

Abstract
The disequilibrium between the production of reactive oxygen (ROS) and nitrogen (RNS) species and their elimination by protective mechanisms leads to oxidative stress. Mitochondria are the main source of ROS as by-products of electron transport chain. Most of the time the intestine responds adequately against the oxidative stress, but with aging or under conditions that exacerbate the ROS and/or RNS production, the defenses are not enough and contribute to developing intestinal pathologies. The endogenous antioxidant defense system in gut includes glutathione (GSH) and GSH-dependent enzymes as major components. When the ROS and/or RNS production is exacerbated, oxidative stress occurs and the intestinal Ca2+ absorption is inhibited. GSH depleting drugs such as DL-buthionine-S,R-sulfoximine, menadione and sodium deoxycholate inhibit the Ca2+ transport from lumen to blood by alteration in the protein expression and/or activity of molecules involved in the Ca2+ transcellular and paracellular pathways through mechanisms of oxidative stress, apoptosis and/or autophagy. Quercetin, melatonin, lithocholic and ursodeoxycholic acids block the effect of those drugs in experimental animals by their antioxidant, anti-apoptotic and/or anti-autophagic properties. Therefore, they may become drugs of choice for treatment of deteriorated intestinal Ca2+ absorption under oxidant conditions such as aging, diabetes, gut inflammation and other intestinal disorders.
Keywords: Transcellular and paracellular Ca2+pathways; DL-buthionine-S,R-sulfoximine; Menadione; Sodium deoxycholate; Lithocholic acid; Ursodeoxycholic acid; Melatonin
Core tip: Glutathione depleting drugs inhibit the intestinal Ca2+ absorption by alteration in the protein expression and/or activity of molecules involved in the transcellular and paracellular Ca2+ pathways through mechanisms of oxidative stress, apoptosis and/or autophagy. Quercetin, melatonin, lithocholic and ursodeoxycholic acids block the effect of those drugs in experimental animals by their antioxidant, anti-apoptotic and anti-autophagic properties. Therefore, they may become drugs of choice for treatment of deteriorated intestinal Ca2+ absorption under oxidant conditions such as aging, diabetes, gut inflammation and other intestinal disorders.
INTRODUCTION
The imbalance between the production of reactive oxygen (ROS) and nitrogen (RNS) species and their elimination by protective mechanisms leads to oxidative stress[1]. This response occurs in various pathophysiological conditions such as aging, inflammation, cardiovascular and neurodegenerative diseases, damaging many components including proteins, DNA/RNA and lipids[2-5]. The cellular dysfunctions caused by excessive ROS and/or RNS might produce loss of energy metabolism, altered cell signaling and cell cycle, gene mutations and impaired cellular transport mechanisms. Taken together, the oxidative stress promotes decreased biological activities, immune activation and inflammation. Moreover, the nutritional stress produced by high-fat and high-carbohydrate diets also generates oxidative stress, which leads to initiation of pathogenic milieu and development of different chronic diseases[6-8]. ROS are also generated by other exogenous sources such as ultraviolet radiation, alcohol consumption, cigarette smoking, ingestion of nonsteroidal anti-inflammatory drugs and infections[9,10]. Ischemia/reperfusion (I/R) injuries also contribute to exacerbating ROS production[11]. ROS are normally produced within the body in small quantities and are involved in the regulation of processes, maintenance of cell homeostasis and functions such as signal transduction, gene expression, and activation of receptors[12]. Mitochondria are one of the most relevant sources of ROS and RNS. The organelles produce ROS and organic peroxides as by-products during the functioning of the electron transport chain (ETC), and, in hypoxic conditions, they also produce nitric oxide (˙NO), one RNS that can further lead to produce reactive aldehydes, malondialdehyde and 4-hydroxynonenal[13,14]. Peroxisomes also play a major role in the cellular ROS and RNS-metabolism, not only because they contain a large number of ROS-producing enzymes, but they also interplay with other organelles, mainly with the mitochondria and endoplasmic reticulum (ER)[15]. The accumulation of unfolded and misfolded proteins in the ER lumen, known as ER stress, activates the unfolded protein response, which enhances the ER capacity for protein folding and modification, attenuates global mRNA translation, and disposes misfolded proteins by ER-associated protein degradation and autophagy. The dysregulated disulfide bond formation and breakage in a stressed ER, may produce ROS accumulation and cause oxidative stress[16].
The small intestine is the main organ of exposure and/or absorption of nutrients, toxic food contaminants and therapeutic drugs. It is also exposed to secreted metabolites and the metabolic products coming from the intestinal bacteria. The alteration of the integrity and/or function of the intestinal epithelium produce a negative impact on the rest of the organism[17]. Therefore, the disequilibrium in the redox state of gut is not only important for its functionality, but also for the entire body. Fortunately, most of the time the intestine responds adequately against the oxidative stress, but with aging or under conditions that exacerbate the ROS and/or RNS production the defenses are not enough, which contribute to developing intestinal pathologies such as inflammatory bowel disease (IBD), gastroduodenal ulcers, colon cancer and others[18-20].
FORMATION OF ROS AND RNS IN THE GUT
ROS are not only highly reactive and continuously produced as by-products of cellular respiration, but are also generated by enzymatic reactions. ROS include radical compounds such as superoxide (O2-˙), hydroxyl radicals (˙HO), lipid hydroperoxides, and reactive nonradical compounds including singlet oxygen (1O2), hydrogen peroxide (H2O2), hypochlorous acid (HOCl) and others[21]. RNS include radical compounds such as ˙NO, nitrogen dioxide (˙NO2), and nonradical compounds such as peroxynitrite (ONOO-) and dinitrogen trioxide (N2O3). Most of these compounds are unstable because of unpaired electrons in the outer electron orbit. When ROS are accumulated, the major cellular antioxidants such as glutathione (GSH) and thioredoxin alter their redox state, and the antioxidant defenses decline.
In the mitochondria, the electron leakage from ETC complexes I and III produces a reduction of molecular oxygen forming O2-˙[22]. In contrast, cytochrome c oxidase (complex IV) is not an important source of ROS. It reduces molecular oxygen to two molecules of H2O through a four-electron reduction[23]. NADPH oxidase, present in the plasma membrane and phagosomes of phagocytes (monocytes, macrophages, neutrophils and eosinophils), also produces an important amount of O2-˙ in the intestine, mainly in conditions of inflammation induced by Helicobacter pylori, IBD, and tumor development[8]. Xantine oxidase (XO) is another enzyme that generates O2-˙ as a by-product by oxidation of hypoxanthine to xanthine, and then to uric acid during purine catabolism. In the I/R of gut, this enzyme produces an enormous quantity of ROS because the oxidation of hypoxanthine is increased[24,25]. XO exists mainly in the small intestine and it may be a major source of ROS in patients during colon surgery[26]. The enzyme is predominantly present as xantine dehydrogenase under physiological conditions, but it can be transformed by proteolysis into XO. In acute pancreatitis, XO is mobilized from the gastrointestinal endothelial cell surface[27]. The enzymes of the XO family share a molybdenum cofactor (Moco), which is a trace element and crucial for life[28]. The reason the mature enterocytes, located at the tip of the microvilli, are more sensitive to I/R than their undifferentiated counterparts located in the villus base seems to be related, at least in part, to the higher expression and activity of XO[29]. The nutritional deficiency in Mo has been associated with high risk of esophageal cancer in populations consuming food grown in molybdenum-poor soil[30]. ROS are also produced in the intestine by other enzymes such as myeloperoxidases, lipoxygenases, ciclooxygenases and transition metals as copper and iron. ˙NO is a weak oxidant generated by oxidation of L-arginine, reaction catalyzed by nitric oxide synthase (NOS). When ˙NO combines with O2-˙, it generates OONO-, which is highly reactive[31]. OONO- provokes enterocyte apoptosis, reduces enterocyte proliferation and interferes with epithelial renewal[32]. ˙NO and OONO- produce stable nitrite and nitrate ions, which can be accumulated in cells leading to form high reactive intermediates, such as ˙NO2 and N2O3. These intermediates may cause nitration and nitrosation of DNA, RNA, proteins and lipids with the consequent dysfunction of these molecules[33].
ENDOGENOUS ANTIOXIDANT DEFENSE SYSTEM IN THE GUT
Any substance or compound that scavenges oxygen free radicals or inhibits the cellular oxidation process is considered an antioxidant[34]. The main non enzymatic antioxidants in gut are GSH and the thioredoxin system. GSH is a tripeptide formed by L-glutamate, L-glycine and L-cysteine, and is present in millimolar concentration (2-10 mmol/L) in all eukaryotic cells. The oxidation of GSH to disulfide of glutathione (GSSG) and subsequent decrease in the GSH/GSSG couple is often a useful indicator of cellular oxidative stress[35]. There are different pools of GSH in the cell. The total cellular GSH/GSSG ratio mainly represents the cytoplasmic GSH/GSSG pool. GSH/GSSG ratios are not in equilibrium with each other in mitochondria, nucleus, the secretory pathway and the extracellular space[36]. Mitochondrial GSH is responsible for 15% to 30% of total GSH[37]. In the ER, the GSH/GSSG ratio ranges between 3/1 and 1/1. It is more oxidized than cytoplasmic GSH/GSSG ratio, which varies between 30/1 to 100/1. GSH in the ER was mainly detected as protein mixed disulfides, which means that it would regulate the activity of redox-active thiol-containing proteins[38]. Protein S-glutathionylation is independently controlled in the cytoplasmic and nuclear compartments and the GSH/GSSG redox potential is probably more reduced in nucleus than in cytoplasm[36]. The cytosolic enzymes γ-glutamylcysteine ligase and GSH synthetase are involved in de novo GSH synthesis, while the regeneration of GSH from GSSG is catalyzed by NADPH-dependent GSSG reductase[39]. In transport epithelial cells as occurs in enterocytes, γ-glutamyltransferase and dipeptidase catalyze the hydrolysis of extracellular GSH to its constituent amino acids[40].
The distribution between nuclear GSH and cytoplasmic GSH is dynamic. The GSH concentration in nucleus is 4 times higher than in cytoplasm during cell cycle and is equal in both compartments when cells are confluent[41]. The intestinal GSH levels depend on the de novo synthesis, regeneration from GSSG and the GSH uptake at the apical membrane[40]. It appears that the cellular GSH/GSSG redox status governs cell transitions from quiescence to that of a proliferative state, as well as the growth arrest, differentiation and apoptosis, not only in the intestine but also in other cells. A reducing redox environment favors proliferation, whereas an oxidized milieu stimulates growth arrest and differentiation[42]. If the redox environment is highly oxidized, it promotes apoptosis or necrosis. Mitochondria are involved in the oxidant-mediated cellular apoptosis[43]. Loss of mitochondrial GSH (mtGSH) produces mitochondrial transition pore opening[44], inhibits ETC, decreases ATP and increases ROS generation, which leads to cell apoptosis[45,46]. mtGSH also preserves intestinal mitochondrial genes and functional integrity[47]. Another GSH pool, the luminal GSH pool, has an important role in the processes of absorption and detoxification as well as in maintenance of mucus fluidity[48-50].
The thioredoxin system is composed of thioredoxin (Trx) and thioredoxin reductases (TrxR). It has a large number of functions in DNA synthesis, defense against oxidative stress and apoptosis or redox signaling. It is located in the cytoplasm, membranes, mitochondria, and in the extracellular space. Oxidized Trxs are reactivated by TrxRs through the reducing power of NADPH[51]. Trx expression is very high in the intestine and has an important role in gut immune response[52]. It has been demonstrated that Trx is involved in redox regulation of human β-defensin 1, a protein with antimicrobial activity[53]. Ulcerative colitis involving Trx as a candidate marker has been revealed by proteomic profiles of colonic biopsies[54].
The major GSH-dependent enzymatic antioxidants in the intestine are superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione-reductase (GR) and catalase (CAT). SOD and CAT provide major antioxidant defenses against ROS[8]. SODs are enzymes that catalyze dismutation of O2-˙ into O2 and H2O2. In humans there are three isoforms of SOD: cytosolic copper and zinc-containing enzyme (Cu2+/Zn2+-SOD), manganese-requiring mitochondrial enzyme (Mn2+-SOD), and an extracellular Cu2+/Zn2+ containing SOD. Mn2+-SOD has an indispensable role in protecting aerobic life from deleterious effects of oxygen. It could be considered as the guardian of the powerhouse. Mn2+-SOD can scavenge O2-˙ generated by the ETC complexes and may be important in preventing ROS-induced inactivation of these complexes[55]. Injuries of the GIT can be prevented by SOD in the gastrointestinal mucosa. Intestinal epithelia from IBD patients have enhanced levels of all three SOD isoforms[56]. H2O2, itself a ROS, is decomposed into water by different enzymes including GPx, CAT and peroxiredoxins[57]. GPx reduces not only H2O2, but also lipid hydroperoxides. In the intestine there are four isoforms of GPx[58]. GPx1 is present in all cell types of the gut, GPx2 is predominantly expressed in the epithelial cells, GPx3 is secreted in plasma, and GPx4 is expressed in epithelial cells and the lamina propria[59]. GPx2 is in the first line of defense against ROS derived from inflammation associated with both pathogenic and nonpathogenic bacteria from the intestine[58]. GR is a ubiquitous enzyme that reduces GSSG to GSH. GR is a NADPH-dependent flavoprotein. Two electrons of reducing power are extracted from NADPH and transferred to reducing GSSG into two molecules of GSH[60]. CAT, which is found mainly in peroxisomes, dismutates H2O2 to H2O and O2. It is present in all human organs and in many pathogens in the GIT to evade host response and survive within the host. In addition, CAT is also expressed in mitochondria and is considered to protect cells from apoptosis[61]. Taken together, all these enzymes and endogenous non enzymatic antioxidants contribute to the equilibrium in the redox state of the intestine under physiological conditions. However, excessive ROS and/or RNS may still lead to oxidative damage to tissue and organs. Hence, the application of antioxidants seems to be a rational therapeutic strategy to prevent or cure diseases involving oxidative stress.
MECHANISMS OF INTESTINAL CALCIUM ABSORPTION
The intestinal Ca2+ absorption is an active process (ATP dependent) that mainly occurs in the small intestine, which is responsible for approximately 90% of overall Ca2+ absorption[62]. The sojourn time in each intestinal segment and the Ca2+ solubility are important factors affecting the intestinal Ca2+ absorption. The solubility depends on the environmental pH. Although the solubility is higher in the stomach because of the acidic pH, this is less relevant because Ca2+ is absorbed in the small and large intestine. It appears that the duodenum is the site with the maximum solubility of Ca2+ because the average pH is 6.0, the lowest of the entire gut[63]. The Ca2+ absorption rate occurs in this order: duodenum > jejunum> ileum[64]. Since the major source of ATP is the mitochondria, the integrity and functionality of these organelles are necessary to produce an appropriate intestinal Ca2+ absorption. Growth, pregnancy and lactation promote the intestinal Ca2+ absorption, while aging decreases cation absorption[65-68]. The efficiency of the intestine to absorb Ca2+ increases not only when the requirements enhance, but also when the intake is low[69]. In other words, the intestinal Ca2+ absorption depends on the physiological needs of Ca2+.
There are two mechanisms of intestinal Ca2+ absorption: transcellular and paracellular. Both mechanisms are regulated by hormones, nutrients and other factors. The transcellular pathway comprises three steps: entry across the brush border membrane (BBM) of the enterocytes, intracellular diffusion from one pole to the other of the epithelial cells and exit through the basolateral membrane (BLM). In the BBM there are Ca2+ epithelial channels, called transient receptor potential vanilloid-family member 6 (TRPV6) and transient receptor potential vanilloid-family member 5 (TRPV5), which are apparently involved in the Ca2+ uptake from the lumen to inside the cell through the BBM[70]. TRPV6 predominates in the intestine, whereas TRPV5 in the kidney. Cav1.3 is another channel from the BBM, which is apparently involved in the active transcellular Ca2+ absorption. TRPV6 would predominate under polarizing conditions between meals, whereas Cav1.3 would play a dominant role under depolarizing conditions during digestion, mainly when diet is plentiful in Ca2+[71]. In contrast, some authors demonstrate that Cav1.3 is not critical for active intestinal Ca2+ absorption in vivo in mice[72]. Calbindin D9k in mammals and calbindin D28k in birds are cytoplasmic proteins that carry the cations as a ferry from the BBM to the BLM[73]. Calbindins also buffer Ca2+, which maintains intracellular Ca2+ concentrations below 10-7 M and prevents cell death by apoptosis. The excess of Ca2+ that occurs when there is a down-regulation of calbindins may trigger apoptosis in the epithelial cells, as shown in different tissues[74,75]. In the BLM, there are two proteins involved in the Ca2+ exit to the lamina propria: the plasma membrane Ca2+-ATPase (PMCA), an ATP-dependent transporter that pumps Ca2+ out of the cytosol[76], and the Na+/Ca2+ exchanger (NCX), whose activity depends on the gradient created by Na+/K+-ATPase[77]. The predominant isoform in the intestine is PMCA1b, mainly located in the caveolae. PMCA1b is responsible for the major Ca2+ exit to the lamina propria. Its expression and activity are higher in villus tip enterocytes than in villus crypt cells, which is in agreement with the hypothesis that mature enterocytes have the greatest ability for transcellular Ca2+ movement[78]. NCX also presents several isoforms, but NCX1 predominates in the intestine[79]. It has a stoichiometry of 3 Na+:1 Ca2+and can function in either a forward mode (Ca2+ extrusion) or in a reversed mode (Ca2+ entry), depending on the Na+ and Ca2+ gradients and the membrane potential[80]. Another novel protein 4.1R, which co-localizes with PMCA1b, could have an important role in the transcellular Ca2+pathway, but its physiological function is not well known[81].
The paracellular Ca2+ pathway occurs through tight junctions (TJ), intercellular structures where plasma membranes of adjacent enterocytes have very close contact. This pathway has been poorly studied, but apparently Claudin (Cldn)-2 and Cldn-12 would be responsible for transporting Ca2+ in the intestine[82]. Ca2+ movement through the TJ is a non-saturable process, which depends on the concentration and the electric gradient across the epithelium. High Ca2+ intakes switch on the paracellular route due to a short sojourn time in the intestine and a down-regulation of proteins involved in the transcellular pathway[83]. It has been observed that the expression of paracellular TJ genes is regulated by the transcellular calbindin protein, suggesting that active and passive Ca2+ transport pathways may work cooperatively[84].
ACTIONS OF PRO-OXIDANTS ON INTESTINAL CALCIUM ABSORPTION
Twenty years ago, we reported that DL-buthionine-S,R-sulfoximine (BSO), an inhibitor of GSH biosynthesis, decreased the intestinal Ca2+ absorption in vitamin D-deficient chicks treated with cholecalciferol. This response was reversed by addition of GSH monoester to the intestinal sac, demonstrating for the first time that the intestinal GSH normal levels are essential for an adequate intestinal Ca2+ absorption. In vitamin D-deficient chicks without treatment, BSO did not affect the Ca2+ transport and the GSH content beyond the low values already triggered by the vitamin deficiency[85]. The activity of intestinal alkaline phosphatase (IAP), an enzyme presumably involved in the intestinal Ca2+ absorption, was also highly reduced by BSO in vitamin D-deficient chicks treated with vitamin D3. The effect of BSO was observed either in vivo or in vitro. BSO did not act directly on IAP, but it caused GSH depletion which led to changes in the redox state of the enterocyte, as evidenced by the ˙HO production and an incremental increase in the protein carbonyl content. Again the reversibility of the BSO effect was demonstrated by addition of GSH monoester to the gut loop[86,87].
Menadione (MEN) or vitamin K3 is another pro-oxidant compound that alters the intestinal Ca2+ absorption via GSH depletion[88]. MEN metabolism involves redox cycling, resulting in the release of ROS. MEN may undergo one or two-electron reduction. When MEN suffers one-electron reduction, there is formation of very unstable semiquinone radicals; they react rapidly with O2 resulting in regeneration of the parent compound and production of O2-˙ yielding H2O2 through enzymatic or spontaneous dismutation. Two-electron reduction of MEN by DT-diaphorase produces hydroquinone, a pathway that constitutes a detoxification mechanism[89]. GSH is the electron donor in both cases, which explains the depletion of the intestinal tripeptide after MEN treatment. GSH depletion triggers oxidative stress as demonstrated by generation of ˙HO and O2-˙ groups and an increase in the protein carbonyl content, which deteriorate the activities of enzymes or proteins involved in the Ca2+ movement from lumen to blood. In fact, the activities of IAP and the plasma membrane Ca2+-ATPase as well as the expression of PMCA1b, calbindin D28k and Cldn-2 were decreased by MEN treatment[88,90]. At the studied doses, the inhibitory action of MEN on intestinal Ca2+ absorption began in half an hour, lasted for several hours and finished after 9 or 10 h of treatment, indicating that the effect was transient, probably because the intestine could reinforce its ability to overcome the oxidative stress[88]. The inhibitory effect of MEN on intestinal Ca2+ absorption implied intestinal mitochondrial dysfunction. As mentioned above, the optimal intestinal Ca2+ absorption needs the integrity of intestinal mitochondria because it is the main source of metabolic energy. MEN caused mtGSH depletion, but it rapidly normalized. However, the mitochondrial membrane potential decreased and, simultaneously, cytochrome c was released from the intermembrane space to the cytoplasm, at least in mature enterocytes, which suggested triggering of apoptosis. In fact, this process was confirmed by DNA fragmentation that occurred in the 30%-40% of enterocytes, without affecting 60%-70% of the absorptive cells. In other words, the inhibitory effect of MEN on intestinal Ca2+ absorption was partial and transient. The activity of two oxidoreductases from the Krebs cycle, malate dehydrogenase and α-ketoglutarate dehydrogenase, was reduced by MEN in 18% and 30%, respectively. This means that the majority of mitochondria remained competent for ATP synthesis, making possible the process of apoptosis[91] and a poor intestinal Ca2+ absorption. MEN not only produced intestinal apoptosis through the mitochondrial pathway, but also by triggering the expression of FAS/FASL/caspase-3[92]. Although an enhancement in the Cu2+/Zn2+-SOD, CAT, GPx and Mn2+-SOD activities could represent cytoprotective mechanisms against the oxidant effects, they were insufficient to avoid an inhibition in the overall process of intestinal Ca2+ absorption[92-94]. The results supported previous data showing alterations in the intracellular thiols and Ca2+ homeostasis, ATP depletion and DNA breakage after toxic MEN concentrations[95-97].
Sodium deoxycholate (NaDOC), a sodium salt of a major hydrophobic bile acid, also inhibits the intestinal Ca2+ absorption at high physiological doses. This inhibition is time and dose dependent. We have demonstrated that PMCA1b decreased by the bile salt and the same occurred with the protein expression of PMCA1b, calbindin D28k and NCX1. NaDOC also produced GSH depletion, as well as ROS generation and mitochondrial swelling, which in turn led to mitochondria mediated apoptosis. Briefly, a single high concentration of NaDOC inhibits intestinal Ca2+ absorption via downregulation of proteins involved in the transcellular pathway, as a result of oxidative stress and apoptosis[98]. Similarly, in a rat model of type 1 Diabetes mellitus induced by streptozotocin, we have also demonstrated oxidative stress in the intestine at early stages of developing of disease, leading to inhibition of the intestinal Ca2 + absorption. Simultaneously, time-dependent adaptive mechanisms triggered an increment in the protein expression of molecules involved in both the transcellular and the paracellular pathways, which contributes to normalizing the intestinal Ca2 + absorption as well as the duodenal redox state[99].
Diets rich in fat also produce redox imbalance and increased oxidative stress in the duodenum causing down-regulation of calbindin D9k, PMCA1b and NCX, and consequently, an inhibitory effect on intestinal Ca2+ absorption[100]. Orihuela et al[101] have found that aluminium interferes with Ca2+ uptake by enterocytes through a decrease in the intestinal GSH level affecting calbindin D function and/or synthesis, which leads to a reduced transcellular Ca2+ absorption. Wu et al[102] have reported that advanced oxidation protein products decrease the expression of Ca2+ transporters in small intestine via the p44/42 MAPK signaling pathway. They consider that these data are relevant to understanding the mechanisms of IBD-associated osteoporosis.
In summary, not only drugs but diet components or pathophysiological conditions that occur with GSH depletion or increased oxidative stress are deleterious for the intestinal Ca2+ absorption because they alter the protein expression and/or activities of molecules involved in the transcellular and/or paracellular Ca2+ pathways. Figure 1 is a schematic representation of the possible mechanisms involved in the inhibition of intestinal Ca2+ absorption caused by oxidative stress.
Figure 1.
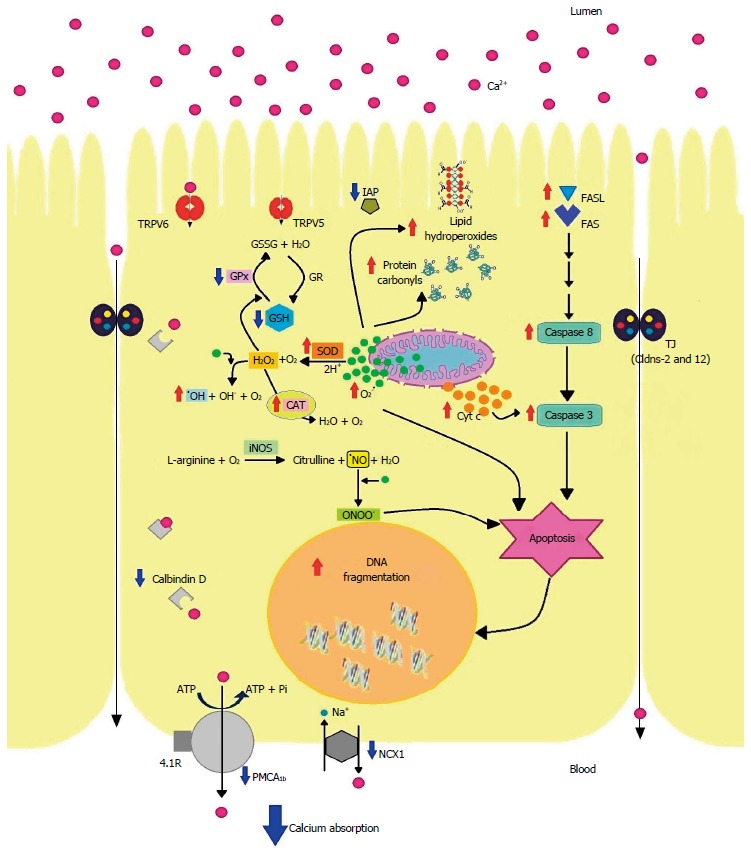
Schematic representation of the possible mechanisms involved in the inhibition of intestinal Ca2+ absorption caused by oxidative stress. TRPV6: Transient receptor potential vanilloid 6; TRPV5: Transient receptor potential vanilloid 5; IAP: Intestinal alkaline phosphatase; GSSG: Disulfide of glutathione; GSH: Glutathione; GPx: Glutathione peroxidase; GR: Glutathione reductase; SOD: Superoxide dismutase; CAT: Catalase; Cyt c: Cytochrome c; Cldns: Claudins; iNOS: Inducible nitric oxide synthase; NCX1: Intestinal Na+/Ca2+ exchanger; PMCA1b: Plasma membrane Ca2+- ATPase 1b; TJ: Tight junctions.
ANTIOXIDANTS AND THEIR MOLECULAR MECHANISMS FOR THE PRESERVATION OF INTESTINAL CALCIUM ABSORPTION
As mentioned earlier, endogenous enzymatic and nonenzymatic compounds defend the cells under oxidant conditions[40]. However, when there is a noticeable shift to the oxidation, they cannot respond adequately. It has been suggested that natural or synthetic compounds would help to overcome the disequilibrium[103,104]. In our laboratory, we have demonstrated that the inhibition of intestinal Ca2+ absorption caused by oxidants, mainly causing GSH depletion, could be either prevented or restored by quercetin[92] (QT, a plant derived flavonoid), melatonin[90,94] (MEL, a natural antioxidant present in humans), lithocholic (LCA)[105] or ursodeoxycholic (UDCA)[106] acids (bile acids less hydrophobic than deoxycholic acid). QT is a polyphenolic flavonoid found in several fruits and vegetables of the human diet[107], mainly highly concentrated in onions, tea and apples[108]. It is a potent scavenger of ROS with various pharmacological properties such as anticancer-activity, anti-virus and anti-inflammatory effects reducing the risk of cardiovascular and renal diseases[109,110]. QT inhibits enzyme systems responsible for the generation of ROS (cyclooxygenase, lipoxygenase and xanthine oxidase)[111], binds to superoxide anions, singlet oxygen and hydroxyl radicals, and as a consequence reduces lipid peroxidation[112], chelates transition metals such as iron and copper[113,114], and inhibits the aldose reductase activity[115]. We could demonstrate that QT protects the intestinal Ca2+ absorption against the inhibition provoked by MEN, but by itself does not affect it. Similarly, QT abolishes the GSH depletion caused by the quinone, but QT alone does not modify the intestinal GSH content. The flavonoid also avoids changes in the mitochondrial membrane permeability and abrogates the activation of FASL/FAS/caspase-3 pathway caused by MEN[92]. Conclusively, QT may be useful to prevent the inhibition of intestinal Ca2+ absorption caused by MEN or other GSH depleting drugs by blocking the oxidative stress and apoptosis. In contrast, the soy isoflavones have shown a lack of dose-responsive on transepithelial Ca2+ transport in human intestinal-like Caco-2 cells[116], although they may reduce bone loss in postmenopausal women, which suggests that they act directly on bone cells.
We have also demonstrated that MEL may also restore the intestinal Ca2+ absorption altered by MEN[94]. MEL is an indolamine that is present in all phyla of multicellular animals[117]. Although its main site of synthesis is the pineal gland, MEL is synthesized in other extracellular sites such as the intestine[118], where the MEL level is 400 times larger than that from the pineal gland[119]. It has been shown that MEL scavenges ROS and protects against the deleterious effects of I/R through a stimulation of certain antioxidant enzymes preserving cellular energy and preventing mitochondrial damage[120,121]. In our study, we have shown that MEL blocks the inhibition of the intestinal Ca2+ absorption caused by MEN, at least in part, by increasing the activity of antioxidant enzymes, returning GSH and protein carbonyl values to control levels, and rescuing the epithelial cells from apoptosis[94]. More recently, we have also proven that MEL not only restores but also prevents the inhibition of intestinal Ca2+ absorption provoked by GSH depleting drugs such as MEN and BSO[90]. MEL restores partially both the transcellular and paracellular Ca2+ pathways altered by the quinone, through dampening the O2-˙ levels without affecting the ˙NO system. MEL was able to return the decreased protein expression of calbindin D28K and Cldn-2 caused by MEN to the control values, but it could not restore the levels of PMCA1b. As MEL has beneficial effects on both Ca2+ transport mechanisms, it might improve the intestinal Ca2+ absorption under conditions of low or adequate Ca intake. The modulation of Ca2+ transporters by MEL has also been reported in pancreatic acinar cells[122] and in pituitary cells[123]. Another mechanism by which MEL blocked the inhibition of intestinal Ca2+ absorption was the attenuation of the mitochondrial dysfunction in the duodenum, which has been also observed to be produced by MEL in other tissues and cells[124-126].
Two other molecules with antioxidant properties such as UDCA and LCA were shown to block the inhibitory effect of NaDOC on intestinal Ca2+ absorption. UDCA and LCA are two bile acids with different solubility, chemical properties and physiological functions[127]. UDCA is a non-toxic hydrophilic bile acid used for treatment of gallstones and primary biliary cirrhosis (PBC)[128]. UDCA is naturally present in humans in a concentration of about 1%-3% of the total bile acid pool. When used in PBC treatment, its concentration increases to 40%-60%, making UDCA the predominant bile acid. The hydrophilicity of bile via UDCA serves to ameliorate cholestasis and minimize toxicity[129]. At the intestinal level, we have shown that UDCA increases the Ca2+ absorption modulating positively the Ca2+ uptake by mature enterocytes, which occurs in part as a result of increasing the vitamin D receptor (VDR) unit numbers[106,130]. When UDCA is simultaneously administered with NaDOC, UDCA avoids the inhibitory effect of NaDOC on intestinal Ca2+ absorption. One of the molecular mechanisms involved in this response is the attenuation of the apoptotic extrinsic pathway triggered by NaDOC. UDCA by itself decreases FAS and FASL protein expression and neither alters caspase 8 protein expression nor caspase 3 activity. In the presence of NaDOC, UDCA avoids the apoptotic effect of NaDOC normalizing the protein expression of FAS, FASL, caspase-8 and the enzyme activity of caspase-3. The NaDOC induced apoptosis is mediated by increment in the NO content and in the iNOS protein expression, effects that were abolished by UDCA. Another molecular mechanism triggered by UDCA is to avoid the enhancement in the LC3 II protein expression and the number of acidic vesicular organelles in the presence of NaDOC. In other words, UDCA avoids efficiently not only NO induced apoptosis, but also autophagy triggered by NaDOC[130].
LCA is a secondary bile acid produced by the intestinal microflora. It binds to VDR[131], has antibacterial activity[132], produces antiproliferative and pro-apoptotic effect on human cancer cell lines[133,134], inhibits proteasome[135], acts as a membrane pore[136] and has anti-aging properties[137]. It is worldwide recognized that 1,25(OH)2D3 is the main stimulator of the intestinal Ca2+ absorption, and both LCA and 1,25(OH)2D3 are VDR ligands, although they have different VDR binding affinity[138]. In a recent study, we have demonstrated that neither the intestinal Ca2+ absorption nor the redox state of enterocytes is changed by LCA alone. Interestingly, LCA did not alter the intestinal Ca2+ absorption but prevented the inhibitory effect of NaDOC[105]. LCA blocked a decrease caused by NaDOC on gene and protein expression of molecules involved in the transcellular pathway of intestinal Ca2+ absorption, ameliorated changes in mitochondrial membrane permeability and abrogated the enhancement in the protein expression of molecules from the apoptotic extrinsic pathway[105]. In addition, the simultaneous treatment of NaDOC and LCA blocked the oxidative stress caused by NaDOC, which indicates that LCA shows antioxidant and antiapoptotic properties in the presence of a pro-oxidant molecule as NaDOC. The functional toxicity of LCA in humans is in question due to the efficient human detoxification[139], therefore, the use of LCA to protect the intestinal Ca2+ absorption under oxidant conditions caused by medications or pathological conditions might become a possible therapeutic strategy.
CONCLUSION
The optimal intestinal Ca2+ absorption is highly dependent on the intactness of intestinal GSH content. GSH depleting drugs such as BSO, MEN or NaDOC trigger oxidative stress, leading to apoptosis and/or autophagy to finally produce inhibition of intestinal Ca2+ absorption. Similarly, pathological conditions associated with intestinal GSH depletion provoke oxidative stress and, hence, inhibition of intestinal Ca2+ absorption, as occurs in type 1 diabetes mellitus. The use of antioxidants could be a therapeutic strategy to protect or to restore the intestinal normal redox state maintaining an adequate intestinal Ca2+ absorption. QT, MEL, UDCA and LCA have been proven to be successful to normalize the Ca2+ transfer from lumen-to-blood in experimental animals under oxidant conditions. Therefore, they could be drugs of choice for the treatment of altered intestinal Ca2+ absorption in pathophysiological conditions such as diabetes, celiac disease, IBD, aging and other disorders associated with intestinal oxidative stress.
ACKNOWLEDGMENTS
Prof. Dr. Nori Tolosa de Talamoni is a Member of Investigator Career from Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET). Solange Guizzardi and Luciana Moine are recipients of Doctoral Fellowships from CONICET.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Argentina
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: No conflicts of interest, financial or otherwise, are declared by the authors.
Peer-review started: January 9, 2017
First decision: February 9, 2017
Article in press: March 30, 2017
P- Reviewer: Lasekan J, Prasad S S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, Rahu N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid Med Cell Longev. 2016;2016:7432797. doi: 10.1155/2016/7432797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boccatonda A, Tripaldi R, Davì G, Santilli F. Oxidative Stress Modulation Through Habitual Physical Activity. Curr Pharm Des. 2016;22:3648–3680. doi: 10.2174/1381612822666160413123806. [DOI] [PubMed] [Google Scholar]
- 3.Panth N, Paudel KR, Parajuli K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv Med. 2016;2016:9152732. doi: 10.1155/2016/9152732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richa R, Yadawa AK, Chaturvedi CM. Hyperglycemia and high nitric oxide level induced oxidative stress in the brain and molecular alteration in the neurons and glial cells of laboratory mouse, Mus musculus. Neurochem Int. 2017;104:64–79. doi: 10.1016/j.neuint.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39:73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- 6.Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016;148:183–193. doi: 10.1016/j.lfs.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Rincón-Cervera MA, Valenzuela R, Hernandez-Rodas MC, Marambio M, Espinosa A, Mayer S, Romero N, Barrera M Sc C, Valenzuela A, Videla LA. Supplementation with antioxidant-rich extra virgin olive oil prevents hepatic oxidative stress and reduction of desaturation capacity in mice fed a high-fat diet: Effects on fatty acid composition in liver and extrahepatic tissues. Nutrition. 2016;32:1254–1267. doi: 10.1016/j.nut.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pang L, Cai Y, Tang EH, Yan D, Kosuru R, Li H, Irwin MG, Ma H, Xia Z. Cox-2 Inhibition Protects against Hypoxia/Reoxygenation-Induced Cardiomyocyte Apoptosis via Akt-Dependent Enhancement of iNOS Expression. Oxid Med Cell Longev. 2016;2016:3453059. doi: 10.1155/2016/3453059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handa O, Majima A, Onozawa Y, Horie H, Uehara Y, Fukui A, Omatsu T, Naito Y, Yoshikawa T. The role of mitochondria-derived reactive oxygen species in the pathogenesis of non-steroidal anti-inflammatory drug-induced small intestinal injury. Free Radic Res. 2014;48:1095–1099. doi: 10.3109/10715762.2014.928411. [DOI] [PubMed] [Google Scholar]
- 11.Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights. 2016;7:11. doi: 10.1007/s13317-016-0083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar S, Pandey AK. Free radicals: health implications and their mitigation by herbals. Br J Med Med Res. 2015;7:438–457. doi: 10.9734/bjmmr/2015/16284. [DOI] [Google Scholar]
- 13.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 14.Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med Cell Longev. 2016;2016:1245049. doi: 10.1155/2016/1245049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front Cell Dev Biol. 2015;3:83. doi: 10.3389/fcell.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Londero AS, Arana MR, Perdomo VG, Tocchetti GN, Zecchinati F, Ghanem CI, Ruiz ML, Rigalli JP, Mottino AD, García F, et al. Intestinal multidrug resistance-associated protein 2 is down-regulated in fructose-fed rats. J Nutr Biochem. 2017;40:178–186. doi: 10.1016/j.jnutbio.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Lam G, Apostolopoulos V, Zulli A, Nurgali K. NADPH oxidases and inflammatory bowel disease. Curr Med Chem. 2015;22:2100–2109. doi: 10.2174/0929867322666150416095114. [DOI] [PubMed] [Google Scholar]
- 19.Moret I, Cerrillo E, Navarro-Puche A, Iborra M, Rausell F, Tortosa L, Beltrán B. [Oxidative stress in Crohn’s disease] Gastroenterol Hepatol. 2014;37:28–34. doi: 10.1016/j.gastrohep.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Balmus IM, Ciobica A, Trifan A, Stanciu C. The implications of oxidative stress and antioxidant therapies in Inflammatory Bowel Disease: Clinical aspects and animal models. Saudi J Gastroenterol. 2016;22:3–17. doi: 10.4103/1319-3767.173753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 22.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Collman JP, Devaraj NK, Decréau RA, Yang Y, Yan YL, Ebina W, Eberspacher TA, Chidsey CE. A cytochrome C oxidase model catalyzes oxygen to water reduction under rate-limiting electron flux. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki M, Joh T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J Clin Biochem Nutr. 2007;40:1–12. doi: 10.3164/jcbn.40.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García-de-la-Asunción J, Barber G, Rus D, Perez-Griera J, Belda FJ, Martí F, García-Granero E. Hyperoxia during colon surgery is associated with a reduction of xanthine oxidase activity and oxidative stress in colonic mucosa. Redox Rep. 2011;16:121–128. doi: 10.1179/174329211X13049558293632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Granell S, Bulbena O, Genesca M, Sabater L, Sastre J, Gelpi E, Closa D. Mobilization of xanthine oxidase from the gastrointestinal tract in acute pancreatitis. BMC Gastroenterol. 2004;4:1. doi: 10.1186/1471-230X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarz G, Belaidi AA. Molybdenum in human health and disease. Met Ions Life Sci. 2013;13:415–450. doi: 10.1007/978-94-007-7500-8_13. [DOI] [PubMed] [Google Scholar]
- 29.Hinnebusch BF, Ma Q, Henderson JW, Siddique A, Archer SY, Hodin RA. Enterocyte response to ischemia is dependent on differentiation state. J Gastrointest Surg. 2002;6:403–409. doi: 10.1016/s1091-255x(01)00076-2. [DOI] [PubMed] [Google Scholar]
- 30.Rasool S, A Ganai B, Syed Sameer A, Masood A. Esophageal cancer: associated factors with special reference to the Kashmir Valley. Tumori. 2012;98:191–203. doi: 10.1700/1088.11929. [DOI] [PubMed] [Google Scholar]
- 31.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guner YS, Ochoa CJ, Wang J, Zhang X, Steinhauser S, Stephenson L, Grishin A, Upperman JS. Peroxynitrite-induced p38 MAPK pro-apoptotic signaling in enterocytes. Biochem Biophys Res Commun. 2009;384:221–225. doi: 10.1016/j.bbrc.2009.04.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song BJ, Akbar M, Abdelmegeed MA, Byun K, Lee B, Yoon SK, Hardwick JP. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014;3:109–123. doi: 10.1016/j.redox.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krinsky NI. Mechanism of action of biological antioxidants. Proc Soc Exp Biol Med. 1992;200:248–254. doi: 10.3181/00379727-200-43429. [DOI] [PubMed] [Google Scholar]
- 35.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 36.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jocelyn PC, Kamminga A. The non-protein thiol of rat liver mitochondria. Biochim Biophys Acta. 1974;343:356–362. doi: 10.1016/0304-4165(74)90099-3. [DOI] [PubMed] [Google Scholar]
- 38.Cuozzo JW, Kaiser CA. Competition between glutathione and protein thiols for disulphide-bond formation. Nat Cell Biol. 1999;1:130–135. doi: 10.1038/11047. [DOI] [PubMed] [Google Scholar]
- 39.Meister A, Tate SS. Glutathione and related gamma-glutamyl compounds: biosynthesis and utilization. Annu Rev Biochem. 1976;45:559–604. doi: 10.1146/annurev.bi.45.070176.003015. [DOI] [PubMed] [Google Scholar]
- 40.Circu ML, Aw TY. Redox biology of the intestine. Free Radic Res. 2011;45:1245–1266. doi: 10.3109/10715762.2011.611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markovic J, Borrás C, Ortega A, Sastre J, Viña J, Pallardó FV. Glutathione is recruited into the nucleus in early phases of cell proliferation. J Biol Chem. 2007;282:20416–20424. doi: 10.1074/jbc.M609582200. [DOI] [PubMed] [Google Scholar]
- 42.Diaz-Vivancos P, de Simone A, Kiddle G, Foyer CH. Glutathione--linking cell proliferation to oxidative stress. Free Radic Biol Med. 2015;89:1154–1164. doi: 10.1016/j.freeradbiomed.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 43.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–2992. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 44.Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Circu ML, Rodriguez C, Maloney R, Moyer MP, Aw TY. Contribution of mitochondrial GSH transport to matrix GSH status and colonic epithelial cell apoptosis. Free Radic Biol Med. 2008;44:768–778. doi: 10.1016/j.freeradbiomed.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. FASEB J. 2002;16:1263–1265. doi: 10.1096/fj.02-0097fje. [DOI] [PubMed] [Google Scholar]
- 47.Circu ML, Moyer MP, Harrison L, Aw TY. Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Radic Biol Med. 2009;47:1190–1198. doi: 10.1016/j.freeradbiomed.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dahm LJ, Jones DP. Secretion of cysteine and glutathione from mucosa to lumen in rat small intestine. Am J Physiol. 1994;267:G292–G300. doi: 10.1152/ajpgi.1994.267.2.G292. [DOI] [PubMed] [Google Scholar]
- 49.Dahm LJ, Jones DP. Rat jejunum controls luminal thiol-disulfide redox. J Nutr. 2000;130:2739–2745. doi: 10.1093/jn/130.11.2739. [DOI] [PubMed] [Google Scholar]
- 50.Snary D, Allen A, Pain RH. Structural studies on gastric mucoproteins: lowering of molecular weight after reduction with 2-mercaptoethanol. Biochem Biophys Res Commun. 1970;40:844–851. [Google Scholar]
- 51.Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun. 2010;396:120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 52.Gasdaska JR, Gasdaska PY, Gallegos A, Powis G. Human thioredoxin reductase gene localization to chromosomal position 12q23-q24.1 and mRNA distribution in human tissue. Genomics. 1996;37:257–259. doi: 10.1006/geno.1996.0554. [DOI] [PubMed] [Google Scholar]
- 53.Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, Buchner J, Schaller M, Stange EF, Wehkamp J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nature. 2011;469:419–423. doi: 10.1038/nature09674. [DOI] [PubMed] [Google Scholar]
- 54.Poulsen NA, Andersen V, Møller JC, Møller HS, Jessen F, Purup S, Larsen LB. Comparative analysis of inflamed and non-inflamed colon biopsies reveals strong proteomic inflammation profile in patients with ulcerative colitis. BMC Gastroenterol. 2012;12:76. doi: 10.1186/1471-230X-12-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK. Manganese superoxide dismutase: guardian of the powerhouse. Int J Mol Sci. 2011;12:7114–7162. doi: 10.3390/ijms12107114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kruidenier L, Kuiper I, van Duijn W, Marklund SL, van Hogezand RA, Lamers CB, Verspaget HW. Differential mucosal expression of three superoxide dismutase isoforms in inflammatory bowel disease. J Pathol. 2003;201:7–16. doi: 10.1002/path.1407. [DOI] [PubMed] [Google Scholar]
- 57.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 58.Chu FF, Esworthy RS, Doroshow JH. Role of Se-dependent glutathione peroxidases in gastrointestinal inflammation and cancer. Free Radic Biol Med. 2004;36:1481–1495. doi: 10.1016/j.freeradbiomed.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Kudva AK, Shay AE, Prabhu KS. Selenium and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2015;309:G71–G77. doi: 10.1152/ajpgi.00379.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnér ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 61.Kim SJ, Cheresh P, Jablonski RP, Morales-Nebreda L, Cheng Y, Hogan E, Yeldandi A, Chi M, Piseaux R, Ridge K, et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic Biol Med. 2016;101:482–490. doi: 10.1016/j.freeradbiomed.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Areco V, Rivoira MA, Rodriguez V, Marchionatti AM, Carpentieri A, Tolosa de Talamoni N. Dietary and pharmacological compounds altering intestinal calcium absorption in humans and animals. Nutr Res Rev. 2015;28:83–99. doi: 10.1017/S0954422415000050. [DOI] [PubMed] [Google Scholar]
- 63.van der Velde RY, Brouwers JR, Geusens PP, Lems WF, van den Bergh JP. Calcium and vitamin D supplementation: state of the art for daily practice. Food Nutr Res. 2014;58 doi: 10.3402/fnr.v58.21796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wasserman RH. Vitamin D and the dual processes of intestinal calcium absorption. J Nutr. 2004;134:3137–3139. doi: 10.1093/jn/134.11.3137. [DOI] [PubMed] [Google Scholar]
- 65.O’Brien KO, Nathanson MS, Mancini J, Witter FR. Calcium absorption is significantly higher in adolescents during pregnancy than in the early postpartum period. Am J Clin Nutr. 2003;78:1188–1193. doi: 10.1093/ajcn/78.6.1188. [DOI] [PubMed] [Google Scholar]
- 66.Zhu Y, Goff JP, Reinhardt TA, Horst RL. Pregnancy and lactation increase vitamin D-dependent intestinal membrane calcium adenosine triphosphatase and calcium binding protein messenger ribonucleic acid expression. Endocrinology. 1998;139:3520–3524. doi: 10.1210/endo.139.8.6141. [DOI] [PubMed] [Google Scholar]
- 67.Liesegang A, Riner K, Boos A. Effects of gestation and lactation on vitamin D receptor amounts in goats and sheep. Domest Anim Endocrinol. 2007;33:190–202. doi: 10.1016/j.domaniend.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Nordin BE, Need AG, Morris HA, O’Loughlin PD, Horowitz M. Effect of age on calcium absorption in postmenopausal women. Am J Clin Nutr. 2004;80:998–1002. doi: 10.1093/ajcn/80.4.998. [DOI] [PubMed] [Google Scholar]
- 69.Tolosa de Talamoni NG. Calcium and phosphorous deficiencies alter the lipid composition and fluidity of intestinal basolateral membranes. Comp Biochem Physiol A Physiol. 1996;115:309–315. doi: 10.1016/s0300-9629(96)00083-7. [DOI] [PubMed] [Google Scholar]
- 70.Cui M, Li Q, Johnson R, Fleet JC. Villin promoter-mediated transgenic expression of transient receptor potential cation channel, subfamily V, member 6 (TRPV6) increases intestinal calcium absorption in wild-type and vitamin D receptor knockout mice. J Bone Miner Res. 2012;27:2097–2107. doi: 10.1002/jbmr.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kellett GL. Alternative perspective on intestinal calcium absorption: proposed complementary actions of Ca(v)1.3 and TRPV6. Nutr Rev. 2011;69:347–370. doi: 10.1111/j.1753-4887.2011.00395.x. [DOI] [PubMed] [Google Scholar]
- 72.Reyes-Fernandez PC, Fleet JC. Luminal glucose does not enhance active intestinal calcium absorption in mice: evidence against a role for Ca(v)1.3 as a mediator of calcium uptake during absorption. Nutr Res. 2015;35:1009–1015. doi: 10.1016/j.nutres.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tolosa de Talamoni N, Perez A, Alisio A. Effect of cholecalciferol on intestinal epithelial cells. Trends in Comparative Biochem & Physiol. 1998;5:179–185. [Google Scholar]
- 74.Liu Y, Porta A, Peng X, Gengaro K, Cunningham EB, Li H, Dominguez LA, Bellido T, Christakos S. Prevention of glucocorticoid-induced apoptosis in osteocytes and osteoblasts by calbindin-D28k. J Bone Miner Res. 2004;19:479–490. doi: 10.1359/JBMR.0301242. [DOI] [PubMed] [Google Scholar]
- 75.Merico V, de Barboza GD, Vasco C, Ponce R, Rodriguez V, Garagna S, Tolosa de Talamoni N. A mitochondrial mechanism is involved in apoptosis of Robertsonian mouse male germ cells. Reproduction. 2008;135:797–804. doi: 10.1530/REP-07-0466. [DOI] [PubMed] [Google Scholar]
- 76.Wasserman RH, Chandler JS, Meyer SA, Smith CA, Brindak ME, Fullmer CS, Penniston JT, Kumar R. Intestinal calcium transport and calcium extrusion processes at the basolateral membrane. J Nutr. 1992;122:662–671. doi: 10.1093/jn/122.suppl_3.662. [DOI] [PubMed] [Google Scholar]
- 77.Ghijsen WE, De Jong MD, Van Os CH. Kinetic properties of Na+/Ca2+ exchange in basolateral plasma membranes of rat small intestine. Biochim Biophys Acta. 1983;730:85–94. doi: 10.1016/0005-2736(83)90320-6. [DOI] [PubMed] [Google Scholar]
- 78.Centeno VA, Díaz de Barboza GE, Marchionatti AM, Alisio AE, Dallorso ME, Nasif R, Tolosa de Talamoni NG. Dietary calcium deficiency increases Ca2+ uptake and Ca2+ extrusion mechanisms in chick enterocytes. Comp Biochem Physiol A Mol Integr Physiol. 2004;139:133–141. doi: 10.1016/j.cbpb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 79.Centeno V, Picotto G, Pérez A, Alisio A, Tolosa de Talamoni N. Intestinal Na(+)/Ca(2+) exchanger protein and gene expression are regulated by 1,25(OH)(2)D(3) in vitamin D-deficient chicks. Arch Biochem Biophys. 2011;509:191–196. doi: 10.1016/j.abb.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 80.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 81.Liu C, Weng H, Chen L, Yang S, Wang H, Debnath G, Guo X, Wu L, Mohandas N, An X. Impaired intestinal calcium absorption in protein 4.1R-deficient mice due to altered expression of plasma membrane calcium ATPase 1b (PMCA1b) J Biol Chem. 2013;288:11407–11415. doi: 10.1074/jbc.M112.436659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y, Yamamoto Y, Wada T, Kojima T, Yokozaki H, et al. Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell. 2008;19:1912–1921. doi: 10.1091/mbc.E07-09-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bronner F. Mechanisms of intestinal calcium absorption. J Cell Biochem. 2003;88:387–393. doi: 10.1002/jcb.10330. [DOI] [PubMed] [Google Scholar]
- 84.Hwang I, Yang H, Kang HS, Ahn C, Hong EJ, An BS, Jeung EB. Alteration of tight junction gene expression by calcium- and vitamin D-deficient diet in the duodenum of calbindin-null mice. Int J Mol Sci. 2013;14:22997–23010. doi: 10.3390/ijms141122997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tolosa de Talamoni N, Marchionatti A, Baudino V, Alisio A. Glutathione plays a role in the chick intestinal calcium absorption. Comp Biochem Physiol A Physiol. 1996;115:127–132. doi: 10.1016/0300-9629(96)00023-0. [DOI] [PubMed] [Google Scholar]
- 86.Marchionatti A, Alisio A, Díaz de Barboza G, Baudino V, Tolosa de Talamoni N. DL-Buthionine-S,R-sulfoximine affects intestinal alkaline phosphatase activity. Comp Biochem Physiol C Toxicol Pharmacol. 2001;129:85–91. doi: 10.1016/s1532-0456(01)00185-5. [DOI] [PubMed] [Google Scholar]
- 87.Tolosa de Talamoni N, Marchionatti AM. Thiol redox balance and the intestinal calcium absorption. Trens in Comparative Biochem & Physiol. 2002;9:175–183. [Google Scholar]
- 88.Marchionatti AM, Díaz de Barboza GE, Centeno VA, Alisio AE, Tolosa de Talamoni NG. Effects of a single dose of menadione on the intestinal calcium absorption and associated variables. J Nutr Biochem. 2003;14:466–472. doi: 10.1016/s0955-2863(03)00078-0. [DOI] [PubMed] [Google Scholar]
- 89.Chiou TJ, Tzeng WF. The roles of glutathione and antioxidant enzymes in menadione-induced oxidative stress. Toxicology. 2000;154:75–84. doi: 10.1016/s0300-483x(00)00321-8. [DOI] [PubMed] [Google Scholar]
- 90.Areco V, Rodriguez V, Marchionatti A, Carpentieri A, Tolosa de Talamoni N. Melatonin not only restores but also prevents the inhibition of the intestinal Ca(2+) absorption caused by glutathione depleting drugs. Comp Biochem Physiol A Mol Integr Physiol. 2016;197:16–22. doi: 10.1016/j.cbpa.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 91.Regula KM, Kirshenbaum LA. Apoptosis of ventricular myocytes: a means to an end. J Mol Cell Cardiol. 2005;38:3–13. doi: 10.1016/j.yjmcc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Marchionatti AM, Pacciaroni A, Tolosa de Talamoni NG. Effects of quercetin and menadione on intestinal calcium absorption and the underlying mechanisms. Comp Biochem Physiol A Mol Integr Physiol. 2013;164:215–220. doi: 10.1016/j.cbpa.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 93.Marchionatti AM, Perez AV, Diaz de Barboza GE, Pereira BM, Tolosa de Talamoni NG. Mitochondrial dysfunction is responsible for the intestinal calcium absorption inhibition induced by menadione. Biochim Biophys Acta. 2008;1780:101–107. doi: 10.1016/j.bbagen.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 94.Carpentieri A, Marchionatti A, Areco V, Perez A, Centeno V, Tolosa de Talamoni N. Antioxidant and antiapoptotic properties of melatonin restore intestinal calcium absorption altered by menadione. Mol Cell Biochem. 2014;387:197–205. doi: 10.1007/s11010-013-1885-2. [DOI] [PubMed] [Google Scholar]
- 95.Di Monte D, Ross D, Bellomo G, Eklöw L, Orrenius S. Alterations in intracellular thiol homeostasis during the metabolism of menadione by isolated rat hepatocytes. Arch Biochem Biophys. 1984;235:334–342. doi: 10.1016/0003-9861(84)90206-6. [DOI] [PubMed] [Google Scholar]
- 96.Sata N, Klonowski-Stumpe H, Han B, Häussinger D, Niederau C. Menadione induces both necrosis and apoptosis in rat pancreatic acinar AR4-2J cells. Free Radic Biol Med. 1997;23:844–850. doi: 10.1016/s0891-5849(97)00064-6. [DOI] [PubMed] [Google Scholar]
- 97.Aherne SA, O’Brien NM. Mechanism of protection by the flavonoids, quercetin and rutin, against tert-butylhydroperoxide- and menadione-induced DNA single strand breaks in Caco-2 cells. Free Radic Biol Med. 2000;29:507–514. doi: 10.1016/s0891-5849(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 98.Rivoira MA, Marchionatti AM, Centeno VA, Díaz de Barboza GE, Peralta López ME, Tolosa de Talamoni NG. Sodium deoxycholate inhibits chick duodenal calcium absorption through oxidative stress and apoptosis. Comp Biochem Physiol A Mol Integr Physiol. 2012;162:397–405. doi: 10.1016/j.cbpa.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 99.Rivoira M, Rodríguez V, López MP, Tolosa de Talamoni N. Time dependent changes in the intestinal Ca2+ absorption in rats with type I diabetes mellitus are associated with alterations in the intestinal redox state. Biochim Biophys Acta. 2015;1852:386–394. doi: 10.1016/j.bbadis.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 100.Xiao Y, Cui J, Shi YH, Sun J, Wang ZP, Le GW. Effects of duodenal redox status on calcium absorption and related genes expression in high-fat diet-fed mice. Nutrition. 2010;26:1188–1194. doi: 10.1016/j.nut.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 101.Orihuela D, Meichtry V, Pizarro M. Aluminium-induced impairment of transcellular calcium absorption in the small intestine: calcium uptake and glutathione influence. J Inorg Biochem. 2005;99:1879–1886. doi: 10.1016/j.jinorgbio.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 102.Wu P, Xie F, Xue M, Xu X, He S, Lin M, Bai L. Advanced oxidation protein products decrease the expression of calcium transport channels in small intestinal epithelium via the p44/42 MAPK signaling pathway. Eur J Cell Biol. 2015;94:190–203. doi: 10.1016/j.ejcb.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 103.Carocho M, Ferreira IC. A review on antioxidants, prooxidants and related controversy: natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem Toxicol. 2013;51:15–25. doi: 10.1016/j.fct.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 104.Lobo V, Patil A, Phatak A, Chandra N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn Rev. 2010;4:118–126. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marchionatti AM, Pérez A, Rivoira MA, Rodríguez VA, Tolosa de Talamoni NG. Lithocholic acid: a new emergent protector of intestinal calcium absorption under oxidant conditions. Biochem Cell Biol. 2017;95:273–279. doi: 10.1139/bcb-2016-0164. [DOI] [PubMed] [Google Scholar]
- 106.Rodríguez V, Rivoira M, Marchionatti A, Pérez A, Tolosa de Talamoni N. Ursodeoxycholic and deoxycholic acids: A good and a bad bile acid for intestinal calcium absorption. Arch Biochem Biophys. 2013;540:19–25. doi: 10.1016/j.abb.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 107.Kumar S, Pandey AK. Chemistry and biological activities of flavonoids: an overview. ScientificWorldJournal. 2013;2013:162750. doi: 10.1155/2013/162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kroon PA, Clifford MN, Crozier A, Day AJ, Donovan JL, Manach C, Williamson G. How should we assess the effects of exposure to dietary polyphenols in vitro? Am J Clin Nutr. 2004;80:15–21. doi: 10.1093/ajcn/80.1.15. [DOI] [PubMed] [Google Scholar]
- 109.Lu Q, Ji XJ, Zhou YX, Yao XQ, Liu YQ, Zhang F, Yin XX. Quercetin inhibits the mTORC1/p70S6K signaling-mediated renal tubular epithelial-mesenchymal transition and renal fibrosis in diabetic nephropathy. Pharmacol Res. 2015;99:237–247. doi: 10.1016/j.phrs.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 110.Gomes IB, Porto ML, Santos MC, Campagnaro BP, Pereira TM, Meyrelles SS, Vasquez EC. Renoprotective, anti-oxidative and anti-apoptotic effects of oral low-dose quercetin in the C57BL/6J model of diabetic nephropathy. Lipids Health Dis. 2014;13:184. doi: 10.1186/1476-511X-13-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ozbek N, Bali EB, Karasu C. Quercetin and hydroxytyrosol attenuates xanthine/xanthine oxidase-induced toxicity in H9c2 cardiomyocytes by regulation of oxidative stress and stress-sensitive signaling pathways. Gen Physiol Biophys. 2015;34:407–414. doi: 10.4149/gpb_2015021. [DOI] [PubMed] [Google Scholar]
- 112.Nabavi SF, Russo GL, Daglia M, Nabavi SM. Role of quercetin as an alternative for obesity treatment: you are what you eat! Food Chem. 2015;179:305–310. doi: 10.1016/j.foodchem.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 113.El Hajji H, Nkhili E, Tomao V, Dangles O. Interactions of quercetin with iron and copper ions: complexation and autoxidation. Free Radic Res. 2006;40:303–320. doi: 10.1080/10715760500484351. [DOI] [PubMed] [Google Scholar]
- 114.Lesjak M, Hoque R, Balesaria S, Skinner V, Debnam ES, Srai SK, Sharp PA. Quercetin inhibits intestinal iron absorption and ferroportin transporter expression in vivo and in vitro. PLoS One. 2014;9:e102900. doi: 10.1371/journal.pone.0102900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Milackova I, Prnova MS, Majekova M, Sotnikova R, Stasko M, Kovacikova L, Banerjee S, Veverka M, Stefek M. 2-Chloro-1,4-naphthoquinone derivative of quercetin as an inhibitor of aldose reductase and anti-inflammatory agent. J Enzyme Inhib Med Chem. 2015;30:107–113. doi: 10.3109/14756366.2014.892935. [DOI] [PubMed] [Google Scholar]
- 116.Cotter AA, Cashman KD. Lack of dose-responsive effect of dietary phyto-oestrogens on transepithelial calcium transport in human intestinal-like Caco-2 cells. Br J Nutr. 2004;91:5–9. doi: 10.1079/BJN20031007. [DOI] [PubMed] [Google Scholar]
- 117.Chu J, Tu Y, Chen J, Tan D, Liu X, Pi R. Effects of melatonin and its analogues on neural stem cells. Mol Cell Endocrinol. 2016;420:169–179. doi: 10.1016/j.mce.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 118.Mukherjee S, Maitra SK. Gut Melatonin in Vertebrates: Chronobiology and Physiology. Front Endocrinol (Lausanne) 2015;6:112. doi: 10.3389/fendo.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bubenik GA. Gastrointestinal melatonin: localization, function, and clinical relevance. Dig Dis Sci. 2002;47:2336–2348. doi: 10.1023/a:1020107915919. [DOI] [PubMed] [Google Scholar]
- 120.Crespo E, Macías M, Pozo D, Escames G, Martín M, Vives F, Guerrero JM, Acuña-Castroviejo D. Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysaccharide-induced multiple organ dysfunction syndrome in rats. FASEB J. 1999;13:1537–1546. [PubMed] [Google Scholar]
- 121.Carpentieri A, Díaz de Barboza G, Areco V, Peralta López M, Tolosa de Talamoni N. New perspectives in melatonin uses. Pharmacol Res. 2012;65:437–444. doi: 10.1016/j.phrs.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 122.Huai J, Shao Y, Sun X, Jin Y, Wu J, Huang Z. Melatonin ameliorates acute necrotizing pancreatitis by the regulation of cytosolic Ca2+ homeostasis. Pancreatology. 2012;12:257–263. doi: 10.1016/j.pan.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 123.Yoo YM, Jeung EB. Melatonin suppresses cyclosporine A-induced autophagy in rat pituitary GH3 cells. J Pineal Res. 2010;48:204–211. doi: 10.1111/j.1600-079X.2010.00744.x. [DOI] [PubMed] [Google Scholar]
- 124.Wang WZ, Fang XH, Stephenson LL, Zhang X, Khiabani KT, Zamboni WA. Melatonin attenuates I/R-induced mitochondrial dysfunction in skeletal muscle. J Surg Res. 2011;171:108–113. doi: 10.1016/j.jss.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 125.Luchetti F, Canonico B, Mannello F, Masoni C, D’Emilio A, Battistelli M, Papa S, Falcieri E. Melatonin reduces early changes in intramitochondrial cardiolipin during apoptosis in U937 cell line. Toxicol In Vitro. 2007;21:293–301. doi: 10.1016/j.tiv.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 126.Pi H, Xu S, Reiter RJ, Guo P, Zhang L, Li Y, Li M, Cao Z, Tian L, Xie J, et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11:1037–1051. doi: 10.1080/15548627.2015.1052208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461–2483. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36 Suppl 1:S3–12. doi: 10.1016/S2210-7401(12)70015-3. [DOI] [PubMed] [Google Scholar]
- 129.Trauner M, Graziadei IW. Review article: mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment Pharmacol Ther. 1999;13:979–996. doi: 10.1046/j.1365-2036.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 130.Rodríguez VA, Rivoira MA, Pérez Adel V, Marchionatti AM, Tolosa de Talamoni NG. Ursodeoxycholic and deoxycholic acids: Differential effects on intestinal Ca(2+) uptake, apoptosis and autophagy of rat intestine. Arch Biochem Biophys. 2016;591:28–34. doi: 10.1016/j.abb.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 131.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 132.do Nascimento PG, Lemos TL, Almeida MC, de Souza JM, Bizerra AM, Santiago GM, da Costa JG, Coutinho HD. Lithocholic acid and derivatives: Antibacterial activity. Steroids. 2015;104:8–15. doi: 10.1016/j.steroids.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 133.Halvorsen B, Staff AC, Ligaarden S, Prydz K, Kolset SO. Lithocholic acid and sulphated lithocholic acid differ in the ability to promote matrix metalloproteinase secretion in the human colon cancer cell line CaCo-2. Biochem J. 2000;349:189–193. doi: 10.1042/0264-6021:3490189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gafar AA, Draz HM, Goldberg AA, Bashandy MA, Bakry S, Khalifa MA, AbuShair W, Titorenko VI, Sanderson JT. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ. 2016;4:e2445. doi: 10.7717/peerj.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dang Z, Lin A, Ho P, Soroka D, Lee KH, Huang L, Chen CH. Synthesis and proteasome inhibition of lithocholic acid derivatives. Bioorg Med Chem Lett. 2011;21:1926–1928. doi: 10.1016/j.bmcl.2011.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Banerjee S, Trivedi GK, Srivastava S, Phadke RS. Proxyl nitroxide of lithocholic acid: a potential spin probe for model membranes. Bioorg Med Chem. 1993;1:341–347. doi: 10.1016/s0968-0896(00)82140-9. [DOI] [PubMed] [Google Scholar]
- 137.Arlia-Ciommo A, Piano A, Svistkova V, Mohtashami S, Titorenko VI. Mechanisms underlying the anti-aging and anti-tumor effects of lithocholic bile acid. Int J Mol Sci. 2014;15:16522–16543. doi: 10.3390/ijms150916522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ishizawa M, Matsunawa M, Adachi R, Uno S, Ikeda K, Masuno H, Shimizu M, Iwasaki K, Yamada S, Makishima M. Lithocholic acid derivatives act as selective vitamin D receptor modulators without inducing hypercalcemia. J Lipid Res. 2008;49:763–772. doi: 10.1194/jlr.M700293-JLR200. [DOI] [PubMed] [Google Scholar]
- 139.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703–722. doi: 10.1081/DMR-200033475. [DOI] [PubMed] [Google Scholar]