

Abstract
A 31-year-old woman with proteinuria, hypocomplementemia, rheumatoid factor, and high serum polyclonal IgM concentration was admitted to our hospital for renal biopsy. She had a past history of two renal biopsies. When she was 12 years old, she developed proteinuria, microscopic hematuria, and hypocomplementemia. She was diagnosed as having ‘IgM nephropathy’ based on minor glomerular abnormalities as determined by light microscopy and IgM and C3 deposition in the mesangial region by immunofluorescence microscopy at the first biopsy. Despite corticosteroid treatment, her proteinuria did not improve and she discontinued regular outpatient checkups. When she was 29 years old and pregnant, she developed preeclampsia and, after delivery, a second renal biopsy was implemented. She was diagnosed as having progressed ‘IgM nephropathy’ with endotheliosis induced by preeclampsia. She was treated with angiotensin II receptor blocker and her proteinuria diminished; however, 1 year after the delivery, she developed proteinuria again, along with microscopic hematuria and hypocomplementemia. A third renal biopsy was conducted at 31 years of age and she was diagnosed as having membranoproliferative glomerulonephritis (MPGN) type I on the basis of diffuse mesangial proliferation, endocapillary hypercellularity with double contour of the capillary wall, and lobular formation in glomeruli, as determined by light microscopy. Immunofluorescence staining demonstrated deposits of C3, C4, C1q, and IgM in the mesangial region and capillary wall. She underwent corticosteroid therapy followed by normalization of urinalysis and serum complement level. Although she had initially been diagnosed with ‘IgM nephropathy’, she was finally diagnosed with secondary MPGN and was successfully treated by corticosteroid therapy.
Keywords: IgM nephropathy, Rheumatoid factor, Foam cell, Preeclampsia, Secondary MPGN
Introduction
Membranoproliferative glomerulonephritis (MPGN) is diagnosed by the pathological findings of the kidney, and is characterized by hypocomplementemia [1]. According to the distribution of electron-dense deposits observed by electron microscopy, this disease is classified as MPGN type I, type II, and type III. MPGN type II is characterized by electron-dense deposits within the basement membrane of glomeruli and is named dense deposit disease; therefore, this type of MPGN is considered as a different type of glomerulonephritis [2, 3]. The most characteristic light microscopic findings of MPGN type I are diffuse global capillary wall thickening and mesangial proliferation, along with lobulation of the glomeruli and double contour of the glomerular basement membrane.
The characteristic pattern of staining in immunofluorescence microscopy is fine to coarse granular or fringe-like staining of C3 along the glomerular capillaries, and C3 is also noted in the glomerular mesangium. C1q, C4, and immunoglobulins such as IgG and IgM are also detected in many, if not most, cases [4, 5]. This corresponds to the prominent subendothelial and mesangial immune complex localization observed by electron microscopy. The pathologic findings indicated that MPGN is an immune complex disease. Secondary MPGN is induced by infection, including hepatitis C and B virus, rheumatologic disease, malignancy, or inherited disease, such as complement deficiency [6, 7]; however, the identity of the nephritogenic antigen is unknown in most patients with idiopathic MPGN. Moreover, not only the pathogenesis, but also the clinical and pathological findings of MPGN, as well as the natural history and response to treatment with glucocorticoids or immunosuppressants, are heterogeneous in this disease. Here, we describe a case of MPGN type I which was first diagnosed as ‘IgM nephropathy’ on the basis of mesangial and capillary IgM and C3 deposits. However, chronic renal injury developed progressively over 20 years and 3 renal biopsies conducted during the clinical course demonstrated different findings at different times, which finally led us to the diagnosis of MPGN type I.
Methods
Serological studies
Levels of serum IgG, IgA, and IgM rheumatoid factors were determined by an indirect solid-phase enzyme immunoassay using Fc fragments of human IgG bound to microwells. Serum rheumatoid factor of the present case was reacted with immobilized IgG–Fc fragments and was detected and quantified by antibodies specific to human IgG, IgA, and IgM (Orgentec Diagnostika, Mainz, Germany). These anti-human IgG, IgA, and IgM antibodies do not possess cross-reactivity with immobilized IgG–Fc fragments.
Case report
A 31-year-old woman with proteinuria, low serum complement concentration, and high polyclonal rheumatoid factor and polyclonal IgM concentrations was admitted to our hospital for renal biopsy to determine the cause of proteinuria.
She had a history of proteinuria (2+), microscopic hematuria (3+), and high serum IgM concentration (1064.2 mg/dl) since she was 12 years old. Rheumatoid factor was positive and cryoglobulin was negative. Continuous hypocomplementemia (C3 64 mg/dl, C4 4.3 mg/dl, CH50 23 U/ml) was detected at that time, but at the age of 13 years, when she underwent her first renal biopsy, serum complement concentration had recovered to within the normal range. This renal biopsy specimen included 24 glomeruli, and light microscopic findings were minor glomerular abnormalities alone, whereas the immunofluorescence microscopic study revealed deposits of C3 and IgM in most of the mesangial lesions and part of the capillary walls (Fig. 1). Electron microscopic observation was not conducted at the first biopsy. She was diagnosed as having ‘IgM nephropathy’ and was treated with 35 mg of prednisolone (0.6 mg/kg), warfarin, and dilazep hydrochloride for 2 years. However, proteinuria (3+) and hematuria (3+) continued and she quit regular outpatient treatment at 18 years old.
Fig. 1.
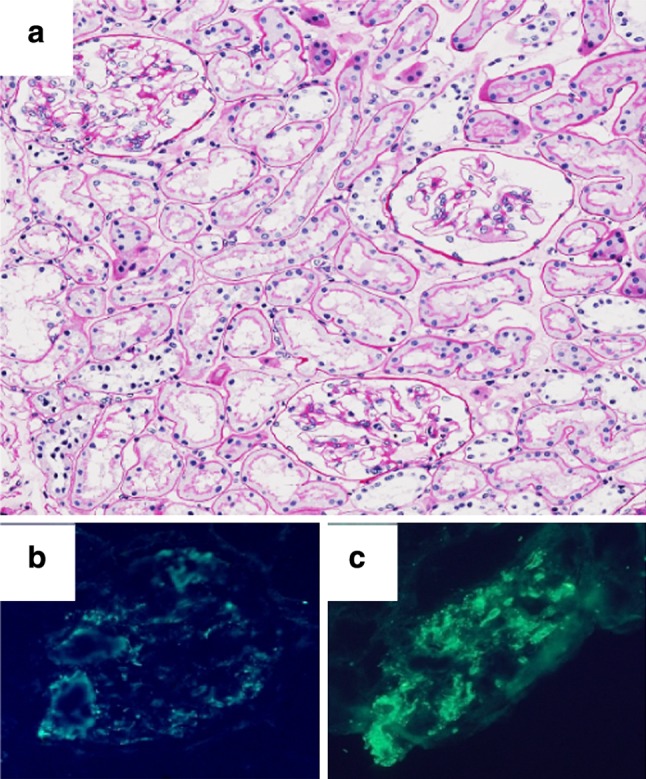
First renal biopsy. a Minor glomerular abnormalities (periodic acid-Schiff, ×100). b, c Diffuse mesangial and partial capillary deposits of IgM (b ×400) and C3 (c ×400)
When she was 29 years old and 9 weeks pregnant, she was diagnosed with proteinuria (2.79 g/day) and started to receive regular outpatient treatment in our hospital. In spite of diet therapy (daily sodium chloride 10 g, protein 60 g), her urine protein was exacerbated and renal function was decreased. At 30 weeks of pregnancy, her blood pressure was over 180/110 mmHg and urine protein was 4.0 g/day. She was diagnosed with preeclampsia, and methyldopa (750 mg/day) was orally administered to control her blood pressure. Cryoglobulin was also detected at the same time. At 33 weeks of pregnancy, hypertension, kidney dysfunction, and proteinuria were, furthermore, exacerbated, despite methyldopa treatment, and, 1 week later, she delivered a baby by Cesarean section. The baby’s birth weight was 1900 g (within ± 1.5 SD) and Apgar score was 7/9. Since hypertension was sustained after the delivery, treatment with valsartan (40 mg), amlodipine (10 mg), and furosemide (40 mg) was started with the discontinuance of lactation. Even though her blood pressure was controlled within the normal range, a nephrotic range of proteinuria (4.8 g/day) continued for 3 weeks after the delivery, and a second renal biopsy was conducted to evaluate renal injury at that time. High serum levels of polyclonal IgM (787 mg/dl) and rheumatoid factor (1100.9 IU/ml) were still positive. This renal biopsy specimen included 24 glomeruli, 3 of which showed global sclerosis. Along with the findings of glomerular capillary endotheliosis characterized by narrowed or obstructed capillary lumina with endothelial cell swelling and hypertrophy, mild proliferation of mesangial cells and increase of mesangial matrix, and diffuse segmental endocapillary hypercellularity with double contour, as well as 20 % interstitial fibrosis and tubular atrophy with foam cells, were also observed in the light microscopic study (Fig. 2a–c). Immunofluorescence staining demonstrated deposits of IgM and fibrinogen in the mesangial region; however, C3 deposits were only scattered (Fig. 2d–f, Table 1). Electron-dense deposits were observed in the subendothelial and mesangial regions (Fig. 2g, h). These findings led us to diagnose a mixture of progressed IgM nephropathy and prolonged preeclampsia.
Fig. 2.

Second renal biopsy. a Glomerular capillary endotheliosis. Capillary lumina were narrowed or obstructed by marked mesangial and endothelial cell swelling and hypertrophy. Global sclerosis of glomeruli and 20 % interstitial fibrosis and tubular atrophy with foam cells (periodic acid silver methenamine–Masson trichrome, ×200). b Endothelial cell swelling indicated by black arrows (periodic acid-Schiff, ×1000). c Doubling of the basement membrane in glomeruli indicated by black arrows (periodic acid silver methenamine, ×1000). d Diffuse mesangial and partial capillary deposit of IgM (×400). e C3c deposit was minimal (×400). f Diffuse mesangial and partial capillary deposit of fibrinogen (×400). g Electron-dense deposit in mesangial region (red arrows, ×2500). h Electron-dense deposit in subendothelial region (red arrows, ×7000)
Table 2.
Laboratory data at the third biopsy
| Hemogram and coagulation | Blood chemistry | LDL-C 137 mg/dl (50–139) |
| WBC 6180/μl (3040–8540) | TP 7.0 g/dl (6.6–8.0) | HDL-C 70 mg/dl (44–96) |
| RBC 387 × 104/μl (378–499 × 104) | Alb 3.9 g/dl (4.1–5.0) | TG 87 mg/dl (32–170) |
| Hb 11.3 g/dl (10.8–14.9) | BUN 17 mg/dl (8–20) | HbA1c 5.4 % (4.3–5.8) |
| Ht 34.6 % (35.6–45.4) | Cre 0.90 mg/dl (0.5–0.8) | Urinalysis and urine chemistry |
| Plt 22.8 × 104/μl (15.0–36.1 × 104) | UA 6.3 mg/dl (2.9–5.2) | Protein 1.28 g/day |
| PT 105 % (PT INR 0.98) | Na 140 mEq/l (138–146) | Selectivity index 0.366 |
| APTT 29.4 s (control 28.3 s) | K 3.7 mEq/l (3.6–4.9) | RBC 30–49/HPF |
| Fibrinogen 341 mg/dl (160–400) | Cl 106 mEq/l (99–109) | WBC 5–9/HPF |
| D–D dimer 1.1 μg/ml (<1.0) | Ca 9.4 mg/dl (8.7–10.0) | Hyaline casts 1-9/HPF |
| Infection | iP 2.8 mg/dl (2.5–4.6) | Ccr 130.25 ml/min/1.73 m2 |
| HBs-Ag (−) | AST 24 IU/l (13–33) | RF 1108.1 IU/ml (<10) |
| HCV-Ab (−) | ALT 27 IU/l (6–27) | IgG RF 481 IU/ml |
| Immunological examinations | LDH 193 IU/l (119–229) | IgM RF 711 IU/ml |
| CRP 0.02 mg/dl (<0.3) | ALP 170 IU/l (115–359) | IgA RF 67 IU/ml |
| IgG 911 mg/dl (870–1700) | γ-GTP 25 IU/l (10–47) | C1q-immune complex (−) |
| IgA 235 mg/dl (110–410) | C3 58.7 mg/dl (65–135) | |
| IgM 657 mg/dl (35–220) | C4 12.9 mg/dl (13–35) | |
| M-protein (−) | CH50 25 U/ml (28–53) | |
| ANA 12.9 index (<20) | ||
| Anti-dsDNA Ab 2 IU/ml (<12) | ||
| Cryoglobulin (−) | ||
| T-Bil 0.3 mg/dl (0.3–0.9) |
Table 1.
Summary of immunofluorescence microscopy at the second biopsy
| IgG | IgA | IgM | C1q | C3c | C4c | Fibrinogen | κ | λ | |
|---|---|---|---|---|---|---|---|---|---|
| Mes | – | Scat | 2+ | – | Scat | Scat | 1+ | 1+ | 1+ |
| Cap | – | – | 1+ | – | – | – | 1+ | – | – |
Mes mesangial region, Cap capillary wall, Scat scattered
After the second renal biopsy, the patient continued to receive regular outpatient treatment with valsartan (80 mg), atorvastatin (10 mg), and dipyridamole (300 mg). Urine protein was less than 1 g/day and cryoglobulin was no longer detected. However, 1 year after the delivery, urine protein had gradually progressed, along with hypocomplementemia; therefore, she was admitted to our hospital for a third renal biopsy.
On physical examination, the patient appeared to be healthy. Her height was 164.6 cm and her weight was 64.9 kg. Her blood pressure was 133/86 mmHg and she had a normal heart rate of 76 beats per minute. No edema was present in her face or extremities, no sign of arthritis was present in her joints, and no apparent physical or neurological abnormalities were observed. Her laboratory data are presented in Table 2. Although proteinuria was not in the nephrotic range, there were clear hypocomplementemia and microscopic hematuria. Serum IgM was polyclonally increased and rheumatoid factors containing all IgG, IgA, and IgM classes were still at high titer. There were no abnormal findings from the hemogram, coagulation testing, and blood chemistry, and there was also no sign of hepatitis B or C virus infection, or specific autoimmune disease.
The third renal biopsy specimen included 26 glomeruli, 6 of which showed global sclerosis. Moreover, 3 of them showed segmental sclerosis, 6 of them showed adhesion, 1 had a fibrocellular crescent, and 3 had collapsed tufts. In addition to moderate proliferation of mesangial cells and increase of mesangial matrix and diffuse segmental endocapillary hypercellularity with double contour, as well as 20 % interstitial fibrosis and tubular atrophy with foam cells, glomeruli were hypertrophic and lobular formation was observed in the light microscopic study (Fig. 3a–d). Immunofluorescence staining demonstrated deposits of C3, C4, C1q, and IgM in the mesangial region and capillary wall; however, their distribution patterns were segmental and unequal (Fig. 3e–g; Table 3). In addition to electron-dense deposits in the subendothelial and mesangial regions, foot process loss of podocytes and mesangial interposition with double contour were also observed (Fig. 3h). Judging from these findings, the patient was finally diagnosed as having MPGN type I.
Fig. 3.

Third renal biopsy. a Chronic lesion was increased compared with that at the second renal biopsy. Glomeruli were hypertrophic and lobular formation was exacerbated (periodic acid-Schiff, ×100). b Interstitial fibrosis and tubular atrophy with foam cells were prominent (periodic acid silver methenamine–Masson trichrome, ×200). c, d Global, moderate mesangial proliferation, segmental endocapillary hypercellularity with double contour, and lobular formation (c periodic acid-Schiff ×400, d periodic acid silver methenamine–Masson trichrome, ×400). e–g Segmental mesangial and partial capillary deposits of IgM (e ×400), C3 (f ×400), and C1q (g ×400). h Electron-dense deposits in the mesangial region (red arrow) and in the subendothelial region (red arrowheads). Scattered dense deposits were also observed in the basement membrane (yellow arrow) and in the subepithelial region. Double contour and mesangial interposition was observed (yellow arrowheads, ×3000)
Table 3.
Summary of immunofluorescence microscopy at the third biopsy
| IgG | IgA | IgM | C1q | C3c | C4c | Fibrinogen | κ | λ | |
|---|---|---|---|---|---|---|---|---|---|
| Mes | – | – | 1+ | Faint | 2+ | Scat | – | 2+ | 2+ |
| Cap | Scat | Scat | 2+ | Faint | 2+ | Faint | 2+ | 2+ | 2+ |
Mes mesangial region, Cap capillary wall, Scat scattered
She was treated with 3 courses of 500 mg of methylprednisolone intravenously for 3 consecutive days, followed by daily oral prednisolone (0.5 mg/kg/day) in addition to valsartan (40 mg), atorvastatin (2.5 mg), and dipyridamole (300 mg). Immediately after methylprednisolone treatment, her serum C4 concentration decreased to undetectable levels (<5 mg/dl) with a mild decrease of C3 (45.1 mg/dl) and CH50 (18 U/ml). After she was discharged from our hospital, the dose of oral prednisolone was gradually decreased and her proteinuria and microscopic hematuria were no longer detected from 2 months after corticosteroid therapy, followed by recovery of serum complement levels and renal function an additional 2 months later. Rheumatoid factor was also decreased to 864.8 IU/ml, especially due to the decrease of IgM rheumatoid factor (IgG RF 443 IU/ml, IgM RF 385 IU/ml, IgA RF 45 IU/ml), along with the decrease of serum IgM concentration (522 mg/dl).
Discussion
We have reported here a case of MPGN type I with a 20-year history and sequential renal biopsies at 3 different times during the development of this disease. Although the patient had initially been diagnosed with ‘IgM nephropathy’, she finally developed MPGN type I and was successfully treated by corticosteroid therapy. A summary of the clinical and pathological findings of these sequential renal biopsies is shown in Table 4.
Table 4.
Summary of the findings of renal pathology
| 1st biopsy | 2nd biopsy | 3rd biopsy | |
|---|---|---|---|
| Time of biopsy | 13 years old Abnormality of urinalysis in regular checkup at school |
29 years old After the delivery of her 1st child |
31 years old Exacerbation of urine protein |
| Urinalysis and renal function | UP (2+) U-RBC >100/HPF Ccr 162.5 ml/min/1.73 m2 |
UP 4.8 g/day U-RBC 5–9/HPF Ccr 103.9 ml/min/1.73 m2 |
UP 1.28 g/day U-RBC 30–49/HPF Ccr 130.3 ml/min/1.73 m2 |
| Hypocomplementemia | Negative, but continuous hypocomplementemia was detected 1 year previously | Negative | Positive, and recovered after corticosteroid therapy |
| Serum IgM (mg/dl) | 1064.2 | 787.0 | 657.0 |
| Rheumatoid factor (IU/ml) | 2030 | 1100.9 | 1108.1 |
| Cryoglobulin | Negative | Positive | Negative |
| Light microscopy | Minor glomerular abnormalities | Diffuse global mild mesangial proliferation and endotheliosis GBM double contour Global sclerosis 3/24 |
Lobular formation GBM double contour Endocapillary hypercellularity Global sclerosis 6/26 |
| Immunofluorescence microscopy | IgM and C3 in the mesangial region and part of the capillary wall | IgM and fibrinogen in the mesangial region and part of the capillary wall | IgM, C3, C4, and C1q in the mesangial region and capillary wall |
| Clinical course | Prednisolone (0.6 mg/kg) Proteinuria and microscopic hematuria continued |
Angiotensin II receptor blocker and proteinuria disappeared | Methylprednisolone pulse and prednisolone (0.5 mg/kg) and proteinuria and hematuria disappeared |
At the first biopsy, this case was diagnosed as IgM nephropathy on the basis of the findings of immunofluorescence microscopy, which showed IgM and C3 deposition, and minor glomerular abnormalities, as determined by light microscopy.
IgM nephropathy was first proposed as a separate disease that presented with nephrotic syndrome or asymptomatic proteinuria, morphologic evidence of mild mesangial alterations, and generalized and diffuse mesangial deposition, primarily of IgM and C3, along with electron-dense deposits in the mesangial region [8–10]. It remains controversial, however, as to whether IgM nephropathy is a separate disease, since scattered IgM deposits can be found with roughly equal incidence in patients with minimal change disease, focal segmental glomerulosclerosis (FSGS), and mesangial proliferative glomerulonephritis (MesPGN) [11], and IgM deposits could be considered as a secondary or nonspecific finding. On the other hand, Myllymäki et al. reported on the precise investigation of 110 IgM nephropathy patients, including 38 with normal or minimal lesions of glomeruli, 37 with mesangial hypercellularity, 40 with mesangial sclerosis, 37 with tubulointerstitial lesions, 28 with vascular disorders, and 17 with obliterated glomeruli. The authors insisted that patients with IgM nephropathy are less likely to respond to immunosuppressive agents than those with minimal change disease; therefore, IgM nephropathy should be categorized as a separate disease. They also reported that, in 11 patients who underwent repeated biopsy because of deterioration in kidney function, exacerbation of proteinuria, and/or poor response to pharmacotherapy, 5 renal pathologies showed FSGS, 5 showed minimal histological lesions, and 1 showed lupus nephritis. Moreover, 3 of 5 patients with FSGS developed end-stage renal failure, but none of the 5 patients with minimal lesions or 1 patient with lupus nephritis showed progressive renal dysfunction, although MPGN lesion was not detected according to the published report [12]. Kishimoto and Arakawa [13] also reported that MesPGN with mesangial IgM deposits demonstrated higher serum IgM level, lower glomerular filtration rate and serum CH50 level, higher level of proteinuria, and higher incidence of adhesion and duplication of capillary loops than MesPGN without mesangial IgM deposits, suggesting that predominant IgM deposition should be referred to as IgM nephropathy with impaired renal function and poor prognosis. In the present case, renal lesions developed and finally demonstrated pathological findings applicable to MPGN, but not FSGS or MesPGN. Considering the past history of hypocomplementemia, we conjecture that this case was MPGN at the onset of the disease, and, during its gradual progression, it showed pathological findings resembling IgM nephropathy. Interestingly, hypocomplementemia and C3 deposits in the capillary walls and mesangial regions were not observed at the second biopsy after preeclampsia. We are tempted to surmise that, at least during her pregnancy, progression of MPGN was suspended, and after the remission of preeclampsia, immunological reaction caused by an unknown antigen flared up again, resulting in hypocomplementemia and exacerbation of proteinuria and hematuria.
This case is also characterized by the presence of foam cells in the interstitium. Interstitial foam cells laden with lipids are a common finding in patients with nephrotic syndrome and hyperlipidemia, as well as in those with Alport syndrome, and generally indicate progression of renal injury and poor prognosis [14, 15].
This case showed continuously high levels of serum polyclonal IgM and rheumatoid factors, which consisted of IgG, IgM, and IgA rheumatoid factors. Considering the existence of these polyclonal rheumatoid factors, it might not be contradictory if type III cryoglobulinemia existed and induced secondary MPGN. However, cryoglobulin was detected during the pregnancy and after the delivery alone, and, at this time, the biopsy specimen indicated glomerular endotheliosis, but not MPGN lesion, as determined by light microscopy. Moreover, immunofluorescence microscopy did not demonstrate C3 deposit, and hypocomplementemia was not detected. Furthermore, in electron microscopy at the third biopsy, microtubular aggregates typical of cryoglobulin deposits [16, 17] were not observed. Taken together, these findings suggest that secondary MPGN due to type III cryoglobulinemia is unlikely; however, this patient had a very low C4 concentration with almost normal or mildly decreased C3 level at both the first biopsy and after methylprednisolone treatment at the third biopsy. In idiopathic MPGN, low serum levels of C3 and CH50 were observed, whereas the C4 level tends to be normal [18]. When MPGN is caused by cryoglobulinemia, however, C4 depression is more prominent than C3 [19]. This case should be diagnosed as secondary MPGN, if not due to cryoglobulinemia, in which the classical complement pathway is involved.
Rossen et al. demonstrated that, in patients with high rheumatoid factor, FITC-labeled human IgG was bound to immune deposits in the glomeruli from some patients with glomerulonephritis, including 3 cases of MPGN. However, 2 of them had systemic lupus erythematosus and this reaction between IgG and deposited immunoglobulins in glomeruli indicated the existence of rheumatoid factors in the glomerular immunodeposits [20]. There was also a report on autologous immune complex deposits in acute poststreptococcal glomerulonephritis [21]. Endoh et al. [22] conducted a comprehensive investigation of the subclasses of serum rheumatoid factors and their relationship with several types of glomerulonephritis, and reported that high titers of IgG and IgA rheumatoid factors were observed in patients with acute poststreptococcal glomerulonephritis, whereas IgG, IgA, and IgM titers were significantly higher in patients with diffuse proliferative lupus nephritis, and, to a lesser extent, with membranous lupus nephritis, IgA nephropathy, and idiopathic membranous nephropathy [23]. Furthermore, human IgG or the Fc portion of IgG bound to immunoglobulin on the glomeruli only in diffuse proliferative lupus nephritis [23]. To the best of our knowledge, the present report should be the first to demonstrate the high titer of IgG, IgA, and IgM rheumatoid factors in a patient with MPGN. However, even though a high concentration of serum polyclonal IgM might affect the development and progression of MPGN, the interaction between IgG polyclonal rheumatoid factor complex formation and the development of MPGN was less likely in this case because IgG deposition was not detected in the renal specimens.
In conclusion, we have reported here a case of MPGN type I with mesangial IgM deposition that developed over 20 years with constantly high levels of serum polyclonal IgM and rheumatoid factor. Although the interaction between the development of MPGN and IgM rheumatoid factor is unclear, corticosteroid therapy improved clinical findings, along with the decrease of serum IgM and rheumatoid factor, suggesting a possible interaction between MPGN and IgM.
Acknowledgments
We thank Keiko Yamagiwa, Kazumasa Sato, Akiko Seino, Kaori Takahashi, and Takae Watanabe for their expert technical assistance. Preliminary data from this study were presented in a poster session at the 41st Eastern Regional Meeting of the Japanese Society of Nephrology, October 2011, Tokyo, Japan.
Conflict of interest
All the authors have declared no competing interest.
References
- 1.West CD, McAdams AJ, McConville JM, Davis NC, Holland NH. Hypocomplementemic and normocomplementemic persistent (chronic) glomerulonephritis; clinical and pathologic characteristics. J Pediatr. 1965;67:1089–1112. doi: 10.1016/S0022-3476(65)80213-X. [DOI] [Google Scholar]
- 2.Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol. 2010;25:1409–1418. doi: 10.1007/s00467-009-1322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: pathogenetic heterogeneity and proposal for a new classification. Semin Nephrol. 2011;31:341–348. doi: 10.1016/j.semnephrol.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Davis BK, Cavallo T. Membranoproliferative glomerulonephritis. Localization of early components of complement in glomerular deposits. Am J Pathol. 1976;84:283–298. [PMC free article] [PubMed] [Google Scholar]
- 5.Zucchelli P, Sasdelli M, Cagnoli L, Donini U, Casanova S, Rovinetti C. Membranoproliferative glomerulonephritis: correlations between immunological and histological findings. Nephron. 1976;17:449–460. doi: 10.1159/000180753. [DOI] [Google Scholar]
- 6.Cameron JS, Turner DR, Heaton J, Williams DG, Ogg CS, Chantler C, et al. Idiopathic mesangiocapillary glomerulonephritis. Comparison of types I and II in children and adults and long-term prognosis. Am J Med. 1983;74:175–192. doi: 10.1016/0002-9343(83)90606-X. [DOI] [PubMed] [Google Scholar]
- 7.Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47:643–656. doi: 10.1038/ki.1995.82. [DOI] [PubMed] [Google Scholar]
- 8.Cohen AH, Border WA, Glassock RJ. Nephrotic syndrome with glomerular mesangial IgM deposits. Lab Invest. 1978;38:610–619. [PubMed] [Google Scholar]
- 9.Border WA. Distinguishing minimal-change disease from mesangial disorders. Kidney Int. 1988;34:419–434. doi: 10.1038/ki.1988.197. [DOI] [PubMed] [Google Scholar]
- 10.O’Donoghue DJ, Lawler W, Hunt LP, Acheson EJ, Mallick NP. IgM-associated primary diffuse mesangial proliferative glomerulonephritis: natural history and prognostic indicators. Q J Med. 1991;79:333–350. [PubMed] [Google Scholar]
- 11.Childhood nephrotic syndrome associated with diffuse mesangial hypercellularity. A report of the Southwest Pediatric Nephrology Study Group. Kidney Int. 1983;24:87–94. [DOI] [PubMed]
- 12.Myllymäki J, Saha H, Mustonen J, Helin H, Pasternack A. IgM nephropathy: clinical picture and long-term prognosis. Am J Kidney Dis. 2003;41:343–350. doi: 10.1053/ajkd.2003.50042. [DOI] [PubMed] [Google Scholar]
- 13.Kishimoto H, Arakawa M. Clinico-pathological characterization of mesangial proliferative glomerulonephritis with predominant deposition of IgM. Clin Exp Nephrol. 1999;3:110–115. doi: 10.1007/s101570050019. [DOI] [Google Scholar]
- 14.Poucell S, Thorner P, Balfe JW, Baumal R. Interstitial foam cells in renal biopsies: an aid in differentiating idiopathic membranoproliferative glomerulonephritis (type I) and membranoproliferative glomerulonephritis associated with systemic lupus erythematosus. Child Nephrol Urol. 1988–1989;9:106–111. [PubMed] [Google Scholar]
- 15.Wu Y, Chen Y, Chen D, Zeng C, Li L, Liu Z. Presence of foam cells in kidney interstitium is associated with progression of renal injury in patients with glomerular diseases. Nephron Clin Pract. 2009;113:c155–c161. doi: 10.1159/000232596. [DOI] [PubMed] [Google Scholar]
- 16.Howell DN, Gu X, Herrera GA. Organized deposits in the kidney and look-alikes. Ultrastruct Pathol. 2003;27:295–312. [PubMed] [Google Scholar]
- 17.Iskandar SS, Herrera GA. Glomerulopathies with organized deposits. Semin Diagn Pathol. 2002;19:116–132. [PubMed] [Google Scholar]
- 18.Kher V, Gulati S. Mesangiocapillary glomerulonephritis. In: Davison AM, Cameron JS, Grünfeld J-P, Ponticelli C, Ritz E, Winearls CG, van Ypersele C, editors. Oxford textbook of clinical nephrology. 3. New York: Oxford University Press; 2005. pp. 523–557. [Google Scholar]
- 19.Nachman PH, Jennette JC, Falk RJ. Primary glomerular disease. In: Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM, editors. Brenner & Rector’s The Kidney. 9. Philadelphia: Elsevier Saunders; 2012. pp. 1101–1191. [Google Scholar]
- 20.Rossen RD, Reisberg MA, Sharp JT, Sucki WN, Schloeder FX, Hill LL, et al. Antiglobulins and glomerulonephritis. Classification of patients by the reactivity of their sera and renal tissue with aggregated and native human IgG. J Clin Invest. 1975;56:427–437. doi: 10.1172/JCI108109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McIntosh RM, García R, Rubio L, Rabideau D, Allen JE, Carr RI, et al. Evidence of an autologous immune complex pathogenic mechanism in acute poststreptococcal glomerulonephritis. Kidney Int. 1978;14:501–510. doi: 10.1038/ki.1978.155. [DOI] [PubMed] [Google Scholar]
- 22.Endoh M, Suga T, Sakai H. IgG, IgA and IgM rheumatoid factors in patients with glomerulonephritis. Nephron. 1985;39:330–335. doi: 10.1159/000183400. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki M, Endoh M, Suga T, Yano N, Kuramoto T, Matsumoto Y, et al. Rheumatoid factors and glomerulonephritis. Clin Exp Immunol. 1990;81:250–255. doi: 10.1111/j.1365-2249.1990.tb03326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]