bbamcr.2016.11.024 [DOI] [PubMed] [Google Scholar]
- 85.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell (2013) 12:329–41. 10.1016/j.stem.2012.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu Z, Wild C, Ding Y, Ye N, Chen H, Wold EA, et al. BH4 domain of Bcl-2 as a novel target for cancer therapy. Drug Discov Today (2016) 21:989–96. 10.1016/j.drudis.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Distelhorst CW, Bootman MD. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: role in Ca2+ signaling and disease. Cell Calcium (2011) 50:234–41. 10.1016/j.ceca.2011.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rong YP, Barr P, Yee VC, Distelhorst CW. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim Biophys Acta (2009) 1793:971–8. 10.1016/j.bbamcr.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis (2013) 4:e632. 10.1038/cddis.2013.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lavik AR, Zhong F, Chang MJ, Greenberg E, Choudhary Y, Smith MR, et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget (2015) 6:27388–402. 10.18632/oncotarget.4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A, Distelhorst CW. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis (2015) 6:e2034. 10.1038/cddis.2015.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deng J, Park D, Wang M, Nooka A, Deng Q, Matulis S, et al. BCL2-BH4 antagonist BDA-366 suppresses human myeloma growth. Oncotarget (2016) 7:27753–63. 10.18632/oncotarget.8513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Han B, Park D, Li R, Xie M, Owonikoko TK, Zhang G, et al. Small-molecule Bcl2 BH4 antagonist for lung cancer therapy. Cancer Cell (2015) 27:852–63. 10.1016/j.ccell.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature (2016) 538:477–82. 10.1038/nature19830 [DOI] [PubMed] [Google Scholar]