

Abstract
Protein-losing enteropathy (PLE) is characterized by loss of serum proteins into the gastrointestinal tract. It may lead to hypoproteinemia and clinically present as protein deficiency edema, ascites, pleural or pericardial effusion and/or malnutrition. In most cases the site of protein loss is the small intestine. Here we present an unusual case of severe PLE in a 55-year old female with a one-year history of recurrent diarrhea, crampy abdominal pain, and peripheral edema. Endoscopy and MRI showed a diffuse inflammatory thickening of the sigmoid colon and the rectum. Surgical resection of the involved colon was performed and the symptoms were significantly resolved. The final histologic evaluation confirmed a diagnosis of a pseudomembranous colitis with cap polyposis-like features. Such a cause of PLE has never been described before.
Keywords: Protein-losing enteropathy, Cap polyposis, Ulcerative colitis, Goblet cells, Pseudomembranes
Core tip: Protein-losing enteropathy (PLE) is characterized by loss of serum proteins into the gastrointestinal tract. The small intestine is the most common site of protein loss. This is an unusual case of severe PLE involving the sigmoid colon and the rectum. Surgical resection was performed and subsequent histologic evaluation confirmed a diagnosis of a pseudomembranous colitis with cap polyposis-like features.
INTRODUCTION
Protein-losing enteropathy (PLE) is a rare condition of excessive serum protein losses into the gastrointestinal tract. The clinical presentation may be variable but commonly consists of peripheral edema secondary to hypoproteinemia. It may also manifest as ascites, pleural and pericardial effusions, associated with gastrointestinal symptoms of diarrhea, abdominal pain, and bloating[1-4]. Along with protein loss, a significant decrease in the level of immunoglobulins and lymphocytes can also occur which may lead to immune deficiency, predisposing the patient to infections[4]. The underlying causes of PLE are divided into erosive gastrointestinal disorders (e.g., inflammatory bowel disease, nonsteroidal anti-inflammatory drug enteropathy, intestinal lymphoma), nonerosive gastrointestinal disorders (e.g., celiac disease, connective tissue disorders, microscopic colitis), and disorders involving increased interstitial pressure (e.g., congestive heart failure, constrictive pericarditis, intestinal lymphangiectasia)[1]. Conventionally, the diagnosis is established by an increased fecal clearance of alpha-1-antitrypsin and its decreased level in the plasma[2]. PLE should be considered in patients with hypoproteinemia against a background of gastrointestinal disease and in whom other causes, such as severe malnutrition, hepatic and renal impairment, as well as cardiac insufficiency, have been excluded[3,4]. In this report we present a case of severe PLE in a 55-year old female with a one-year history of recurrent diarrhea, crampy abdominal pain, and peripheral edema. The sigmoid colon and the rectum were identified as specific sites of protein loss. Surgical resection was performed and subsequent histologic evaluation confirmed a diagnosis of pseudomembranous colitis with cap polyposis-like features.
CASE REPORT
A 54-year old Caucasian female was referred to our hospital for a one-year history of recurrent diarrhea of up to 15 times per day with nocturnal component. The stool was described as loose, mucilaginous, and non-bloody. The diarrhea was associated with moderate, crampy abdominal pain and weight loss. Her appetite was intact and she denied anorexia, febrile episodes or fecal incontinence. She was previously hospitalized for the same complaint and was managed as ulcerative colitis. The symptoms, however, remained unresolved. Her previous medical, family, social, and travel history was unremarkable with no record of allergies or antibiotic intake.
On admission, physical examination revealed a weight of 50 kg, height of 170 cm, and normal vital signs. Cardiovascular and pulmonary findings were unremarkable. There was no abdominal distention, no palpable mass, no tenderness, and with normoactive bowel sounds. The liver and spleen were not enlarged. There was marked pitting edema of both lower extremities. No superficial lymph nodes were palpable. Neurological status was normal. Laboratory investigations revealed leukocyte count of 11.5 × 103/μL [4.0-10.0], thrombocytes of 590 × 103/μL [140-400], total serum protein was 45 g/L [64-83], albumin was 25 g/L [35-52], IgG was 3.21 g/L [7-16], and diamine oxidase was 2 U/mL [> 10]. Serum electrolytes, liver and pancreatic enzymes, renal and thyroid function tests, coagulation tests, and C-reactive protein were unremarkable. Urinalysis showed neither proteinuria nor hematuria. Celiac disease serology was negative. ECG, chest X-ray, and thoracic CT findings were normal. Stool examination was unremarkable and no bacteria or viruses were found in the stool culture. Abdominal MRI demonstrated diffuse wall thickening at the level of the sigmoid colon and rectum (Figure 1). There was no evidence of malignancy.
Figure 1.
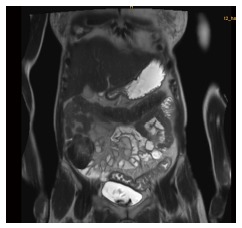
MRI-Scan of the abdomen showing a long-segment bowel wall thickening in the distal parts of the colon.
Esophago-gastro-duodenoscopy (EGD) and capsule endoscopy of the small intestine revealed no pathological findings and no evidence of Helicobacter pylori. Colonoscopy showed continuous, diffuse mucosal inflammation (Figure 2) from the anocutaneous line upward for approximately 40 cm with a sharp demarcation of normally-appearing mucosa. There were no ulcers or polyps. The endoscopic findings were suggestive of ulcerative colitis. However, the patient was unresponsive to prednisolone, mesalamine, cyclosporine, and infliximab. The hypoalbuminemia and decreased IgG remained constant. Albumin infusions resulted in transient improvement of symptoms.
Figure 2.
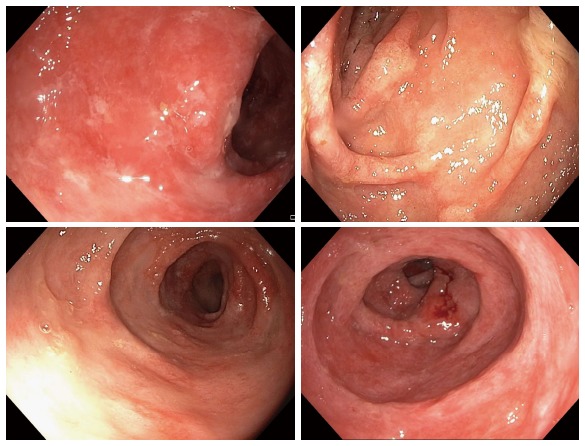
Diffuse inflammation of the colonic mucosa spreading contiuously. The mucosa appeared to be covered by a protein-rich exudate.
After a 6-mo period of unsuccessful immunosuppressive therapy the patient was readmitted due to worsening symptoms including increased stool frequency and peripheral edema. On laboratory examinations the leucocyte count was 10.3 × 103/μL [4.0-10.0], thrombocytes 741 × 103/μL [140-400], hemoglobin 11.4 g/L [12-16], total protein 32 g/L [64-83], albumin 16 g/L [35-52], IgG 4.04 g/L [7-16], and C-reactive protein 5 mg/L [< 5]. Microbiological analyses of the stool specimens did not reveal signs of infection. To rule out immune deficiency disorders peripheral and mucosal lymphocytes were analyzed. Results showed a normal CD4/CD8-ratio of 1.69 (absolute counts of CD4+ T-cells: 1026/μL; CD8+ T-cells: 608/μL). There was no indication of T-cell deficiency. The percentages of naïve CD4+ T-cells (CD45RA+/CD4+), regulatory T-cells (CD25+ FOXP3+/CD4+, and NK-cells (CD56+CD3-/lymphocytes) in the peripheral blood were normal, as well as the percentages of IL-17 or IFN-y producing CD4+ T-cells. The only abnormality was a high percentage of CD25- FOXP3+ CD4+ T-cells: 10% in the peripheral blood and 5% in the mucosa. PCR analyses for IgH and TCRγ rearrangements using BIOMED-2 primer revealed no clonal B-cell or T-cell population. EGD and colonoscopy were repeated and showed the same findings as previously described.
Multiple biopsy samples, surgical colectomy, and mucosectomy specimens revealed unusual histologic changes involving the mucosa from the distal descending colon to the proximal rectum (Figures 3 and 4): Hematoxylin and eosin stained sections cut from formalin-fixed, paraffin-embedded tissue blocks showed confluent erosions due to a widespread surface epithelial injury. The surface was covered by a thick adherent pseudomembranous layer admixed with inflammatory cells (Figure 3A). A striking architectural disarray of the subjacent mucosa was reminiscent of the pattern seen in cap polyposis. However, the mucosal changes diffusely involved the large bowel without formation of polypoid structures. A distortion and cystic dilatation involved the basal and middle parts of the colonic crypts that were predominantly lined by hyperplastic goblet cells and contained abundant periodic acid-Schiff (PAS)+ mucus and cell detritus (Figure 3B and C). In some crypts, an eosinophilic epithelium forming a mild serrated pattern was observed and was the reason for further molecular analyses (Figure 3D). The epithelium of the upper parts of the dilated crypts was clearly attenuated as demonstrated by anti-cytokeratin 20 immunohistochemistry (IH; Figure 4A). The adjacent superficial lamina propria was replaced by a layer of granulation tissue that contained numerous ectatic capillaries, erythrocyte extravasates, CD68+ macrophages, CD138+ plasma cells, myeloperoxidase+ neutrophilic granulocytes, and scattered eosinophils. Granulocytes predominated in the deeper parts of the lamina propria but rarely infiltrated the crypt epithelium. Additional IH for IgA, IgG, IgM, IgD, Kappa, Lambda, CD20, CD3, CD4, CD8, Ki67, and p53 were evaluated as follows: Plasma cells with polytypic light chain expression were abundant in the granulation tissue and the upper third of the lamina propria but rarely observed in the deeper mucosa. Only few CD20+ lymphocytes without any follicles were present. The number of CD3+ T-cells was considerably reduced to about 2 to 4 cells per high power field and rarely infiltrated the crypt epithelium (Figure 4B). The ratio of CD4:CD8 was normal. No aberrant Ki67+ or p53 labelling of the crypt epithelium was noted. Infectious agents could not be detected by IH for mycobacteria and CMV, chromogen in situ-hybridisation for EBV-encoded RNA or Ziehl-Neelson-, Gram-, PAS or Grocott stains.
Figure 3.
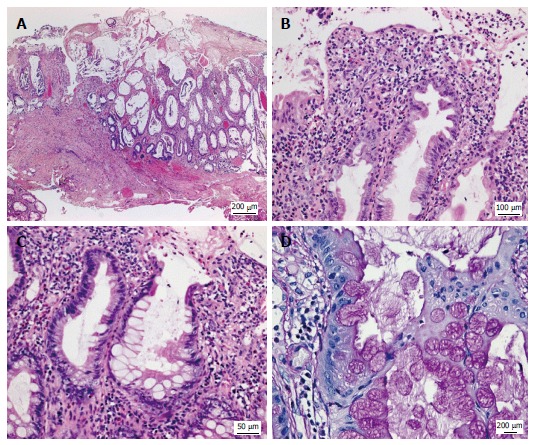
Histologic appearance of the colonic mucosa. A: Erosive lesions covered by a thick pseudomembranous layer; damage of the surface epithelium and architectural disarray of the mucosa showing prominent cystic dilatation of crypts; B: Foci of mildly serrated epithelium; C, D: Abundant hyperplastic goblet cells associated with a strikingly increased accumulation of intracellular and extracellular mucus. A-C: Hematoxylin and eosin; D: Periodic acid-Schiff reaction.
Figure 4.
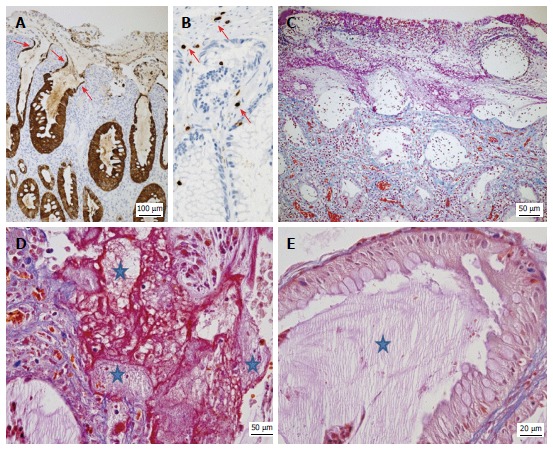
Typical features of protein-losing colitis demonstrated by immunohistochemistry and special stains. A: Strong cytokeratin 20 expression (brown) by hyperplastic and attenuated (arrows) epithelial cells; B: Detection of rare CD3+ T-cells (brown, arrows) in the lamina propria and epithelium; C: Acid fuchsin orange G-stain (AFOG) stain demonstrating proteins (red) in the apical part of the denuded crypts; the granulation tissue and the pseudomembranes covering the surface, but not in the deeper parts of the mucosa; D, E: AFOG stain combined with a PAS reaction highlighting the presence of both proteins (red) and admixed mucus (pale pink, stars); E: Abundant pale pink mucus (star) but no significant proteins in the crypts lined by hyperplastic goblet cells.
To further localize the source of the protein loss, we performed the acid fuchsin-orange G-stain (AFOG) that highlighted abundant proteins as red extracellular material especially in the pseudomembranes and superficial parts of the mucosa (Figure 4C). A combination of PAS and AFOG staining (Figure 4D and E) methods allowed us to visualize mucus simultaneously with protein deposits. Red-appearing proteins were detected within the granulation tissue in the vicinity of capillaries, the lumina of the superficial denuded crypts, the erosive lesions, and the thick pseudomembranes (Figure 4D). Here the red-staining proteins were admixed with pale pink-appearing mucous (Figure 4D). By contrast, the cytoplasm of the goblet cells and the adjacent lumina of the dilated crypts mainly contained pinkish mucous and cellular debris but were generally devoid of a protein-rich exudate (Figure 4E).
Since the architecture of scattered crypts showed a mild serrated pattern reminiscent of serrated adenoma/polyps further mutational analyses were done by next generation sequencing but did not reveal mutations in BRAF (exon 15), EGFR (exons 18, 19, 20, 21), KRAS (exons 2, 3, 5), NRAS (exon 2, 3), KIT (exons 9, 11, 13, 17), and PDGFRA (exon 18).
Surgical resection of the affected part of the colon and the proximal rectum sparing the distal 8 cm was performed to preserve the anal sphincter. Descendostomy was done as a transient measure. This surgical procedure resulted in a significant reduction of protein loss and normalization of serum albumin and IgG (Figure 5). The patient tolerated the procedure well and had nearly no abdominal symptoms. Following the reanastomosis of the descending colon and distal part of the rectum the protein loss remained compensated but the symptoms recurred, though to a considerably lesser degree. Repeated endoscopy showed involvement of proctitis in the remaining < 8 cm of the rectum. Therefore, a complete rectal mucosectomy was performed in two sessions.
Figure 5.
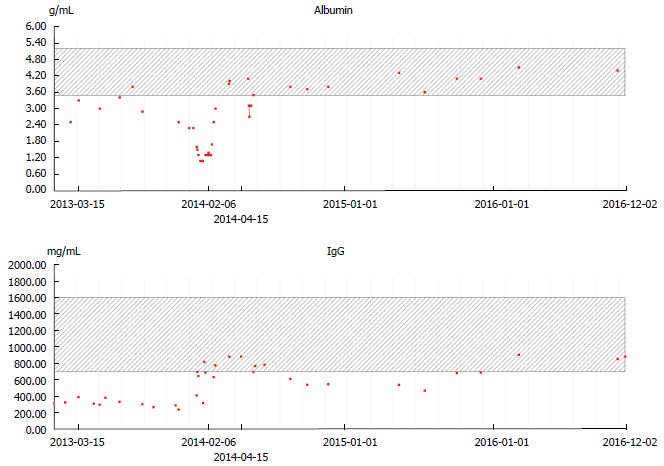
Albumin (upper panel) and IgG (lower panel) levels during the course of the disease. Complete protein loss remission after surgical resection (February 6, 2014).
DISCUSSION
Here we describe a very unusual case of a PLE where the site of protein loss was solely the distal colon. This protein loss was so marked that protein deficiency edema developed and very low concentrations of albumin and IgG were observed. The underlying cause was a diffuse inflammation in the rectum and sigmoid colon with goblet cell hyperplasia, protein rich pseudomembranes, and cap polyposis-like features.
Upon initial diagnostic evaluation most known causes of PLE could immediately be ruled out including intestinal infections, celiac disease, eosinophilic gastroenteritis, microscopic colitis, intestinal lymphangiectasia, heart or liver diseases, and amyloidosis. Histology and analysis of peripheral and mucosal lymphocytes excluded lymphoma, immune deficiency, and malignancy. Upper endoscopy including histology and capsule endoscopy revealed a completely normal upper gastrointestinal tract. The distal colon was recognized as site of protein loss after exclusion of other possibilities. The macroscopic aspect was quite similar to ulcerative colitis as was the type of colonic manifestation: continuous involvement beginning in the distal rectum. Consequently, the disease was initially diagnosed as an “atypical form of ulcerative colitis.” After a nearly complete unresponsiveness to escalating immunosuppressive therapy such therapeutic attempts were discontinued and a reevaluation was performed.
Serrated adenoma-like lesion were ruled out in our patient using methods recently described by Kovaleva et al[5]. Histology revealed lesions that were compatible or even typical for cap polyposis (CP) as summarized by Ng et al[6]. Since the first description by Williams et al[7] CP has been recognized as a rare type of inflammatory pseudo-polyps found mainly in the distal colon. Typical clinical symptoms of which are mucilaginous diarrhea and crampy abdominal pain. Bloody diarrhea or protein-losing enteropathy may also occur[6]. On endoscopy, the surface of the polyps appears covered by a cap of fibrinopurulent exudate. Histology shows dilatation, elongation, and distortion of crypts which may have a serrated-type like pattern. The cap is formed by an inflammatory granulation tissue which is covered by a layer of protein-rich exudate. The peculiarity of our case was the diffuse and continuous involvement of approximately 40 cm of the distal colon. No distinct polyps could be found. Generally, cap polyps manifest as polypoid lesions with intervening normal mucosa[6,8]. However, contiguous polyps have been described and it is not uncommon that CP is often misinterpreted as chronic inflammatory bowel disease[9,10]. This was the case in our patient. After complete unresponsiveness to immunosuppressive therapy, the reevaluation of the case ultimately led to a more accurate interpretation of endoscopy and histology. The clinical presentation, endoscopy, and histology suggest that this case may represent a hitherto undescribed variant of cap polyposis.
According to the PAS/AFOG double staining the intestinal protein loss of our patient predominantly resulted from capillary leakage within the erosive lesions and the granulation tissue. The hyperplastic goblet cells were no relevant source of the intestinal protein loss. While in the lung the main component is MUC5AC mucin (as is the case in the stomach), the intestinal mucin is mainly composed of MUC2 mucin, among others[11]. In cap polyposis non-sulfated mucins were found, as well as an abnormal expression of genes for MUC4, MUC3, and MUC5AC[12]. For the lung there is literature indicating that albumin and IgG are part of the secreted mucus[13,14]. However, to the best of our knowledge neither albumin nor IgG were proven components of intestinal mucus. On histology, it could be shown that the affected epithelia are covered with a broad layer of proteins other than mucins. Arimura et al[15] found that in CP pore-forming claudin-2 was abnormally expressed and redistributed, whereas pore-sealing claudin-7 was downregulated. Since claudins are important components of the tight-junctions[16], it may be inferred that an imbalance between pore-forming and pore-sealing claudins is the biochemical background for loss of serum proteins in CP.
The patient was not affected by any type of congenital or acquired systemic immunodeficiency. However, the T-cell depletion restricted to the affected parts of the colorectal mucosa is highly unusual both for the normal intestinal tract and especially for inflammatory bowel diseases. The reduction of CD3+ -T-cells associated with a relative increase in CD25- FoxP3+ regulatory T-cells (10% in the peripheral blood, normal < 1%, 5% in mucosal biopsies, no normal range available) implies a local perturbation of the mucosal immunity and homeostasis. In active systemic lupus erythematosus a higher percentage of this type of regulatory T-cells has been observed in peripheral blood and a relation to disease activity was suggested[17,18]. The depletion of resident T- cells with a putative impact on IgA secretion by plasma cells may have contributed to a persistent damage of the intestinal barrier integrity and an increased vulnerability to enteropathogens. The predominantly superficial infiltrates which we observed are generally considered to be more typical for an infectious colitis than for a chronic inflammatory bowel disease. We cannot completely exclude the possibility that the disease was initially triggered by pathogens and enhanced by the immunosuppressive therapy initially given under the suspected diagnosis of ulcerative colitis. However, no intestinal pathogen was found during the entire clinical course.
There is no generally accepted therapy for CP. The most successful therapy seems to be endoscopic polypectomy[6]. However, there are several papers which reported a beneficial effect of corticosteroids[19], infliximab[20,21], or therapy of a concomitant infection with Helicobacter pylori[22]. In serious cases, surgical resection is the preferred therapy. In our patient resection of the affected parts of the large intestine resulted in resolution of the symptoms and complete stop of protein loss.
Taken together we report an unusual case of pseudomembranous colitis with cap- polyposis like features and continuous involvement of the rectum and sigmoid colon resulting in severe protein-losing enteropathy. Resection of the affected parts of the colon led to a complete remission of the protein loss.
COMMENTS
Case characteristics
The patient presented with mucilaginous diarrhea, crampy abdominal pain, and pitting edema.
Clinical diagnosis
The patient was in a reduced general condition and had bilateral pitting edema.
Differential diagnosis
After the diseases of the small intestine were excluded, an atypical form of colitis of the distal colon was identified as the underlying cause.
Laboratory diagnosis
Laboratory evaluation showed a marked reduction of serum albumin and serum IgG.
Imaging diagnosis
MRI and endoscopy revealed a diffuse inflammation of the distal part of the colon.
Pathological diagnosis
Histology showed mucus-filled crypts arranged in a serrated pattern with goblet cell hyperplasia and pseudomembranes consisting of serum proteins.
Treatment
Since medical therapy was not possible a resection of the affected parts of the colon was performed leading to a reversal of the protein loss.
Related reports
The clinical presentation and histological findings suggest protein loss due to a pseudomembranous colitis with histological lesions similar to cap polyposis. However, polyps were not present.
Term explanation
Cap polyposis is a rare form of intestinal polyps whose tip is covered by pseudomembranes consisting of secreted serum proteins.
Experiences and lessons
This is the first description of a pseudomembranous colitis with histological features similar to cap polyposis, but with a continuous spread in the colon without evidence of polyps.
Peer-review
This case report has received generally positive reviews with suggestions to improve diagnostic reasoning.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Germany
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: Ethics Commission, Universitätsklinikum Freiburg, 79106 Freiburg, Germany.
Informed consent statement: The patient whose clinical course was described gave a written informed consent for this publication.
Conflict-of-interest statement: There was no conflict of interest.
Peer-review started: February 3, 2017
First decision: February 23, 2017
Article in press: April 12, 2017
P- Reviewer: Li HD, Taleban S, Markovic BS S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Umar SB, DiBaise JK. Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol. 2010;105:43–9; quiz 50. doi: 10.1038/ajg.2009.561. [DOI] [PubMed] [Google Scholar]
- 2.Braamskamp MJ, Dolman KM, Tabbers MM. Clinical practice. Protein-losing enteropathy in children. Eur J Pediatr. 2010;169:1179–1185. doi: 10.1007/s00431-010-1235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milovic V, Grand RJ. Protein-losing gastroenteropathy - UpToDate. Available from: https://www.uptodate.com/contents/protein-losing-gastroenteropathy.
- 4.Aslam N, Wright R. Protein-Losing Enteropathy: Background, Pathophysiology, Etiology. (2016) Available from: http://emedicine.medscape.com/article/182565-overview.
- 5.Kovaleva V, Geissler AL, Lutz L, Fritsch R, Makowiec F, Wiesemann S, Hopt UT, Passlick B, Werner M, Lassmann S. Spatio-temporal mutation profiles of case-matched colorectal carcinomas and their metastases reveal unique de novo mutations in metachronous lung metastases by targeted next generation sequencing. Mol Cancer. 2016;15:63. doi: 10.1186/s12943-016-0549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng KH, Mathur P, Kumarasinghe MP, Eu KW, Seow-Choen F. Cap polyposis: further experience and review. Dis Colon Rectum. 2004;47:1208–1215. doi: 10.1007/s10350-004-0561-8. [DOI] [PubMed] [Google Scholar]
- 7.Williams GT, Bussey HJ, Morson BC. Inflammatory ‘cap polyps’ of the large intestine. Br J Surg. 1985;72:S133. doi: 10.1002/bjs.1800721355. [DOI] [Google Scholar]
- 8.Li JH, Leong MY, Phua KB, Low Y, Kader A, Logarajah V, Ong LY, Chua JH, Ong C. Cap polyposis: a rare cause of rectal bleeding in children. World J Gastroenterol. 2013;19:4185–4191. doi: 10.3748/wjg.v19.i26.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kini GP, Murray I, Champion-Young J, Lau M, Katta V, Thorn M, Schultz MP. Cap polyposis mistaken for Crohn’s disease: case report and review of literature. J Crohns Colitis. 2013;7:e108–e111. doi: 10.1016/j.crohns.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Batra S, Johal J, Lee P, Hourigan S. Cap Polyposis Masquerading as Inflammatory Bowel Disease in a Child. J Pediatr Gastroenterol Nutr. 2016 doi: 10.1097/MPG.0000000000001343. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. 2010;12:319–330. doi: 10.1007/s11894-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buisine MP, Colombel JF, Lecomte-Houcke M, Gower P, Aubert JP, Porchet N, Janin A. Abnormal mucus in cap polyposis. Gut. 1998;42:135–138. doi: 10.1136/gut.42.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rogers DF. Physiology of airway mucus secretion and pathophysiology of hypersecretion. Respir Care. 2007;52:1134–146; discussion 1134-146;. [PubMed] [Google Scholar]
- 14.Lai HY, Rogers DF. Mucus hypersecretion in asthma: intracellular signalling pathways as targets for pharmacotherapy. Curr Opin Allergy Clin Immunol. 2010;10:67–76. doi: 10.1097/ACI.0b013e328334643a. [DOI] [PubMed] [Google Scholar]
- 15.Arimura Y, Isshiki H, Hirayama D, Onodera K, Murakami K, Yamashita K, Shinomura Y. Polypectomy to eradicate cap polyposis with protein-losing enteropathy. Am J Gastroenterol. 2014;109:1689–1691. doi: 10.1038/ajg.2014.227. [DOI] [PubMed] [Google Scholar]
- 16.Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta. 2008;1778:588–600. doi: 10.1016/j.bbamem.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Zhang B, Zhang X, Tang FL, Zhu LP, Liu Y, Lipsky PE. Clinical significance of increased CD4+CD25-Foxp3+ T cells in patients with new-onset systemic lupus erythematosus. Ann Rheum Dis. 2008;67:1037–1040. doi: 10.1136/ard.2007.083543. [DOI] [PubMed] [Google Scholar]
- 18.Bonelli M, Savitskaya A, Steiner CW, Rath E, Smolen JS, Scheinecker C. Phenotypic and functional analysis of CD4+ CD25- Foxp3+ T cells in patients with systemic lupus erythematosus. J Immunol. 2009;182:1689–1695. doi: 10.4049/jimmunol.182.3.1689. [DOI] [PubMed] [Google Scholar]
- 19.Chang HS, Yang SK, Kim MJ, Ye BD, Byeon JS, Myung SJ, Kim JH. Long-term outcome of cap polyposis, with special reference to the effects of steroid therapy. Gastrointest Endosc. 2012;75:211–216. doi: 10.1016/j.gie.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Bookman ID, Redston MS, Greenberg GR. Successful treatment of cap polyposis with infliximab. Gastroenterology. 2004;126:1868–1871. doi: 10.1053/j.gastro.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Kim ES, Jeen YT, Keum B, Seo YS, Chun HJ, Um SH, Kim CD, Ryu HS. Remission of cap polyposis maintained for more than three years after infliximab treatment. Gut Liver. 2009;3:325–328. doi: 10.5009/gnl.2009.3.4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akamatsu T, Nakamura N, Kawamura Y, Shinji A, Tateiwa N, Ochi Y, Katsuyama T, Kiyosawa K. Possible relationship between Helicobacter pylori infection and cap polyposis of the colon. Helicobacter. 2004;9:651–656. doi: 10.1111/j.1083-4389.2004.00273.x. [DOI] [PubMed] [Google Scholar]