

Abstract
A 46-year-old Japanese male with a past medical history of microscopic hematuria presented with nausea, vomiting, and abdominal pain for which he had been diagnosed with rapidly progressive glomerulonephritis with a peak serum creatinine of 6.6 mg/dL and anti-glomerular basement membrane antibody of 214 EU. Light microscopy showed cellular crescent formation, and immunofluorescence illustrated both linear staining of IgG along the glomerular basement membrane and granular staining of IgA and C3 in the mesangial area; however, the PAS staining of mesangial expansions and mesangial proliferations were not observed. Clinical and histological findings suggested anti-glomerular basement membrane glomerulonephritis with mesangial IgA deposition, suggesting IgA nephropathy, a rare condition.
Keywords: Anti-glomerular basement membrane glomerulonephritis, IgA nephropathy, Rapidly progressive glomerulonephritis
Introduction
Rapidly progressive glomerulonephritis (RPGN) is characterized clinically by a rapid progression to end-stage renal disease and histologically by profuse epithelial proliferation, often with epithelial crescents. RPGN can be classified into three categories: pauci-immune type, anti-glomerular basement membrane (GBM) type, and immune complex type. Of these categories, anti-GBM glomerulonephritis can occur in all ages of patients and is pathologically and clinically the most severe form of glomerulonephritis [1]. While it does not usually occur along with another disease having an additional distinctive pattern of glomerular immunofluorescence staining, some concurrent processes include ANCA disease and membranous nephropathy [2]. The following case, therefore, illustrates the rare occurrence of anti-GBM glomerulonephritis with mesangial IgA deposition, suggesting IgA nephropathy.
Case report
A 46-year-old Japanese male with significant past medical history of well-controlled hypertension, diabetes mellitus, and microscopic hematuria with intermittent proteinuria for 3 years presented with nausea, vomiting, and lower abdominal pain. He reported fevers and chills 1 week prior, while denying upper respiratory symptoms, arthralgias, and myalgias. Physical examination showed a blood pressure of 134/83 mmHg, heart rate of 84 beats per minute, normal respiratory rate range, and body temperature of 37.5 °C. His head, cardiac, pulmonary, abdominal, and neurologic examinations did not reveal any apparent abnormalities. In addition, no purpura, rash, joint tenderness, effusion, edema, or oral and genital ulceration had been found.
The laboratory data (Table 1) showed normocytic normochromic anemia (hemoglobin 11.9 g/dL), leukocytosis (WBC 17,350/mm3), elevated C-reactive protein (16.7 mg/dL), and renal failure (BUN 64.9 mg/dL and creatinine 5.2 mg/dL). His medical record proved that his serum creatinine level was 0.7 mg/dL 1 month prior to this episode. Urinalysis showed proteinuria (2.98 g/gCr), hematuria (>100 red blood cells/high power field), and waxy casts. Elevated levels of IgA (640 mg/dL, normal range 90–400) and anti-GBM antibody (214 EU, normal range 0–9) had also been found. Serology for human immunodeficiency virus types 1 and 2, hepatitis B, hepatitis C, PR3-ANCA, MPO-ANCA, and cryoglobulin was negative. Serum C3, C4, and CH50 were within the normal ranges. Computed tomography showed normal sized kidneys and no pulmonary abnormalities. While his creatinine level continued to increase to 6.6 mg/dL, a renal biopsy revealed 1 of 16 glomeruli with global sclerosis, while the remainder showed cellular crescents. Light micrograph of weak magnification showed diffuse crescentic formation and few residual glomerular tufts. Significant mesangial expansion was not detected (Fig. 1). The basement membrane was disrupted in light microscopy (Fig. 2); no apparent mesangial proliferation had been observed. The diffuse tubular and interstitial infiltrates were composed of monocytes and neutrophils.
Table 1.
Laboratory tests at presentation
| Laboratory tests | |
|---|---|
| CBC | |
| WBC | 17,350/μL |
| RBC | 395 × 106/μL |
| Hb | 11.9 g/dL |
| Ht | 34 % |
| Plt | 38.1 × 104/μL |
| Chemistry | |
| Sodium | 134 mEq/dL |
| Potassium | 3.7 mEq/dL |
| Chlorine | 105 mEq/dL |
| BUN | 64.9 mg/dL |
| Creatinine | 5.24 mg/dL |
| AST | 19 mU/mL |
| ALT | 38 mU/mL |
| ALP | 632 mU/mL |
| LDH | 173 mU/mL |
| T-bil | 0.6 mg/dL |
| TP | 6.4 g/dL |
| CRP | 16.7 mg/dL |
| Immune-related | |
| IgG | 1,465 mg/dL |
| IgA | 640 mg/dL |
| IgM | 116 mg/dl |
| Cryoglobulin | − |
| C3 | 149 mg/dL |
| C4 | 35.1 mg/dL |
| CH50 | 66 U/mL |
| Anti-nuclear antibody | − |
| Anti-GBM antibody | 214 EU |
| PR3-ANCA | − |
| MPO-ANCA | − |
| Anti-DNA antibody | − |
| Urinalysis | |
| Protein | 313 mg/dL |
| Occult blood | 3+ |
| WBC | 1–4/HPF |
| RBC | >100/HPF |
Fig. 1.
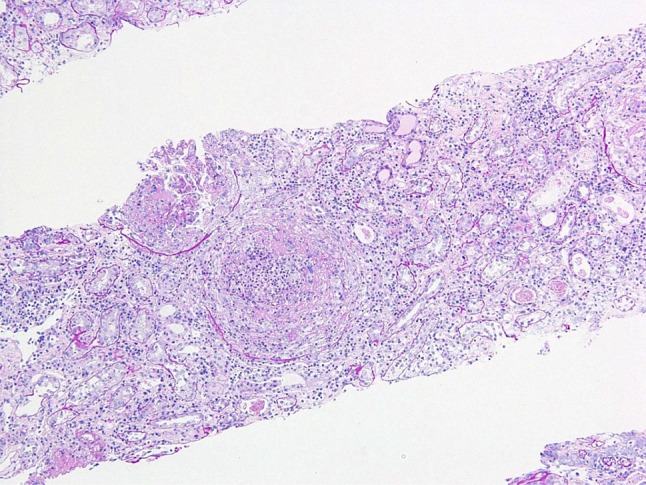
Light micrograph of weak magnification shows diffuse crescentic formation and few residual glomerular tufts. Significant mesangial expansion was not detected. PAS (×10)
Fig. 2.
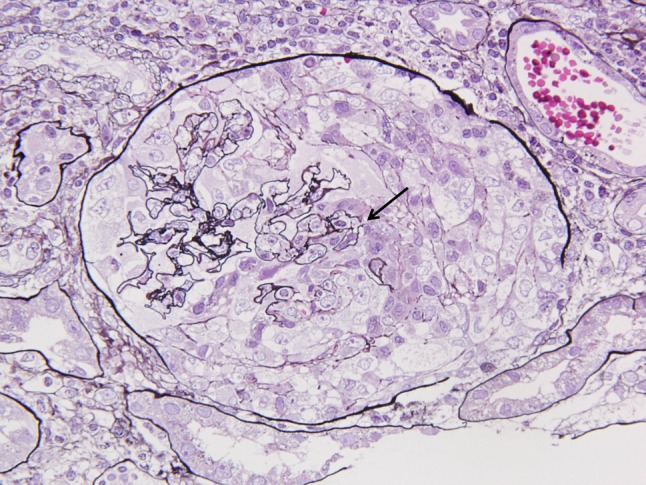
Glomerulus showed cellular crescents and the basement membranes were fragmented (arrow). PAS (×100)
By immunofluorescence study, linear staining for IgG along the GBM had been observed and was accompanied by granular staining of IgA and C3 in the mesangial area (Fig. 3). Fibrinogen was detected in the urinary space, indicating that the GBM was severely damaged. The tissue submitted for electron microscopy contained one glomerulus showing the wrinkling and irregularity of the GBM (Fig. 4). Electron-dense deposits (EDD), even in the mesangial area, could not be detected. Although EDD could not be demonstrated, according to the diffuse mesangial IgA and C3 staining, the diagnosis of anti-GBM glomerulonephritis with mesangial IgA deposition had been established.
Fig. 3.

Two staining patterns from the patient: linear immunofluorescence for IgG along the glomerular capillary walls (left) and mesangial staining for IgA (right)
Fig. 4.

Glomerulus showing the wrinkling and irregularity of the GBM (×1,200)
Intravenous infusion of methyl prednisolone (500 mg/day for three successive days) followed by oral prednisolone (60 mg/day) was started. Plasma exchange was not completed because of an observed allergic reaction of a rash during the first day of therapy. Although the anti-GBM antibody gradually decreased, his renal function did not recover, and he underwent hemodialysis (Fig. 5). His clinical condition and course progressed as a typical case of anti-GBM glomerulonephritis.
Fig. 5.

Clinical course of the patient
Discussion
Relatively few glomerular diseases exist in which dominant IgA deposits occur. Major differential diagnoses for the above include primary IgA nephropathy, Henoch–Schönlein purpura (HSP), and secondary IgA nephropathy due to liver disease (hepatic IgA nephropathy). HSP and hepatic IgA nephropathy are indistinguishable from isolated IgA nephropathy on renal histology. Clinical information is very important to differentiate HSP and hepatic IgA nephropathy, but some differences exist between them. For example, capillary wall staining for IgA and endocapillary proliferation are more frequently found in HSP [3]; however, this finding does not allow the pathologic differentiation of a specific case of HSP from IgA nephropathy. As in primary IgA nephropathy, the glomerular deposits of IgA are frequently accompanied by deposits of IgG and/or IgM; more than half of cases of hepatic IgA have been reported with C1q deposition [4]. This patient showed no purpura, liver dysfunction, or findings histologically consistent with HSP and hepatic IgA, so IgA nephropathy was suspected, although the residual glomerular findings were limited.
The merger of two or more clinical or pathological categories is seen comparatively often in renal diseases. The reason for the concomitant occurrence is unclear; they can be a cause and effect, a new, separate category, or a coincidence. Between 10 and 38 % of patients with anti-GBM antibody disease have positive ANCA at the time of diagnosis [5]; these patients have a poorer prognosis and higher incidence of lung hemorrhage [6]. In patients who have membranous nephropathy before the onset of anti-GBM disease, the pathogenic link may be the release of immunogenic GBM antigens by membranous nephropathy that induces an anti-GBM immune response [7]. On the other hand, IgA nephropathy patients with positive ANCA may represent a distinct clinical entity with a diverse, more exaggerated clinical and histological picture [8]. Combined IgA nephropathy and membranous nephropathy does not appear to have an appreciable negative impact on renal outcome [9]. The coexistence of anti-GBM glomerulonephritis and IgA nephropathy is quite rare, as evidenced by only four English case reports. Savige et al. [10] reported a case of anti-GBM glomerulonephritis complicating an episode of previously documented IgA-positive mesangioproliferative glomerulonephritis, while Cui et al. described a case of anti-GBM glomerulonephritis showing glomerular deposits of IgA(3+) and IgG(1+) along the glomerular capillary walls and in the mesangium [11].
Trpkov et al. fully reported the recurrence of anti-GBM glomerulonephritis after transplantation with de novo IgA nephropathy, demonstrating both linear IgG and mesangial IgA stainings in immunofluorescence and mesangial immune complexes in electron microscopy. The relationship between anti-GBM glomerulonephritis and IgA nephropathy, unfortunately, remains unclear. One hypothesis is that the IgA-related immune complex promotes immunologic and inflammatory events, resulting in conformational changes of the GBM and in exposure of the GBM antigens, thereby, increasing the subliminal anti-GBM antibody production [12]. Because of the rarity of these concurrent conditions, however, this hypothesis is difficult to prove. Fischer and Lager [13] described that, in a series of renal biopsy specimens from 80 patients of anti-GBM glomerulonephritis, one patient had granular mesangial IgA and C3 deposits due to coexisting IgA nephropathy. In crescentic anti-GBM glomerulonephritis, the average creatinine level at the time of diagnosis is 9.7 mg/dL [1], higher than any other type of glomerular disease. Given the severity and rapidity of this disease, the number of glomeruli free from destruction is very limited. For this reason, the pathological feature of IgA glomerulonephritis may not be observed, even when IgA glomerulonephritis precedes it, thereby, possibly causing a lack of case reports describing the combination of anti-GBM glomerulonephritis and IgA nephropathy as compared to the number of ANCA with IgA nephropathy. In this reported case, the creatinine level at biopsy was 6.6 mg/dL, while the other case reports had levels of 15.2 mg/dL (1,347 μmol/L) [11] and 8.14 mg/dL (720 μmol/L) [12]. In addition, the length of time to diagnosis is relatively shorter than the average time reported by Jennette [1]. The anti-GBM nephritis, therefore, may have occurred in our patient with underlying IgA nephropathy, but it is difficult to prove because of the severe and extensive damage to the glomeruli due to the anti-GBM nephritis. Immunofluorescence microscopy, which demonstrated prominent, globular deposits of IgA accompanied by C3 in the mesangium, and his history of hematuria for 2 years, however, may support that this patient has had IgA nephropathy, but it is difficult to diagnose it correctly because the PAS staining of mesangial expansions and mesangial proliferations were not observed. We speculate that the cause could be attributed to the severe and rapid destruction of the glomeruli by an anti-GBM antibody.
In order to understand further the coexistence of IgA nephropathy and anti-GBM glomerulonephritis, more case reports are needed. The above-mentioned reports and case series [10, 11, 13] did not describe patients’ courses precisely, and only a single case after renal transplantation was fully described [12]. In conclusion, this article reported on anti-GBM glomerulonephritis with mesangial IgA deposition in detail and suggested that IgA nephropathy could trigger anti-GBM nephritis.
References
- 1.Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164–1177. doi: 10.1046/j.1523-1755.2003.00843.x. [DOI] [PubMed] [Google Scholar]
- 2.Jennette JC, Nickeleit V. Anti-glomerular basement membrane glomerulonephritis and Goodpasture’s syndrome. In: Jenette JC, Olson JL, Schwartz MM, Silva FG, editors. Heptinstall’s pathology of the kidney. 6. Philadelphia: Lippincott Williams and Wilkins; 2007. pp. 626–629. [Google Scholar]
- 3.Davin JC, Ten Berge IJ, Weening JJ. What is the difference between IgA nephropathy and Henoch–Schönlein purpura nephritis? Kidney Int. 2001;59:823–834. doi: 10.1046/j.1523-1755.2001.059003823.x. [DOI] [PubMed] [Google Scholar]
- 4.Haas M. IgA nephropathy and Henoch–Schönlein purpura nephritis. In: Jennette JC, Olson JL, Schwartz MM, Silva FG, editors. Heptinstall’s pathology of the kidney. 6. Philadelphia: Lippincott-Williams and Wilkins; 2007. pp. 424–486. [Google Scholar]
- 5.Levy JB, Hammad T, Coulthart A, Dougan T, Pusey CD. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int. 2004;66:1535–1540. doi: 10.1111/j.1523-1755.2004.00917.x. [DOI] [PubMed] [Google Scholar]
- 6.Niles JL, Böttinger EP, Saurina GR, Kelly KJ, Pan G, Collins AB, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med. 1996;156:440–445. doi: 10.1001/archinte.1996.00440040118013. [DOI] [PubMed] [Google Scholar]
- 7.Jennette JC, Lamanna RW, Burnette JP, Wilkman AS, Iskander SS. Concurrent antiglomerular basement membrane antibody and immune complex mediated glomerulonephritis. Am J Clin Pathol. 1982;78:381–386. doi: 10.1093/ajcp/78.3.381. [DOI] [PubMed] [Google Scholar]
- 8.Bantis C, Stangou M, Schlaugat C, Alexopoulos E, Pantzaki A, Memmos D, et al. Is presence of ANCA in crescentic IgA nephropathy a coincidence or novel clinical entity? A case series. Am J Kidney Dis. 2010;55:259–268. doi: 10.1053/j.ajkd.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 9.Stokes MB, Alpers CE. Combined membranous nephropathy and IgA nephropathy. Am J Kidney Dis. 1998;32:649–656. doi: 10.1016/S0272-6386(98)70031-9. [DOI] [PubMed] [Google Scholar]
- 10.Savige JA, Dowling J, Kincaid-Smith P. Superimposed glomerular immune complexes in anti-glomerular basement membrane disease. Am J Kidney Dis. 1989;14:145–153. doi: 10.1016/s0272-6386(89)80190-8. [DOI] [PubMed] [Google Scholar]
- 11.Cui Z, Zhao MH, Wang SX, Liu G, Zou WZ, Wang HY. Concurrent antiglomerular basement membrane disease and immune complex glomerulonephritis. Ren Fail. 2006;28:7–14. doi: 10.1080/08860220500461195. [DOI] [PubMed] [Google Scholar]
- 12.Trpkov K, Abdulkareem F, Jim K, Solez K. Recurrence of anti-GBM antibody disease twelve years after transplantation associated with de novo IgA nephropathy. Clin Nephrol. 1998;49:124–128. [PubMed] [Google Scholar]
- 13.Fischer EG, Lager DJ. Anti-glomerular basement membrane glomerulonephritis: a morphologic study of 80 cases. Am J Clin Pathol. 2006;125:445–450. doi: 10.1309/nptp-4ukv-7ju3-elmq. [DOI] [PubMed] [Google Scholar]