

Abstract
Background and purpose
Significant effects on clinical/neuroradiological disease activity have been reported in patients with relapsing–remitting multiple sclerosis treated with delayed‐release dimethyl fumarate (DMF) in phase III DEFINE/CONFIRM trials. We conducted a post hoc analysis of integrated data from DEFINE/CONFIRM to evaluate the effect of DMF on achieving no evidence of disease activity (NEDA) in patients with relapsing–remitting multiple sclerosis.
Methods
The analysis included patients randomized to DMF 240 mg twice daily, placebo or glatiramer acetate (CONFIRM only) for ≤2 years. A time‐to‐event method was used to estimate the percentage of patients achieving NEDA. Clinical NEDA (no relapses/no 12‐week confirmed disability progression) was analysed in the intention‐to‐treat (ITT) population. Neuroradiological (no new/newly enlarging T2 hyperintense lesions/no gadolinium‐enhancing lesions) and overall NEDA (clinical and neuroradiological NEDA) were analysed in the magnetic resonance imaging (MRI) cohort.
Results
The ITT and MRI populations comprised 1540 and 692 patients, respectively. The percentage of patients with clinical NEDA (ITT population) and neuroradiological NEDA (MRI cohort) was higher with DMF versus placebo over 2 years [clinical NEDA: 38.9% relative reduction; hazard ratio (HR), 0.61; 95% confidence interval (CI), 0.52‐0.72; P < 0.0001; neuroradiological NEDA: 40.0% relative reduction; HR, 0.60; 95% CI, 0.49–0.73; P < 0.0001]. The percentage of patients achieving overall NEDA (MRI cohort) was also higher with DMF (26%) versus placebo (12%) over 2 years, with a relative risk reduction of 42.7% (HR, 0.57; 95% CI, 0.48–0.69; P < 0.0001).
Conclusions
A significantly higher percentage of patients treated with DMF achieved NEDA status over 2 years compared with placebo.
Keywords: clinical relapse, dimethyl fumarate, disability progression, disease monitoring, magnetic resonance imaging, no evidence of disease activity, relapsing–remitting multiple sclerosis
Short abstract
Click here to view the accompanying paper in this issue.
Introduction
As new and more efficacious treatments are developed for multiple sclerosis (MS), the composite outcome, no evidence of disease activity (NEDA), is being increasingly evaluated as a measure of therapeutic response 1, 2, 3. The term NEDA evolved by consensus from a similar term (‘freedom from disease activity’) and was first reported in the MS literature in a 2009 post hoc analysis of AFFIRM, a pivotal trial involving the disease‐modifying therapy natalizumab 1, 4. Patients with NEDA (n = 242) in AFFIRM had significantly smaller median percentage decreases in brain parenchymal fraction and significantly better results in performance measures of cognition, walking speed and upper extremity function over 2 years compared with patients without NEDA (n = 662), suggesting a likely association between NEDA, brain atrophy and functional outcomes 5.
The current definition of NEDA encompasses a combination of three related measures of disease activity: no relapses, no confirmed disability progression (CDP) sustained for 12 weeks as measured on the Expanded Disability Status Scale (EDSS) and no magnetic resonance imaging (MRI) disease activity, defined as no new gadolinium‐enhancing (Gd+) lesions and no new/newly enlarging T2 hyperintense lesions 4.
Delayed‐release dimethyl fumarate (DMF, also known as gastro‐resistant DMF) is indicated for the treatment of patients with relapsing or relapsing–remitting forms of MS. Treatment with DMF demonstrated significant improvements in clinical and neuroradiological outcomes compared with placebo and a favourable benefit–risk profile over a 2‐year period in two pivotal phase III studies (DEFINE/CONFIRM) in relapsing–remitting MS (RRMS) 6, 7. Sustained effects of DMF were reported in a long‐term extension of these studies (ENDORSE) 8, 9.
In view of the emergence of NEDA as a key treatment goal in the management of patients with MS, we conducted a post hoc analysis of integrated data from DEFINE/CONFIRM to evaluate the effect of DMF on the percentage of patients with clinical and/or brain neuroradiological NEDA using a commonly accepted definition for NEDA.
The benefits of performing integrated analyses of the DEFINE and CONFIRM studies have been noted elsewhere 10. Briefly, integrated analyses enable a more precise estimation of a treatment's effect than can be obtained from individual studies. Moreover, they allow for investigation of the consistency of a treatment's effect in subgroups of patients, and enable the evaluation of clinical and neuroradiological outcomes with reduced variability due to the greater number of patients analysed.
Methods
Study designs and patients
DEFINE (ClinicalTrials.gov NCT00420212) and CONFIRM (ClinicalTrials.gov NCT00451451) were multi‐centre, randomized, double‐blind, placebo‐controlled, parallel‐group clinical trials evaluating DMF as monotherapy for RRMS. Methodological details for the two studies have been described previously 6, 7. In brief, patients enrolled in DEFINE were randomized (1:1:1) to receive DMF 240 mg two (BID) or three (TID) times daily or matching placebo for up to 2 years. In CONFIRM, patients were randomized (1:1:1:1) to receive DMF 240 mg BID or TID or matching placebo, or an active reference comparator [glatiramer acetate (GA) 20 mg once daily] for 2 years. Each study year for DEFINE/CONFIRM comprised 48 weeks. Key eligibility criteria included age 18–55 years, RRMS diagnosis per McDonald criteria 11, EDSS score 0–5.0 12 and ≥1 clinically documented relapse within 12 months before randomization with a prior brain MRI demonstrating lesion(s) consistent with MS or ≥1 Gd+ lesion observed on brain MRI within 6 weeks before randomization. Key exclusion criteria were progressive forms of MS, other significant illnesses or pre‐specified laboratory abnormalities, MS relapse or prior corticosteroid treatment within 50 days before randomization, treatment with GA within the previous 3 months (DEFINE) or at any time (CONFIRM) and treatment with interferon‐α or interferon‐β within 3 months before randomization. The studies were approved by central and local ethics committees and conducted according to the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent before study entry.
Primary endpoints were the percentage of patients relapsed at 2 years in DEFINE (secondary endpoint in CONFIRM) and annualized relapse rate at 2 years in CONFIRM (secondary endpoint in DEFINE). Additional endpoints at 2 years included time to 12‐week CDP as measured by EDSS, number of new/newly enlarging T2 hyperintense lesions and number of Gd+ lesions on brain MRI. MRI endpoints were examined at weeks 24, 48 and 96 in both studies in a subset of patients (MRI cohort) who were enrolled at sites with MRI capabilities.
No evidence of disease activity analysis
Post hoc integrated NEDA analysis over 2 years was performed in the DEFINE/CONFIRM studies. Data pooling was possible due to the similarities of the two studies (DEFINE and CONFIRM) 6, 7, 10. Both the baseline characteristics and the treatment effect were homogeneous across the studies. Clinical NEDA was assessed in the intention‐to‐treat (ITT) population, and neuroradiological NEDA and overall NEDA were assessed in the MRI cohort.
Clinical NEDA was defined as no relapses and no 12‐week CDP as measured by EDSS score (>1.0‐point increase from a baseline score of >1.0 or >1.5‐point increase from a baseline score of 0, sustained for ≥12 weeks). Relapses were confirmed by an independent neurology evaluation committee. Patients with no new/newly enlarging T2 hyperintense and no Gd+ lesions were considered to have neuroradiological NEDA. Patients were defined as having overall NEDA if they did not experience clinical or neuroradiological NEDA during the respective time periods.
Statistical analysis
Post hoc NEDA data were analysed and presented at specific intervals based on the scheduled visits in the study. For clinical NEDA, it was possible to estimate the probability of achieving NEDA at all time points over the 2 years because the exact time of clinical events (relapse and CDP) were known. However, for the occurrence of MRI events, it was only known that MRI lesions were present or absent at the time of the assessment (i.e. the exact time of the events was unknown). Therefore, for the analyses of overall and neuroradiological NEDA, the time of relapse occurrence and CDP were organized into intervals determined by the scheduled assessment visits (0–6 months, 6 months–1 year and 1–2 years) of the MRI endpoints. The probability of achieving overall and neuroradiological NEDA over 2 years was estimated based on these intervals.
The estimated percentage of patients with clinical NEDA was derived using the Kaplan–Meier product limit method, which was based on time to first relapse or CDP (whichever occurred first). A Cox proportional hazard model adjusted for baseline EDSS score (≤2, >2), number of relapses at baseline, age (≤40, >40 years) and region was used to calculate the overall 2‐year hazard ratios (HRs) and corresponding P values for clinical NEDA.
A discrete time‐to‐event method was used to analyse the data for neuroradiological and overall NEDA. Unlike traditional methods of analyses that ignore patients’ differential follow‐up times and make unverifiable assumptions about NEDA status of censored patients, the time‐to‐event method allows for incorporation of different follow‐up times of patients and for appropriate handling of censoring and treatment discontinuation. This method allows for incorporation of patients’ data into the analyses for as long as they are known to be in the study and at risk of an event. It also allows the pattern of NEDA over time to be reflected and ensures that the analyses are based on the ITT principle 13.
The estimated percentage of patients with neuroradiological and overall NEDA was based on life‐table estimates of survival probability. A patient event (relapse, CDP, new/newly T2 hyperintense or new Gd+ lesions) was counted for the interval for which the patient was known to be in the study and hence at risk (e.g. a patient who discontinued before the end of the 0–6‐month interval and who had no subsequent MRI data only contributed to the first interval in the estimation). At the end of each interval, the percentage of patients achieving NEDA status was based on the number of patients at risk at the beginning of each interval.
A complementary log–log model 14 adjusting for region, baseline relapses (<1, >1), baseline EDSS score (≤2, >2), baseline T2 hyperintense lesion volume and presence of Gd+ lesions was used to calculate the overall 2‐year HR and corresponding P values for neuroradiological and overall NEDA.
Sensitivity analyses were also performed using the traditional method that ignores the event time and makes unverifiable assumptions about NEDA status for patients who discontinue without experiencing an event. Odds ratios and associated P values were calculated using a logistic regression model adjusted for age, region and corresponding baseline measures.
Results from patients treated with DMF 240 mg BID (approved dose in the USA and European Union 15), GA and placebo are reported. This post hoc analysis was not designed to test the superiority or non‐inferiority of DMF to GA.
Results
Patients
The ITT population for DEFINE, CONFIRM and the integrated analysis [including patients in the placebo, DMF 240 mg BID (hereafter referred to as DMF) and GA groups] included 818, 1072 and 1540 patients, respectively; the MRI cohort included 356, 511 and 692 patients, respectively.
Baseline demographic and disease characteristics were generally well balanced among placebo and treatment groups and between studies, as described previously 6, 7, 10. Mean (SD) patient age in DEFINE/CONFIRM was 37.7 (9.2) years for placebo and 37.9 (9.2) years for DMF; approximately 70% of patients were female. Mean time since first MS symptoms was 7.1–8.5 years. Patients experienced a mean (SD) of 1.3 (0.7) relapses in the year before study enrolment. Mean (SD) EDSS score was 2.5 (placebo, 1.2; DMF, 1.3) and mean (SD) number of Gd+ lesions at baseline was slightly lower for DMF vs. placebo [1.9 (5.0) vs. 2.2 (5.9), respectively], whereas mean T2 hyperintense lesion volume at baseline was similar [11.1 (12.1) vs. 10.4 (11.4), respectively]. Baseline demographic and patient disease characteristics were similar between the ITT and MRI cohorts.
No evidence of disease activity
The relative risk reduction in clinical disease activity with DMF treatment compared with placebo was 45.2%, 28.9% and 38.9% in DEFINE, CONFIRM and the integrated analysis, respectively {HR [95% confidence interval (CI)]: 0.55 (0.44–0.68), P < 0.0001 for DEFINE; 0.71 (0.56–0.91), P = 0.0064 for CONFIRM; 0.61 (0.52–0.72), P < 0.0001 for the integrated analysis} (Fig. 1). Kaplan–Meier estimates showed a higher percentage of patients with clinical NEDA in the DMF treatment group compared with placebo over 2 years for DEFINE, CONFIRM and the integrated analysis (Fig. 2 for integrated analysis, other analyses not shown).
Figure 1.
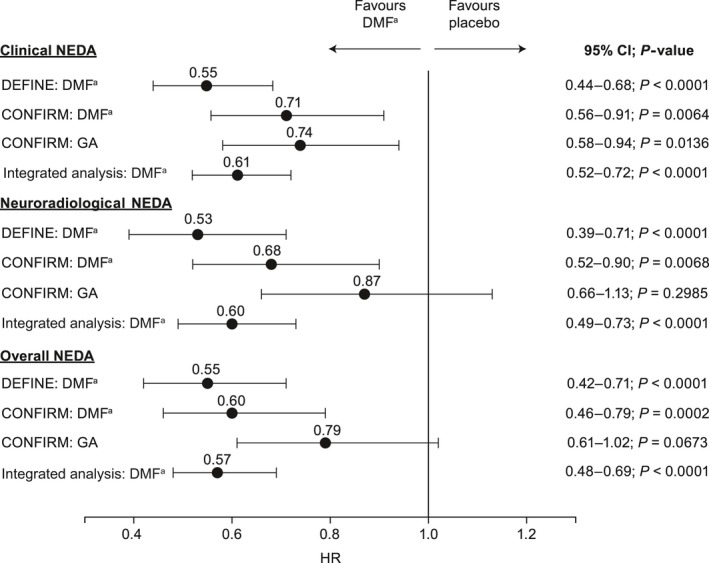
Impact of delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) on clinical, neuroradiological and overall no evidence of disease activity (NEDA) over the 2‐year study period. CI, confidence interval; GA, glatiramer acetate; HR, hazard ratio.
Figure 2.
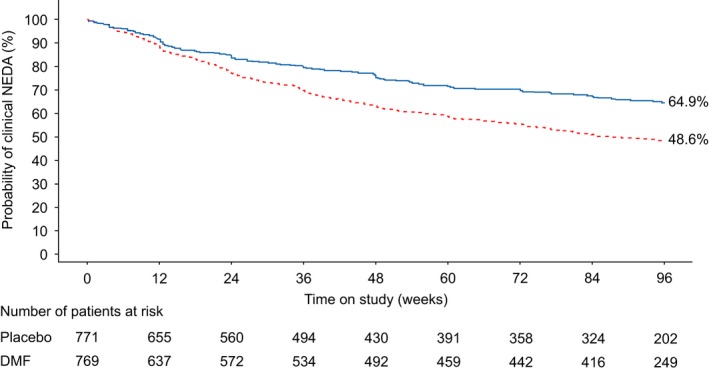
Kaplan–Meier estimates of the percentage of patients treated with delayed released dimethyl fumarate (DMF, also known as gastro‐resistant DMF) with clinical no evidence of disease activity (NEDA) (intention‐to‐treat population) in the integrated analysis of DEFINE and CONFIRM over 2 years. P values are based on the log‐rank test.  , DMF;
, DMF;  , placebo. Hazard ratio (95% CI): DMF vs. placebo = 0.61 (0.52–0.72); P value: DMF vs. placebo = <0.0001. [Colour figure can be viewed at wileyonlinelibrary.com].
, placebo. Hazard ratio (95% CI): DMF vs. placebo = 0.61 (0.52–0.72); P value: DMF vs. placebo = <0.0001. [Colour figure can be viewed at wileyonlinelibrary.com].
Over the 2‐year study period, the percentage of patients with neuroradiological NEDA was higher with DMF treatment compared with placebo in DEFINE, CONFIRM and the integrated analysis [HR (95% CI): 0.53 (0.39–0.71), P < 0.0001 for DEFINE; 0.68 (0.52–0.90), P = 0.0068 for CONFIRM; 0.60 (0.49–0.73), P < 0.0001 for the integrated analysis] (Fig. 1). This represents a reduction in the risk of new/newly enlarging T2 hyperintense and Gd+ lesions with DMF treatment relative to placebo of 47.2% (DEFINE), 31.7% (CONFIRM) and 40.0% (integrated analysis). Based on life‐table estimates, the percentages of patients with neuroradiological NEDA from baseline to 2 years were also higher with DMF treatment compared with placebo for all three analyses (Fig. 3 for integrated analysis, other analyses not shown).
Figure 3.
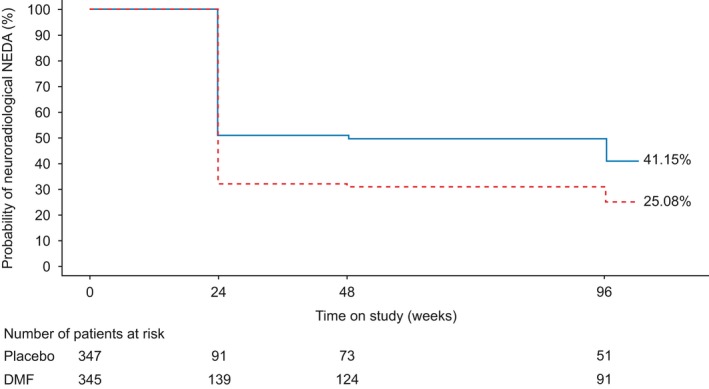
Life‐table estimates of the percentage of patients treated with delayed‐release dimethyl fumarate (DMF, also known as gastro‐resistant DMF) with neuroradiological no evidence of disease activity (NEDA) (magnetic resonance imaging population) in the integrated analysis of DEFINE and CONFIRM over 2 years. P values and confidence intervals (CIs) are based on complementary log–log model, adjusting for region, baseline T2 volume and baseline gadolinium‐enhancing lesions.  , DMF;
, DMF;  , placebo. Hazard ratio (95% CI): DMF vs. placebo = 0.60 (0.49–0.73); P value: DMF vs. placebo = <0.0001. [Colour figure can be viewed at wileyonlinelibrary.com].
, placebo. Hazard ratio (95% CI): DMF vs. placebo = 0.60 (0.49–0.73); P value: DMF vs. placebo = <0.0001. [Colour figure can be viewed at wileyonlinelibrary.com].
Over the 2‐year study period, the percentage of patients with overall NEDA was higher with DMF treatment compared with placebo [31% vs. 13%, HR (95% CI), 0.55 (0.42–0.71), P < 0.0001 for DEFINE; 20% vs. 9%, HR (95% CI), 0.60 (0.46–0.79), P = 0.0002 for CONFIRM; 26% vs. 12%, HR (95% CI), 0.57 (0.48–0.69), P < 0.0001 for integrated analysis]. The relative reductions in risk of relapse, CDP or new/newly enlarging T2 hyperintense and Gd+ lesions with DMF treatment compared with placebo were 45.3% (DEFINE), 39.5% (CONFIRM) and 42.7% (integrated analysis) (Fig. 1). Based on life‐table estimates, the percentage of patients with overall NEDA favoured DMF treatment over the 2 years for all three analyses (Fig. 4 for integrated analysis; other analyses not shown).
Figure 4.
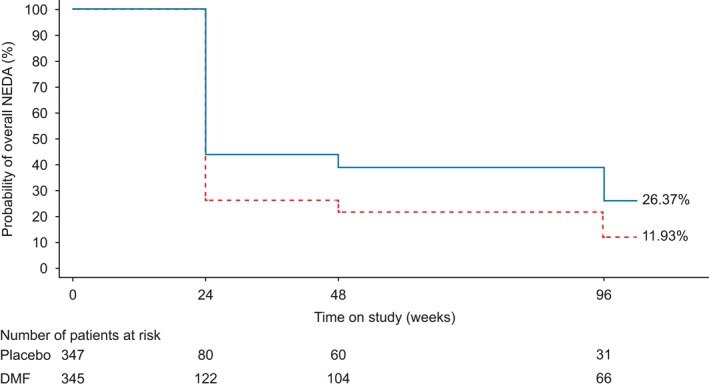
Life‐table estimates of the percentage of patients treated with delayed‐release dimethyl fumarate (DMF, also known as gastro‐resistant DMF) with overall no evidence of disease activity (NEDA) (magnetic resonance imaging population) in the integrated analysis of DEFINE and CONFIRM over 2 years. P values and confidence intervals (CIs) are based on complementary log–log model, adjusting for region, baseline relapse, baseline disability progression status, baseline T2 volume and baseline gadolinium‐enhancing lesions.  , DMF;
, DMF;  , placebo. Hazard ratio (95% CI): DMF vs. placebo = 0.57 (0.48–0.69); P value: DMF vs. placebo = <0.0001. [Colour figure can be viewed at wileyonlinelibrary.com].
, placebo. Hazard ratio (95% CI): DMF vs. placebo = 0.57 (0.48–0.69); P value: DMF vs. placebo = <0.0001. [Colour figure can be viewed at wileyonlinelibrary.com].
Similar to DMF treatment, the percentages of patients with clinical, neuroradiological and overall NEDA at the time points analysed were higher with GA treatment compared with the placebo group in CONFIRM. Over the 2‐year study period, the relative risk reduction of relapse with GA over placebo was statistically significant for clinical NEDA only [HR (95% CI), 0.74 (0.58–0.94), P = 0.0136] (Fig. 1).
Results from the sensitivity analyses were consistent with the primary NEDA analysis. A significantly higher percentage of patients treated with DMF compared with placebo had clinical, neuroradiological and overall NEDA over 2 years of treatment in DEFINE, CONFIRM and the integrated analysis. In addition, all odds ratios for DMF were >1.0, therefore favouring DMF treatment over placebo (Figs [Link], [Link], [Link]). In CONFIRM, a higher percentage of patients treated with GA achieved clinical, neuroradiological and overall NEDA over 2 years compared with patients treated with placebo.
Discussion
The results of this post hoc analysis of integrated data from DEFINE/CONFIRM suggest that DMF has a significant beneficial effect on clinical, MRI and overall NEDA in patients with RRMS. A higher percentage of patients treated with DMF achieved clinical NEDA in the ITT population, and higher percentages of patients achieved neuroradiological and overall NEDA (i.e. clinical and neuroradiological NEDA) in the MRI population throughout a 2‐year period compared with placebo. These findings are also consistent with individual results from DEFINE/CONFIRM, which showed a significantly higher percentage of patients receiving DMF with clinical, neuroradiological and overall NEDA compared with placebo during a 2‐year period. The GA arm was only included in the CONFIRM study as a reference comparator, not to test the superiority or non‐inferiority of DMF to GA treatment, and thus a comparison of DMF versus GA was not addressed in the present analysis. Sensitivity analysis using the actual percentages of patients with NEDA is consistent with the primary analysis results.
This integrated analysis provided an increased sample size (1540 patients in the ITT population and 692 in the MRI cohort), robust data and reduced variability in estimating the DMF treatment effect compared with the individual studies. By integrating the data from the DEFINE and CONFIRM studies, a more precise estimation of DMF efficacy was obtained than when analysed in either study alone. Additionally, as the different endpoints of NEDA represent distinct characteristics of the MS disease manifestation, this analysis demonstrates the therapeutic effect of DMF across these endpoints without the need to adjust for multiplicity of tests. Available data from a longitudinal MS cohort, analysed independently of disease‐modifying therapy, suggest that NEDA status at 2 years may have prognostic value in the longer term 16. The analysis included 219 patients with RRMS enrolled in the CLIMB study, which had a minimum of 7 years of prospective follow‐up (including yearly brain MRI and biannual clinic visits). The positive predictive value of NEDA to predict no progression at 7 years was 78.3% at 2 years, whereas additional follow‐up at 3–5 years was associated with only minor improvement in the positive predictive value 16. However, real‐world clinics may include longer titration methods to improve tolerability and long‐term adherence to DMF 17.
Limitations of the current study include the post hoc nature of the analyses, the relatively short follow‐up time of 2 years and the reduced number of patients who underwent MRI examination due to study design. The number of patients in the MRI population was less than half the number of patients in the ITT population (692 vs. 1540, respectively), the latter being the basis for the clinical NEDA analysis. Criticism has also been levelled at the current definition of NEDA, which is considered by some experts to be centred around clinical activity and much less towards disease progression or worsening, and overly driven by MRI parameters 18, 19 that are less informative about clinical worsening or progression. With ongoing advances in clinical practice, it is likely that the definition of NEDA will continue to evolve 4, 19. Nonetheless, NEDA as currently defined remains an important measure for therapeutic benefit as the effectiveness of treatment for MS continues to improve.
In conclusion, analysis of NEDA provides further information on the clinical benefits achievable with DMF treatment in patients with RRMS. Analysis of data from the extension study may facilitate better understanding of the prognostic importance of NEDA and its components.
Disclosure of conflicts of interest
E.H. has received honoraria from Actelion, Bayer HealthCare, Biogen, Genzyme, GlaxoSmithKline, Merck Serono, Novartis, Receptos, Sanofi‐Aventis and Teva; research support from Biogen; and is supported by the PRVOUK‐P26/LF1/4, program of the Ministry of Education, Czech Republic. G.G. has received honoraria from AbbVie Biotherapeutics Inc., Bayer HealthCare, Biogen, Canbex, FivePrime, Genzyme, GlaxoSmithKline, GW Pharma, Merck Serono, Novartis, Protein Discovery Laboratories, Roche, Synthon, Teva Neuroscience, UCB and Vertex; research grant support from Biogen, Ironwood, Merck Serono, Merz and Novartis; and compensation from Elsevier as co‐chief editor of Multiple Sclerosis and Related Disorders. R.G. has received honoraria from Bayer HealthCare, Biogen, Merck Serono, Novartis and Teva Neuroscience; research support from Bayer HealthCare, Biogen, Merck Serono, Novartis and Teva Neuroscience; and compensation from Sage for serving as editor of Therapeutic Advances in Neurological Disorders. R.J.F. has received consultant fees from Actelion, Biogen, MedDay, Novartis, Questcor, Teva and Xenoport; served on advisory committees for Actelion, Biogen and Novartis; and received research grant funding from Novartis. L.K.'s institution (University Hospital, Basel) received in the last 3 years and used exclusively for research support: steering committee, advisory board and consultancy fees from Actelion, Addex, Bayer HealthCare, Biogen, Biotica, Genzyme, Lilly, Merck, Mitsubishi, Novartis, Ono Pharma, Pfizer, Receptos, Sanofi‐Aventis, Santhera, Siemens, Teva, UCB and Xenoport; speaker fees from Bayer HealthCare, Biogen, Merck, Novartis, Sanofi‐Aventis and Teva; support of educational activities from Bayer HealthCare, Biogen, CSL Behring, Genzyme, Merck, Novartis, Sanofi‐Aventis and Teva; royalties from Neurostatus Systems GmbH; and grants from Bayer HealthCare, Biogen, the European Union, Merck, Novartis, Roche, Roche Research Foundations, Swiss MS Society and the Swiss National Research Foundation. J.T.P. has received consulting fees from Acorda, Biogen, Genentech, Genzyme, Merck Serono, Sanofi‐Aventis and Xenoport. M.O. is an employee of and holds stock/stock options in Biogen. J.L.M. performed the majority of the work while an employee of Biogen, Cambridge, MA; J.L.M.'s current affiliation is with Alexion Pharmaceuticals, New Haven, CT.
Supporting information
Figure S1. Percentage of patients treated with delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) with clinical no evidence of disease activity (NEDA) [no measured clinical activity was defined as having no relapses and no 12‐week confirmed disability progression as measured by the Expanded Disability Status Scale (EDSS)] at 6 months, 1 and 2 years of treatment in (a) DEFINE, (b) CONFIRM and (c) the integrated analysis (sensitivity analysis, intention‐to‐treat population). Odds ratios are shown with 95% confidence intervals in parentheses; P values for comparisons versus placebo are from a logistic regression model adjusted for baseline EDSS score, age (≤40, >40 years), region and number of relapses in the year before study entry.
Figure S2. Percentage of patients treated with delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) with neuroradiological no evidence of disease activity (NEDA) [no measured brain magnetic resonance imaging (MRI) disease activity was defined as no new/newly enlarging T2 hyperintense lesions and no gadolinium‐enhancing (Gd+) lesions] at 6 months, 1 and 2 years of treatment in (a) DEFINE, (b) CONFIRM and (c) the integrated analysis (sensitivity analysis, MRI cohort). Odds ratios are shown with 95% confidence intervals in parentheses; P values for comparisons versus placebo are from a logistic regression model adjusted for region, baseline volume of T2 hyperintense lesions and baseline number of Gd+ lesions.
Figure S3. Percentage of patients treated with delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) with overall no evidence of disease activity (NEDA) [no measured overall disease activity was defined as having no measured clinical and no measured brain magnetic resonance imaging (MRI) disease activity] at 6 months, 1 and 2 years of treatment in (a) DEFINE, (b) CONFIRM and (c) the integrated analysis (sensitivity analysis, MRI cohort). Odds ratios are shown with 95% confidence intervals in parentheses; P values for comparisons versus placebo are from a logistic regression model adjusted for baseline Expanded Disability Status Scale score, baseline age (≤40, >40 years), study, region, number of relapses in the year before study entry, baseline T2 hyperintense lesion volume and baseline number of gadolinium‐enhancing lesions.
Acknowledgements
We thank Dr Karl E. Peace for contributing to the development of this article. Biogen provided funding for medical writing support in the development of this article. Julie Adkins, from Complete Medical Communications, wrote the first draft of the manuscript based on input from authors. Ana Antaloae from Excel Scientific Solutions incorporated feedback from authors and Kristen DeYoung from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the article to the authors. The authors had full editorial control of the article and provided their final approval of all content. Biogen funded the study.
See editorial by Lanzillo on page 661.
References
- 1. Havrdova E, Galetta S, Hutchinson M, et al Effect of Natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing‐Remitting Multiple Sclerosis (AFFIRM) study. Lancet Neurol 2009; 8: 254–260. [DOI] [PubMed] [Google Scholar]
- 2. Arnold DL, Calabresi PA, Kieseier BC, et al Effect of peginterferon beta‐1a on MRI measures and achieving no evidence of disease activity: results from a randomized controlled trial in relapsing‐remitting multiple sclerosis. BMC Neurol 2014; 14: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nixon R, Bergvall N, Tomic D, Sfikas N, Cutter G, Giovannoni G. No evidence of disease activity: indirect comparisons of oral therapies for the treatment of relapsing‐remitting multiple sclerosis. Adv Ther 2014; 31: 1134–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giovannoni G, Turner B, Gnanapavan S, Offiah C, Schmierer K, Marta M. Is it time to target no evident disease activity (NEDA) in multiple sclerosis? Mult Scler Relat Disord 2015; 4: 329–333. [DOI] [PubMed] [Google Scholar]
- 5. Rudick R, Fisher E, Goodman A, et al No evident disease activity (NEDA): associations with brain atrophy and functional outcomes in patients from the AFFIRM study. Presented at: 2014 CMSC ACTRIMS Annual Meeting; 30 May 2014; Dallas, TX.
- 6. Fox RJ, Miller DH, Phillips JT, et al Placebo‐controlled phase 3 study of oral BG‐12 or glatiramer in multiple sclerosis. N Engl J Med 2012; 367: 1087–1097. [DOI] [PubMed] [Google Scholar]
- 7. Gold R, Kappos L, Arnold DL, et al Placebo‐controlled phase 3 study of oral BG‐12 for relapsing multiple sclerosis. N Engl J Med 2012; 367: 1098–1107. [DOI] [PubMed] [Google Scholar]
- 8. Gold R, Giovannoni G, Phillips JT, Fox RJ, Zhang A, Marantz JL. Sustained effect of delayed‐release dimethyl fumarate in newly diagnosed patients with relapsing‐remitting multiple sclerosis: 6‐year interim results from an extension of the DEFINE and CONFIRM studies. Neurol Ther 2016; 5: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gold R, Giovannoni G, Phillips J, et al Longer‐term follow‐up of the efficacy of delayed‐release dimethyl fumarate in newly diagnosed patients with RRMS: integrated analysis of DEFINE, CONFIRM, and ENDORSE. Neurology 2016; 86(16 suppl): P3.033. [Google Scholar]
- 10. Viglietta V, Miller D, Bar‐Or A, et al Efficacy of delayed‐release dimethyl fumarate in relapsing‐remitting multiple sclerosis: integrated analysis of the phase 3 trials. Ann Clin Transl Neurol 2015; 2: 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Polman CH, Reingold SC, Edan G, et al Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 2005; 58: 840–846. [DOI] [PubMed] [Google Scholar]
- 12. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444–1452. [DOI] [PubMed] [Google Scholar]
- 13. Okwuokenye M. Discrete time‐to‐event and score‐based methods with application to composite endpoint for assessing evidence of disease activity‐free. Presented at: Biopharmaceutical Applied Statistics Symposium XXII; 2 November 2015; Washington, DC.
- 14. Prentice R, Gloeckler L. Regression analysis of grouped survival data with application to breast cancer data. Biometrics 1978; 34: 57–67. [PubMed] [Google Scholar]
- 15. Tecfidera (dimethyl fumarate) [prescribing information]. Cambridge, MA: Biogen, 2015. [Google Scholar]
- 16. Rotstein DL, Healy BC, Malik MT, Chitnis T, Weiner HL. Evaluation of no evidence of disease activity in a 7‐year longitudinal multiple sclerosis cohort. JAMA Neurol 2015; 72: 152–158. [DOI] [PubMed] [Google Scholar]
- 17. Theodore Phillips J, Erwin AA, Agrella S, et al Consensus management of gastrointestinal events associated with delayed‐release dimethyl fumarate: a delphi study. Neurol Ther 2015; 4: 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bevan CJ, Cree BA. Disease activity free status: a new end point for a new era in multiple sclerosis clinical research? JAMA Neurol 2014; 71: 269–270. [DOI] [PubMed] [Google Scholar]
- 19. Kappos L, De Stefano N, Freedman MS, et al Inclusion of brain volume loss in a revised measure of ‘no evidence of disease activity’ (NEDA‐4) in relapsing‐remitting multiple sclerosis. Mult Scler 2016; 22: 1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Percentage of patients treated with delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) with clinical no evidence of disease activity (NEDA) [no measured clinical activity was defined as having no relapses and no 12‐week confirmed disability progression as measured by the Expanded Disability Status Scale (EDSS)] at 6 months, 1 and 2 years of treatment in (a) DEFINE, (b) CONFIRM and (c) the integrated analysis (sensitivity analysis, intention‐to‐treat population). Odds ratios are shown with 95% confidence intervals in parentheses; P values for comparisons versus placebo are from a logistic regression model adjusted for baseline EDSS score, age (≤40, >40 years), region and number of relapses in the year before study entry.
Figure S2. Percentage of patients treated with delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) with neuroradiological no evidence of disease activity (NEDA) [no measured brain magnetic resonance imaging (MRI) disease activity was defined as no new/newly enlarging T2 hyperintense lesions and no gadolinium‐enhancing (Gd+) lesions] at 6 months, 1 and 2 years of treatment in (a) DEFINE, (b) CONFIRM and (c) the integrated analysis (sensitivity analysis, MRI cohort). Odds ratios are shown with 95% confidence intervals in parentheses; P values for comparisons versus placebo are from a logistic regression model adjusted for region, baseline volume of T2 hyperintense lesions and baseline number of Gd+ lesions.
Figure S3. Percentage of patients treated with delayed‐release dimethyl fumarate (DMF) (adelayed‐release dimethyl fumarate, also known as gastro‐resistant DMF) with overall no evidence of disease activity (NEDA) [no measured overall disease activity was defined as having no measured clinical and no measured brain magnetic resonance imaging (MRI) disease activity] at 6 months, 1 and 2 years of treatment in (a) DEFINE, (b) CONFIRM and (c) the integrated analysis (sensitivity analysis, MRI cohort). Odds ratios are shown with 95% confidence intervals in parentheses; P values for comparisons versus placebo are from a logistic regression model adjusted for baseline Expanded Disability Status Scale score, baseline age (≤40, >40 years), study, region, number of relapses in the year before study entry, baseline T2 hyperintense lesion volume and baseline number of gadolinium‐enhancing lesions.