

Abstract
5‐Hydroxymethylfurfural (HMF) is a versatile intermediate in biomass conversion pathways. However, the notoriously unstable nature of HMF imposes challenges to design selective routes to chemicals such as furan‐2,5‐dicarboxylic acid (FDCA). Here, a new strategy for obtaining furans is presented, bypassing the formation of the unstable HMF. Instead of starting with glucose/fructose and thus forming HMF as an intermediate, the new route starts from uronic acids, which are abundantly present in many agro residues such as sugar beet pulp, potato pulp, and citrus peels. Conversion of uronic acids, via ketoaldonic acids, to the intermediate formylfuroic acid (FFA) esters, and subsequently to FDCA esters, proceeds without formation of levulinic acid or insoluble humins. This new route provides an attractive strategy to valorize agricultural waste streams and a route to furanic building blocks without the co‐production of levulinic acid or humins.
Keywords: furandicarboxylic acid, isomerization, levulinic acid, oxidation, uronic acids
Introduction
One of the most promising examples of biobased building blocks is furan‐2,5‐dicarboxylic acid (FDCA), which has many potential applications in polyesters, polyamides, and plasticizers.1 The majority of research for the production of FDCA focusses on the use of hexose sugars (i.e., d‐glucose and d‐fructose), which are first dehydrated to the intermediate 5‐hydroxymethylfurfural (HMF),2 followed by oxidation to FDCA (Scheme 1). Currently, the main challenge for the efficient synthesis of furans from sugars is the intrinsic instability of HMF. Under the acidic aqueous conditions applied, HMF is easily rehydrated to form levulinic acid and formic acid.2, 3 Furthermore, insoluble humins can be formed in high amounts.4 All these side reactions do not only lead to loss of the desired HMF, but the valorization of levulinic acid and humin side products is required to make the overall route economically viable.5 Alternative routes have been developed, in which more stable HMF ethers are formed.6 However, humin formation still occurs, and alkyl levulinates are formed as byproducts.5
Scheme 1.
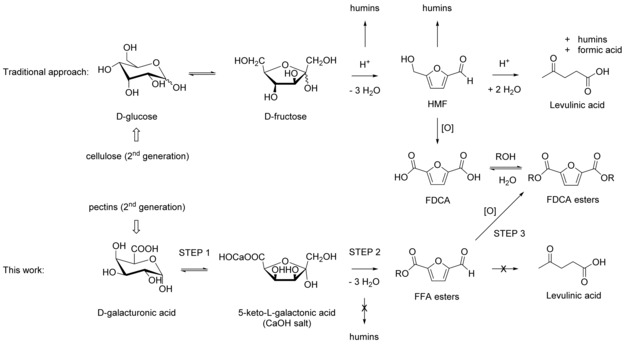
Comparison of the synthesis of FDCA esters using the traditional approach starting from glucose with the alternative route starting from uronic acids.
A second disadvantage of using HMF as an intermediate is related to the feedstock used to make the HMF. Currently, the feedstock is made from glucose and/or fructose, which both interfere with food production. For example, commercial d‐fructose resources are high‐fructose syrups, which are generally produced from starch‐containing food crops such as corn.5 To eliminate the conflict with food production, a route to FDCA should be developed starting from nonfood lignocellulosic feedstocks such as wood or grasses, or preferably from the residues of agro‐food production. This prompted us to explore the potential of obtaining FDCA esters through an alternative approach, not starting from glucose/fructose and circumventing the intrinsic instability of HMF.
We propose that 2‐formyl‐5‐furoic acid esters (FFA esters) can be used as alternatives to HMF (Scheme 1). We hypothesize that, owing to the lack of primary alcohol functional groups in FFA esters, they are more stable than HMF because acidic dehydration pathways and subsequent formation of levulinic acid are blocked.2, 3 Catalytic oxidation of FFA esters would yield the desired FDCA esters.
Scheme 1 shows an overview of the new proposed route to FDCA esters. If FFA esters are the desired intermediates towards FDCA esters, a higher oxidation state of the carbohydrate starting materials is also required. 5‐Keto‐aldonic acids (5‐KAs) are the retrosynthetic precursor for FFA esters (analogous to the dehydration of d‐fructose to HMF). Uronic acids would then be the retrosynthetic precursors for 5‐KAs (analogous to the isomerization of d‐glucose to d‐fructose). Both FDCA and FDCA esters can be used in polyester synthesis (currently the largest potential application area of FDCA), analogous to the use of terephthalic acid and dimethyl‐terephthalate, which are both used in the industrial synthesis of polyethylene terephthalate (PET).7
The proposed route is also implementable on an industrial scale. Uronic acids are present in various large agro‐residue streams. d‐Galacturonic acid is the most abundant uronic acid and is the main constituent of pectin. Pectin is found in large amounts in sugar beet pulp (contains 900 000 t d‐galacturonic acid worldwide),8 chicory pulp,9 fruit peels such as citrus peels (375 000 t d‐galacturonic acid worldwide),10 potato peels/pulp (200 000 t d‐galacturonic acid in the USA11; 7100 t in the UK),12 and coffee pulp (ca. 30 000 t d‐galacturonic acid worldwide).13 As an example, efforts are being undertaken to scale up the commercial production of d‐galacturonic acid from sugar beet pulp.14
d‐Glucuronic acid is the second important readily available uronic acid, found in xanthan gum,15 gum arabic,16 and wheat bran,17 and is one of the main pectin constituents in various types of soft‐ and hardwoods.18 Three other uronic acids are present in nature, for example, d‐mannuronic acid, l‐guluronic acid, and l‐iduronic acid in macroalgae.15, 19 However, these have to be obtained by hydrolysis of alginates, which is currently still challenging. Therefore, these uronic acids are not yet readily available.20
Overall, there is already a significant stream of non‐edible uronic acids available (>1.5 Mt d‐galacturonic acid alone), which can serve as 2nd generation nonfood‐based starting materials for furans such as FDCA. Here, we present a new catalytic 3‐step route to 2nd generation FDCA: 1) Isomerization of uronic acids to their corresponding 5‐KAs. 2) Acid‐catalyzed cyclodehydration to FFA esters. 3) Oxidation to FDCA esters.
Results and Discussion
Isomerization (step 1)
As discussed above, the first step in our proposed process is the isomerization of uronic acids to their corresponding 5‐KAs (step 1 in Scheme 1). This reaction can be performed by different microorganisms and enzymes,21 as has been shown for the conversion of d‐galacturonic acid to 5‐keto‐l‐galactonic acid (IUPAC name: d‐arabino‐hex‐5‐ulosonic acid).22 In addition, chemical isomerization of uronic acids to 5‐KAs in an alkaline medium has also been reported.23 This is the so‐called Lobry de Bruyn–van Ekenstein transformation.24 Unfortunately, these isomerization methods result in equilibrium mixtures that contain only a limited fraction of the desired product (e.g., d‐galacturonic acid/5‐keto‐l‐galactonic acid=34:66).21c To shift the equilibrium to the desired products, we adapted a procedure reported by Ehrlich and Guttmann,25 who showed that 5‐keto‐l‐galactonic acid could be selectively precipitated by Ca2+. These authors used poorly soluble Ca(OH)2, which has the disadvantage of working in very dilute reaction mixtures. Here, we used a more soluble calcium precursor (CaCl2), which allowed us to use more concentrated solutions. This resulted in an increase of the yield of 5‐keto‐l‐galactonic acid from 46 % using Ca(OH)2 to 82 % using CaCl2.
The second important uronic acid, d‐glucuronic acid, was isomerized in the presence of CaCl2 to afford 5‐keto‐d‐mannonic acid (IUPAC name: d‐lyxo‐Hex‐5‐ulosonic acid) in 42 % isolated yield after filtration of the reaction mixture. Thin layer chromatography (TLC) analysis of the filtrate revealed the presence of residual product, apparently caused by the higher aqueous solubility of 5‐keto‐d‐mannonic acid. The use of CaCl2 has the advantage of selectively obtaining one of the isomers. Further optimization is needed to maximize the yield.
Out of the five naturally occurring uronic acids, only three different 5‐KAs can be prepared (for a detailed explanation, see Figure S1 in the Supporting Information). Two of those 5‐KAs were prepared as discussed above. The third one, 5‐keto‐d‐gluconic acid, (IUPAC name: d‐xylo‐Hex‐5‐ulosonic acid) is commercially available because it is one of the intermediates in the production of vitamin C through fermentation.26 Therefore, all three possible 5‐KAs are available for the second step, the cyclodehydration to FFA esters.
Cyclodehydration (step 2)
Keto‐aldonic acid versus uronic acid
The second step in our process is the cyclodehydration of 5‐KAs to FFA esters (step 2 in Scheme 1). Table 1 displays an overview of the yield of the desired Me‐FFA starting from the three 5‐KAs (entries 1–4) and uronic acids (entries 5–7). 5‐Keto‐d‐gluconic acid (K salt) was reacted in methanolic HCl to afford the expected Me‐FFA in 43 % crude yield (Table 1, entry 1), thereby confirming the previous results reported by Votocek and Malachta (40 % crude yield).27 The same conditions were applied to 5‐keto‐l‐galactonic acid (CaOH salt), which afforded Me‐FFA in 50–65 % crude yield (Table 1, entries 2 and 3). This yield is significantly higher than previously reported yields (11 % after purification) by Stutz and Deuel,28 and more in line with the results obtained with 5‐keto‐d‐gluconic acid (K salt). Also, 5‐keto‐d‐mannonic acid (mixed Ca/Na salt; Table 1, entry 4) could be converted to the desired product, although in a somewhat lower yield (17 %). We did not study the influence of the cation (K/ Ca). These initial results show that the conversion of all three 5‐KAs to FFA‐esters is possible.
Table 1.
Cyclodehydration of various 5‐keto‐aldonic acids and uronic acid derivatives in MeOH/HCl.[a]
| Entry | Substrate | Reaction time [h] | Crude Me‐FFA yield[b] [mol %] |
|---|---|---|---|
| 1 | 5‐keto‐d‐gluconic acid (K salt) | 72 | 43 |
| 2 | 5‐keto‐l‐galactonic acid (CaOH salt) | 72 | 50 |
| 3 | 5‐keto‐l‐galactonic acid (CaOH salt) | 24 | 65 |
| 4 | 5‐keto‐d‐mannonic acid (Ca/Na salt) | 24 | 17 |
| 5 | d‐galacturonic acid (monohydrate) | 24 | 0 |
| 6 | d‐galacturonic acid (Na salt) | 24 | <1 |
| 7 | d‐glucuronic acid | 24 | <1 |
[a] Conditions: substrate (1.0 g) in 3n MeOH/HCl (10 mL), 65 °C. [b] Isolated yield of the product after extraction without purification (purity≈90–100 % based on GC and NMR analyses).
To verify if the isomerization of uronic acids to 5‐KAs (step 1 in Scheme 1) is a necessary step; therefore, the conversion of uronic acids was investigated under the same reaction conditions. No Me‐FFA was formed starting from d‐galacturonic acid, the parent uronic acid of 5‐keto‐l‐galactonic acid [Table 1, entries 5 (monohydrate form) and 6 (Na salt)]. Instead of Me‐FFA, methyl (methyl galactosid)uronates were the only detectable products (Figure S4). Similar results were obtained when starting from d‐glucuronic acid, the parent uronic acid of 5‐keto‐d‐mannonic acid, that is, no yield of Me‐FFA was observed (Table 1, entry 7).
To summarize, the (unoptimized) conversion of all three possible 5‐KAs afforded Me‐FFA in reasonable yields (17–65 %). However, the parent uronic acids furnished <1 % Me‐FFA under the same reaction conditions, showing that the isomerization of the aldose to the ketose species is required to achieve an effective cyclodehydration.
Clearly, the ring conformation of the carbohydrate has a marked influence on the Me‐FFA yield. The uronic acids exist mainly in the six‐membered ring pyranose form in water (d‐galacturonic acid: ca. 90 %).29 This prevents direct cyclodehydration to five‐membered furans, instead leading to undesired degradation.30 However, the related 5‐keto‐l‐galactonic acid adapts a furanose conformation (five‐membered ring) almost exclusively (>99 %).31 The other two 5‐KAs also prefer a five‐membered ring conformation (>99 % for 5‐keto‐d‐mannonic acid32 and 80–89 % for 5‐keto‐d‐gluconic acid). The remainder is in the open chain or lactone form32, 33. The effect of the ring conformation on reactivity has been observed before for the conversion of glucose and fructose to HMF. The highest yields of HMF were achieved starting from d‐fructose, which has a higher preference for the furanose form (five‐membered ring) in aqueous solution (ca. 25 %), whereas only 1 % of d‐glucose is in the furanose form.34 Although still under debate, it is thought that owing to its furanose configuration, d‐fructose dehydrates more efficiently to HMF, which is also a five‐membered ring.2, 35 It is also believed that the fructofuranose form results in a decrease in competitive pathways during cyclodehydration.36 Here, we found a similar effect for the conversion of uronic acids (six‐membered ring) versus KAs (five‐membered ring).
Influence of the nature of the acid
The experiments reported in Table 1 were all performed in methanolic HCl. However, for practical reasons, we wanted to switch to acids that are less corrosive, less volatile, and easier to dose. The screening of such acids [H2SO4, H3PO4, methane sulfonic acid (MSA), and p‐toluenesulfonic acid (p‐TSA)] showed that the best initial results were obtained using MSA (6–12 equiv.). The next part of this investigation discusses the most important reaction parameters.
Table 2 shows an overview of all results of the conversion of 5‐keto‐l‐galactonic acid analogues under varying reaction conditions. The following sub‐sections will discuss the most important factors including 5‐KA versus fructose conversion, the stability of the intermediates, mass balance, the influence of water, and the influence of solvent. All products used in subsequent experiments were isolated and purified using silica column chromatography to facilitate fair comparisons.
Table 2.
Cyclodehydration of 5‐keto‐l‐galactonic acid (analogues) (5‐KG) and product stability in various alcohols.[a]
| Entry | Substrate | Alcohol | Water removal[b] | t [h] | T [°C] | Intended product | Purified yield[c] [mol %] |
|---|---|---|---|---|---|---|---|
| 1 | 5‐KG.CaOH | MeOH | no | 24 | 65 | Me‐FFA | 49 |
| 2 | 5‐KG.CaOH | MeOH | yes | 24 | 65 | Me‐FFA | 48 |
| 3 | 5‐KG.CaOH | MeOH | +6 equiv. H2O | 24 | 65 | Me‐FFA | 38 |
| 4 | Me‐FFA | MeOH | no | 24 | 65 | Me‐FFA | 78 |
| 5 | HMF | MeOH | no | 24 | 65 | HMF | <2[d] |
| 6 | d‐Fructose | MeOH | no | 24 | 65 | HMF | <1[d] |
| 7 | 5‐KG.CaOH | EtOH | no | 18 | 78 | Et‐FFA | 50 |
| 8 | 5‐KG.CaOH | EtOH | yes | 18 | 78 | Et‐FFA | 55 |
| 9 | 5‐KG.CaOH | n‐PrOH | no | 24 | 97 | Pr‐FFA | 43 |
| 10 | 5‐KG.CaOH | n‐PrOH | yes | 24 | 97 | Pr‐FFA | 45 |
[a] Conditions: substrate (12 mmol) in MeOH (100 mL), and of MSA (12 equiv.). [b] Active water removal through Soxhlet setup (yes/no) or deliberate addition of water. [c] Isolated yield of product after silica gel chromatography (purity >99 %). [d] Me‐levulinate was isolated as the main product (entry 5: 35 mol %; entry 6: 39 mol %) and significant formation of insoluble humins was observed.
1. ‐Keto‐aldonic acid versus fructose conversion
To determine the advantage of the FFA esters versus the HMF route with respect to the formation of products and side products, we compared the conversion of 5‐keto‐l‐galactonic acid (Table 2, entry 1) with the conversion of d‐fructose (Table 2, entry 6). Starting from 5‐keto‐l‐galactonic acid, Me‐FFA was isolated in 49 % purified yield after flash column chromatography (Table 2, entry 1). The crude product already had good purity (>90 %); no levulinic acid (ester) was present, and the amount of insoluble humins in the reaction mixture was very limited (<5 wt %, too little to isolate during workup). This was in contrast to a reaction starting from d‐fructose that afforded Me‐levulinate and humins as the main products instead of the desired HMF (Table 2, entry 6).
Stability of the intermediates
These results prompted us to investigate if the stability of Me‐FFA and HMF under our conditions could explain the differences in product yield. Me‐FFA was stirred for 24 h in MeOH/MSA under reflux to afford a clear, light‐yellow reaction mixture. After workup, Me‐FFA was recovered in 78 % yield (Table 2, entry 4). For comparison, HMF was exposed to the same conditions (Table 2, entry 5). The HMF reaction mixture turned black within 30 min, and after workup (after 24 h) no HMF was recovered. The main products were Me‐levulinate (35 mol %) and insoluble humins. Figure 1 shows a picture of both reaction mixtures to illustrate the difference in stability further. Hence, the presence of substituents with a high oxidation state (aldehyde and carboxylic acid) on the FFA furan ring significantly reduced the propensity for acid‐catalyzed hydration and subsequent formation of levulinic acid and humins compared to HMF.2
Figure 1.

Appearance of reaction mixtures during stability tests. Left: Me‐FFA (after 24 h reaction time). Right: HMF (after 30 min reaction time). Reaction conditions: Substrate (12 mmol), methanol (100 mL), MSA (12 equiv.), reflux.
Mass balance
Although we did not observe any levulinic acid formation from the 5‐KAs (by NMR, GC–MS, or HPLC analysis), the isolated yields were less than 100 % at full conversion. This indicates that side products were formed. To identify these products and obtain insight into the competitive reaction pathways, alternative (non‐aqueous) work‐up procedures of the crude reaction mixtures were applied (Figures S5–S7).37, 38 NMR and HPLC–MS analyses indicated that, apart from the desired FFA esters, the reaction mixture consisted of a complex mixture of glycosides. These glycosides can be further valorized, for example, by transacetalization with higher alcohols to produce surfactants. A full characterization of this complex mixture falls outside of the scope of the present study but will be part of future work.
Influence of water
Because water is inevitably formed as a co‐product during the cyclodehydration, we investigated the influence of water on the conversion, Me‐FFA selectivity, and humin formation. From 1 mol 5‐KA (CaOH salt) a maximum of 6 mol water is formed (step 2 in Scheme 1). We investigated the influence of water on the cyclodehydration in two ways: First, by deliberately adding water to the reaction mixture, and second, by actively removing water during the reaction (Table 2, entries 1–3).
The addition of 6 equiv. of water to the substrate resulted in a decrease in the Me‐FFA yield from 49 to 38 % (Table 2, entries 1 and 3). Moreover, visual inspection of the reaction mixture indicated that the amount of insoluble humins also increased (visual observation only because accurate gravimetric analysis was hampered by the presence of salts). Although water and methanol do not form an azeotrope, we tried to actively remove water by applying a Soxhlet setup filled with 3 Å molecular sieves. However, the purified yield of Me‐FFA did not change (48–49 %, Table 2, entries 1 and 2) in methanol.
Influence of solvent
We investigated the influence of removing water by using two higher alcohols, ethanol and propanol. Because these alcohols do form azeotropes with water, the water removal was expected to be more efficient in the Soxhlet setup. Another effect of changing to other solvents under reflux was the increased reaction temperature.
5‐Keto‐l‐galactonic acid (CaOH salt) was reacted in ethanol with 12 equiv. of MSA for 18 h, with and without active water removal (Table 2, entries 7 and 8). The yields of purified Et‐FFA (50–55 %) of both reactions were slightly higher compared to the reactions in methanol (48–49 % Me‐FFA, Table 2, entries 1 and 2), indicating an advantageous effect of temperature. The increased yield during active water removal in ethanol indicates the detrimental effect of water on the selectivity of the dehydration reaction.
To further investigate the effect of solvent on the conversion of 5‐keto‐l‐galactonic acid, 1‐propanol was used. Without active water removal, Pr‐FFA was isolated in 43 % purified yield (Table 2, entry 9). Removal of the water slightly increased the purified yield of Pr‐FFA to 45 % (Table 2, entry 10).
The cyclodehydration of all 5‐KAs afforded the desired FFA esters in moderate‐to‐good isolated yields (17–55 %). The best yields were obtained in ethanol with continuous removal of water. Although further optimization is required, the isolated yield after column chromatography (55 %) was already quite high and competitive with HMF yields starting from glucose (realistic process yields are approximately 55 %).39
Oxidation (step 3)
The final step in the new route to FDCA was the catalytic oxidation of FFA esters to FDCA esters (step 3 in Scheme 1). We investigated the effectiveness of oxidation using Au/C, a base, and O2 at RT. The Au/C catalyst contained 1.3 wt % Au with an average Au particle size of 2.2 nm, and was prepared through cationic adsorption (Figure S3). The FFA esters were oxidized according to a previously reported procedure.40 Me‐FFA and Et‐FFA were oxidized in methanol, using 10 mol % NaOMe as a base. The reactions were performed at RT using a 1:344 Au/substrate ratio, and 5 bar O2 overpressure. Both Me‐FFA and Et‐FFA gave full conversion to FDCA dimethyl ester in >99% selectivity within 22 h.
The main advantage of FFA ester oxidation compared to the oxidation of HMF is that the oxidation of the primary alcohol of HMF is often the most challenging.41 This primary alcohol is not present in FFA esters, and only the more reactive aldehyde functionality needs to be oxidized, which can be performed under very mild conditions.
Conclusions
In conclusion, we have shown that abundantly available uronic acids (potentially >1.5 Mt a−1 from 2nd generation nonfood feedstock) can be converted into furan‐2,5‐dicarboxylic acid (FDCA) dimethyl ester through a new 3‐step catalytic route, in an overall unoptimized isolated yield of 45 %, which is competitive with routes starting from glucose. In the first step, uronic acids are isomerized in alkaline water in the presence of Ca2+, leading to the selective precipitation of 5‐keto‐aldonic acids (5‐KAs) at RT. In the second step, 5‐KAs were converted into 2‐formyl‐5‐furoic acid (FFA) esters through a mild cyclodehydration in alcoholic solvents in the presence of an acid catalyst. Control experiments showed that uronic acids did not afford FFA esters as products, proving the necessity of the isomerization step. During the conversion of 5‐KA to FFA esters, no levulinic acid and only a limited amount of insoluble humins were formed. In the third and final step, FFA esters were converted to FDCA dimethyl ester through a mild Au‐catalyzed oxidation reaction at RT, using only O2 as the oxidant. Hereby, we demonstrated that this new route from agro‐residue derived feedstocks such as sugar beet pulp or citrus peels is an attractive strategy for the production of 2nd generation FDCA and its derivatives.
Experimental Section
Materials
The following chemicals, solvents, and gasses were used without further purification: acetic acid (100 %, GPR RECTAPUR, VWR Chemicals); activated carbon (SX1G, Norit); ammonia solution (ACS reagent, Sigma–Aldrich); ammonium nitrate (>99.0 %, Sigma–Aldrich); calcium chloride (dehydrated, purum, >97 %, Sigma–Aldrich); calcium oxide (fine powder, puriss., Riedel‐de Haën); Celite 545 (Sigma–Aldrich); chloroform (HPLC, stabilized with ethanol, min. 99.9 %, Actu‐All Chemicals); diethyl ether, (for analysis, Merck); ethanol (absolute, 99.9 %, VWR Chemicals); ethyl acetate (pure, Acros Organics); d‐(−)‐fructose (>99.0 %, Merck); d‐galacturonic acid monohydrate (>99 %, gift from Royal Cosun), d‐galacturonic acid sodium salt monohydrate (purum >98.0 %, Fluka); d‐glucuronic acid (>98 %, Sigma–Aldrich); gold(III)chloride trihydrate (ACS reagent, Sigma–Aldrich); 5‐(hydroxymethyl)furfural (99 %, Sigma–Aldrich); 5‐keto‐d‐gluconic acid potassium salt (98 %, International Laboratory USA); magnesium sulfate (dried, with ≈1–2 mol water of hydration, Alfa Aesar); methanesulfonic acid (>99.5 % Sigma–Aldrich); methanol (for analysis, Merck); methanolic HCl, (3 n, Supleco); molecular sieves (3 Å, beads, 8–12 mesh, Aldrich); oxygen (5.0, Linde Gas Benelux); nitric acid (p.a., 65 %, Sigma–Aldrich); petroleum ether (ACS reagent, boiling range 40–60 °C, Acros Organics); phosphoric acid (>85 %, p.a., Sigma–Aldrich); 1‐propanol (ACS reagent, >99.5 %, Sigma–Aldrich); pyridine (ACS reagent, 99.0 %, Sigma–Aldrich); Sicapent (with indicator, Merck); silica gel 60 (0.040–0.063 mm, 230–400 mesh, Alfa Aesar); sodium hydrogen carbonate (99 %, Alfa Aesar); sodium hydroxide (pellets, reagent grade, >98 %, anhydrous, Sigma–Aldrich); sodium methoxide (Sigma–Aldrich); sulfuric acid (98 %, p.a., Merck), p‐toluenesulfonic acid monohydrate (98.5 %, ACS reagent, Sigma–Aldrich).
Analytical Equipment
TLC was performed using Silica gel 60 F254 plates (500 Glass plates, 2.5×7.5 cm, Merck). All carbohydrates were analyzed using the following procedure. Mobile phase: ethyl acetate/pyridine/water/acetic acid 5:5:1:3. An aqueous solution of the compound was applied on the plate. A few drops of HCl were added to calcium salts that do not dissolve in pure water. The TLC plate was developed, dried, developed a second time, dried again, and stained with 5 % sulfuric acid in ethanol. Heating with a heat gun showed the compounds as black dots. FFA esters/dimethyl‐FDCA: Mobile phase: ethyl acetate/petroleum ether 1:9. TLC plates were developed and dried, and the products were detected under UV light (254 nm). HMF and methyl‐levulinate: Mobile phase: petroleum ether/ethyl acetate 7:3. The plate was colored using a permanganate stain followed by gentle heating.
GC–MS analyses were performed using an Interscience TraceGC Ultra GC with an AS3000 II autosampler. Products were separated on a Restek GC column (Rxi‐5 ms 30 m×0.25 mm×0.25 μm). Helium was used as the carrier gas with a flow of 1 mL min−1, and a split flow of 20 mL min−1. Temperature program: hold 3 min at 50 °C, ramp 7.5 °Cmin−1, final temperature 330 °C. An Interscience TraceDSQ II XL quadrupole mass selective detector (EI, mass range 35–500 Dalton, 150 ms sample speed) was used for detection.
NMR spectra were recorded on a Bruker Advance III spectrometer operating at 400.17 MHz (1H) and 100.62 MHz (13C) in CDCl3 (99.9 at % D, Aldrich), [D6]DMSO [99.5 at % D, containing 0.03 % v/v trimethylsilane (TMS), Aldrich], MeOD‐D4 (≥99.8 at % D, containing 0.03 % v/v TMS, Aldrich) or D2O (99.9 at % D, Aldrich). D2O solutions were adjusted to the desired pH value with DCl (35 wt % solution in D2O, 99 atom % D, Aldrich) or NaOD (40 wt % solution in D2O, 99+at % D, Aldrich). Chemical shifts are quoted in parts per million (ppm) referenced to the appropriate solvent peaks or 0 ppm for TMS.
Elemental analysis (carbon, hydrogen, calcium and sodium) was measured by Mikroanalytisches Laboratorium KOLBE, Mülheim an der Ruhr, Germany.
TEM and high‐angle annular dark‐field imaging (HAADF) measurements were performed with a Philips Tecnai 20 FEG instrument, operating at 200 kV. The catalyst was used without further treatment and was dispersed in ethanol followed by deposition on a carbon‐coated copper grid. The particle size distribution was determined by measuring the dimensions of 913 particles using Image‐J software.
XRD patterns were recorded using a Philips PC‐APD diffractometer with a CuKα1 anode operating at 40 mA and 40 kV, and a 15 mm Ni foil monochromator. The patterns were recorded between 2 θ=4 and 50° using a step‐size of 0.02° and a counting time of 5 s.
Literature procedure for the preparation of 5‐keto‐l‐galactonic acid mono‐basic calcium salt25
Calcium oxide (11.18 g, 0.199 mol) was suspended in demineralized water (10 L) and stirred for 10 min in an open beaker. A small amount of undissolved particles was removed by filtration, yielding a colorless clear solution. d‐galacturonic acid monohydrate (40.01 g, 0.198 mol) was then added under stirring to give a clear, slightly yellow solution. After three days at RT, the formed crystals were collected by suction filtration. The initially transparent crystals were dried to a constant weight under vacuum at 40 °C in the presence of Sicapent. The resulting 5‐keto‐l‐galactonic acid mono‐basic calcium salt was obtained as a bright yellow powder (22.9 g, yield 46 mol %). TLC (ethyl acetate/pyridine/water/acetic acid 5:5:1:3): 5‐Keto‐l‐galactonic acid gave a single spot (R f 0.39), whereas the starting d‐galactonic acid gave a single spot at a lower R f value (0.32). 13C NMR (100.62 MHz, D2O): δ=62.44 (6β), 63.15 (6α), 66.78 (6γ), 70.43 (4β), 71.55 (4γ), 71.87 (3γ), 72.55 (3β), 72.66 (3α), 73.10 (2γ), 78.09 (4α), 80.08 (2α), 80.88 (2β), 103.52 (5β), 105.68 (5α), 175.26 (1α), 175.53 (1β), 178.41 (1γ), 213.16 ppm (5γ). The NMR spectrum was in agreement with previously reported values.31 Elemental analysis calcd (%) for C6H10CaO8 (250.22): C 28.8, H 4.03, Ca 16.02, O 51.15; found: C 29.9, H 3.27, Ca 16.49, Na 0.15, O 50.0; XRD: see Figure S2
Improved method for the preparation of 5‐keto‐l‐galactonic acid mono‐basic calcium salt
Calcium chloride (52.4 g, 0.472 mol) and d‐galacturonic acid monohydrate (100 g, 0.471 mol) were dissolved in demineralized water (1.6 L) and stirred for 15 min in a 2.5 L beaker. A small amount of undissolved particles was removed by filtration, yielding a slightly yellow clear solution. A solution of sodium hydroxide (38 g, 0.95 mol) in water (200 mL) was added dropwise over 30 min under continuous stirring. After the addition of approximately 50 % of the sodium hydroxide solution, the solution became more yellow, and solids appeared. After complete addition of the sodium hydroxide, the mixture was stirred for an additional 6 h at RT. The solid material was collected by suction filtration, washed with water, and dried to a constant weight under vacuum at 40 °C over Sicapent. The resulting 5‐keto‐l‐galactonic acid mono‐basic calcium salt was obtained as a bright yellow powder (96.7 g, yield 82 mol %). TLC (ethyl acetate/pyridine/water/acetic acid 5:5:1:3) was identical to the material prepared through the literature procedure (see above). Elemental analysis calcd (%) for C6H10CaO8 (250.22): C 28.8, H 4.03, Ca 16.02, O 51.15; found: C 30.1, H 3.17, Ca 16.53, Na 0.2, O 50.14; XRD: in agreement with the material prepared using the literature procedure (see Figure S2)
Preparation of 5‐keto‐d‐mannonic acid
Calcium chloride (7.18 g, 64.7 mmol) and d‐glucuronic acid monohydrate (15 g, 64.7 mmol) were dissolved in demineralized water (240 mL) and stirred for 15 min in a 500 mL beaker to give a clear slightly yellow solution. A solution of sodium hydroxide (2.59 g, 64.7 mmol) in water (30 mL) was added dropwise over 15 min under continuous stirring. White solids started to appear after the addition of the first drops of sodium hydroxide. After complete addition of the sodium hydroxide, the milky mixture was stirred for three days at RT. The solid material was collected by suction filtration, washed with water, and dried to a constant weight under vacuum at 40 °C over Sicapent. The resulting 5‐keto‐d‐mannonic acid was obtained as a bright yellow powder (6.82 g, yield≈17 mol %). TLC (ethyl acetate/pyridine/water/acetic acid 5:5:1:3): Both the precipitated 5‐Keto‐d‐mannonic acid and the filtrate gave single spots (R f 0.53), whereas the starting d‐glucuronic acid gave a single spot at a lower R f value (0.38). 13C NMR (100.62 MHz, CDCl3): δ=62.91 (6β), 62.93 (6α), 75.90 (4α), 78.00 (4α), 79.58 (4β), 80.61 (2α), 81.31 (3β), 81.72 (2β), 102.45 (5α), 105.33 (5β), 177.78 (1β), 178.42 (1α). The NMR (of the Na form after ion exchange) is in agreement with previously reported values.32 Elemental analysis found (%) for a mix of C6H10CaO8 (250.22) and C6H9NaO7 (216.12): C 26.2, H 3.4, Ca 12.4, Na 2.8, O 55.2; XRD: see Figure S2
General literature procedure for the cyclodehydration of various 5‐keto‐aldonic acids and uronic acid derivatives in MeOH/HCl to Me‐FFA28a
The following general procedure was used for the experiments reported in Table 1: Round bottom flasks (50 mL) were equipped with magnetic stirring bars and reflux condensers with a N2 inlet. The flasks were charged with a substrate (4 mmol) and methanolic HCl (3n solution, 10 mL). The substrates dissolved rapidly to give clear colorless solutions. The reaction mixtures were heated to reflux (65 °C) for 24–72 h. The resulting clear red–brown solutions were allowed to cool to RT, and then demineralized water (10 mL) was added under stirring. After 45 min the aqueous mixtures were extracted with diethyl ether (3×20 mL). The combined organic layers were then washed with brine until the pH value of the water layer was neutral. The organic layers were dried over MgSO4, filtered, and concentrated at 40 °C under vacuum using a rotary evaporator. The resulting methyl 5‐formyl‐2‐furoate was obtained as a red–brown oil. This red–brown oil tended to crystallize within a few minutes after cooling to RT. The products were analyzed by 1H/13C NMR spectroscopy and GC–MS.
Most crude products consisted of a mixture of the free aldehyde (major product) and the dimethyl acetal (minor product) owing to incomplete hydrolysis of the aldehyde dimethyl acetals under these workup conditions. For initial identification, the products were separated by silica gel column chromatography (petroleum ether/ethyl acetate 8:2) to give the pure compounds as slightly yellow crystalline solids.
Methyl 5‐formyl‐2‐furoate (free aldehyde): TLC (ethyl acetate/petroleum ether 1:9): R f (0.19); 1H NMR (400.17 MHz, CDCl3): δ=9.82 (1 H, s), 7.29–7.26 (2 H, m), 3.96 ppm (3 H, s); 13C NMR (100.62 MHz, CDCl3): δ=52.54, 118.63, 118.75, 147.62, 153.87, 158.38, 178.92 ppm. 1H NMR (400.17 MHz, [D6]DMSO): δ=9.75 (1 H, s), 7.62 (1 H, d, J=3.8 Hz), 7.49 (1 H, d, J=3.8 Hz), 3.88 ppm (3 H, s); MS (GC–MS, 70 eV): m/z (%)=154 (55) [M +], 123 (100), 109 (2), 95 (24), 67 (6), 53 (5), 39 (23). NMR (CDCl3) spectroscopy and MS corresponded to the values reported by Khusnutdinov et al.42 and the NMR in [D6]DMSO corresponded to the values reported by Shackelford et al.43 (the two furan protons overlap in deuterated chloroform, whereas they give two separate signals in deuterated DMSO)
Methyl 5‐formyl‐2‐furoate (dimethyl acetal): TLC (ethyl acetate/petroleum ether 1:9): R f (0.38); 1H NMR (400.17 MHz, CDCl3): δ=7.16 (1 H, d, J=3.5 Hz), 6.54 (1 H, d, J=3.5 Hz), 5.46 (1 H, s), 3.89 (3 H, s), 3.37 ppm (6 H, s). 13C NMR (100.62 MHz, CDCl3): δ=51.84, 53.02, 97.52, 110.32, 118.33, 144.27, 155.03, 158.95 ppm. MS (GC–MS, 70 eV): m/z (%)=200 (5) [M +], 169 (100), 153 (4), 141 (10), 123 (20), 109 (3), 95 (3), 82 (3), 75 (10), 67 (3), 59 (7), 53 (4), 39 (8). Values correspond to those previously reported by Khusnutdinov et al.42
Improved general procedure for the preparation of 5‐formyl‐2‐furoate esters
The following general procedure was used for the experiments reported in Table 2. A 250 mL round bottom flask was equipped with a magnetic stirring bar and placed on a DrySyn heating plate. The flask was equipped with a Soxhlet setup. In experiments with active water removal, the Soxhlet setup was filled with dried 3 Å molecular sieves (20 g). In experiments without active water removal, the Soxhlet setup was not filled with molecular sieves. The round bottom flask was charged with the desired substrate (12 mmol) and 100 mL alcohol (MeOH, EtOH, or nPrOH). The Soxhlet setup was filled with another 70 mL of the same alcohol. Acid catalyst (H2SO4, p‐TSA, or MSA) was added, and the reaction mixture was heated to reflux under stirring for the desired time period. The reaction mixture was allowed to cool to RT and concentrated to 50 mL at 40 °C by using a rotary evaporator. Demineralized water (150 mL) was added, and the mixture was stirred for 45 min to hydrolyze the acetals to the free aldehyde. The aqueous mixture was extracted with diethyl ether (4×50 mL). The combined organic layers were then washed with sat. NaHCO3 and brine until the pH value of the water layer was neutral. The organic layer was dried over MgSO4, filtered, and concentrated at 40 °C under vacuum using a rotary evaporator. The products were purified by silica gel column chromatography (petroleum ether/ethyl acetate (8:2) and analyzed by 1H/13C NMR and GC–MS.
Ethyl 5‐formyl‐2‐furoate (from ethanol as the solvent): TLC (ethyl acetate/petroleum ether 1:9): R f (0.22); 1H NMR (400.17 MHz, CDCl3): δ=9.77 (1 H, s), 7.24 (1 H, d, J=3.8 Hz), 7.23 (1 H, d, J=3.8 Hz), 4.38 (2 H, q, J=7.1 Hz), 1.36 ppm (3 H, t, J=7.1 Hz); 13C NMR (100.62 MHz, CDCl3): δ=14.28, 61.96, 118.56, 119.05, 148.04, 153.87, 158.08, 179.06 ppm. MS (GC–MS, 70 eV): m/z (%)=168 (22) [M +], 140 (70), 139 (53), 123 (20), 95 (35), 67 (7), 39 (38). 1H NMR values correspond to those previously reported by Dawson et al.44
Propyl 5‐formyl‐2‐furoate (from propanol as the solvent): TLC (ethyl acetate/petroleum ether 1:9): R f (0.28); 1H NMR (400.17 MHz, CDCl3): δ=9.82 (1 H, s), 7.29 (1 H, d, J=3.7 Hz), 7.27 (1 H, d, J=3.7 Hz), 4.32 (2 H, t, J=6.8 Hz), 1.88–1.73 (2 H, m), 1.02 ppm (3 H, t, J=7.7 Hz). 13C NMR (100.62 MHz, CDCl3): δ=14.11, 21.91, 67.28, 118.39, 118.79, 147.91, 153.78, 158.03, 178.96 ppm; MS (GC–MS, 70 eV): m/z (%)=182 (3) [M +], 153 (3), 141 (51), 140 (47), 139 (49), 123 (100), 95 (22), 67 (7), 39 (40).
Methyl levulinate (from d‐fructose and HMF): TLC (petroleum ether/ethyl acetate 7:3): R f (0.50); 1H NMR (400.17 MHz, CDCl3): δ=3.65 (3 H, s), 2.72 (2 H, t, J=6.4 Hz), 2.54 (2 H, t, J=6.4 Hz), 2.18 ppm (3 H, s); MS (GC–MS, 70 eV): m/z (%)=130 (2) [M +], 115 (26), 99 (28), 88 (14), 71 (10), 58 (15), 55 (26), 43 (100). 1H NMR values corresponded to those reported by Morandi et al.,45 and GC–MS values corresponded to those reported by Hengne et al.46
Preparation of tetraammine gold(III)nitrate (catalyst precursor)
The following method was adapted from two previously reported procedures.47 Ammonium nitrate (95 g, 1.19 mol) was dissolved in water (70 mL) in a 300 mL Erlenmeyer flask. The mixture was stirred and allowed to warm to RT to give a clear solution (pH 4.5). Using a plastic spoon (Caution: do not use a metal spoon), gold(III)chloride trihydrate (1 g, 2.54 mmol) was added to the solution. The solution changed from colorless to yellow/orange and the pH value dropped to pH 1.3. Under stirring, conc. ammonia was carefully dropped in the solution until the pH value increased to pH 5. Although Mason and Gray47a reported precipitation of the desired product at pH 5, we did not observe any precipitation after 30 min. The pH value was increased to pH 7 by the addition of conc. ammonia and a white precipitate formed immediately, as reported by Skibsted and Bjerrum47b (Caution: care must be taken not to increase the pH value too much because explosive gold salts might be formed)47a The white suspension was allowed to stand at 7 °C for 16 h, after which the precipitate was collected by filtration. The residue was washed with a small amount of ice water followed by diethyl ether. The product was dried at RT and transported to a 100 mL beaker. Water was added under stirring until the salt was almost completely dissolved (35 mL water was needed). The remaining particles were removed by filtration through a microfilter. The clear colorless solution (pH 6) was cooled in an ice bath and conc. nitric acid (5 mL) was added. A white precipitate formed immediately in the acidic mixture (pH<1). The precipitate was collected by filtration, washed with a small amount of ice water, and washed with ethanol. The product was dried under vacuum at 40 °C to a constant weight to give 476 mg white product (42 mol %).
Preparation of Au/C catalyst through cationic adsorption48
Step 1 (cationic adsorption): Activated carbon (SX1G, 10 g) was placed in a 100 mL beaker and water (40 mL) was added. The suspension was stirred for 30 min until the carbon was completely wetted. In a separate 500 mL beaker, tetraammine gold(III)nitrate (334 mg, 0.763 mmol) was dissolved in 300 mL water (2.55 mmol L−1). Under fast stirring with an overhead stirrer, the previously prepared carbon suspension was added to the gold solution as fast as possible. The resulting suspension was stirred for 2 h at RT. The suspension was filtered through a paper filter, and the carbon was washed with demineralized water. The carbon with adsorbed gold was dried under vacuum at 40 °C in the presence of Sicapent.
Step 2 (reduction): A quartz plug‐flow reactor was filled with 1.6 g of the material prepared in step 1. The reactor was flushed with argon, and a hydrogen flow (8 mL min−1) was applied. The sample was heated to 400 °C with a ramp of 5 °C min−1 and was kept at this temperature for 4 h. The gas flow was switched to argon and the reactor was allowed to cool to RT. The gold loading was determined by elemental analysis (1.3 wt % gold) and the gold particle size distribution was determined by TEM/HAADF (average gold particle size 2.2 nm, see the Supporting Information).
Preparation of dimethyl‐FDCA38
75 mL Parr MRS5000 pressure reactors were charged with magnetic stirring bars, the substrate (either Me‐FFA or Et‐FFA, 2 mmol), methanol (20 mL), and sodium methoxide (43 mg, 0.2 mmol) to give clear solutions. The 1.3 wt % Au/C catalyst (88 mg, 5.8 μmol Au) was added, the reactors were closed, flushed three times with oxygen, and then pressurized at 5 bar. Stirring was started (600 rpm) and the reactions were allowed to proceed at RT. After 19 h, the reactors were opened, and the reaction mixtures were filtered through filter paper to remove the catalyst. Methanol was removed under reduced pressure to give yellow/brown solids. The remaining sodium methoxide was removed by taking up the reaction mixtures in chloroform (product dissolves whereas NaOMe does not dissolve) followed by filtration through Pasteur pipettes filled with cotton wool. The clear yellow solutions were concentrated to give dimethyl‐FDCA as slightly yellow crystalline solids (365–368 mg, yield 99 %). R f (0.32); 1H NMR (400.17 MHz, CDCl3): δ=7.22 (2 H, s), 3.94 ppm (6 H, s). 13C NMR (100.62 MHz, CDCl3): δ=52.33, 118.41, 146.64, 158.37 ppm; MS (GC–MS, 70 eV): m/z (%)=184 (32) [M +], 153 (100), 139 (1), 125 (6), 113 (1), 95 (8), 82 (2), 69 (6), 59 (9), 53 (4), 38 (9). The NMR and MS spectra were in agreement with previously reported values.49
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was co‐financed by TKI‐funding from the Topconsortia for Knowledge & Innovation (TKI's) of the Ministry of Economic Affairs in The Netherlands, and the companies Royal Cosun, RefrescoGerber, Arkema, and North Seaweed. The authors would like to thank W. Vogelzang, BSc. for fruitful discussions that led to initiating of this research strategy, Dr. A. E. Frissen for the NMR and XRD analyses, Alniek van Zeeland, B.Sc. for the HPLC‐MS analyses, and Hans Meeldijk (Utrecht University) for TEM/HAADF imaging.
F. van der Klis, J. van Haveren, D. S. van Es, J. H. Bitter, ChemSusChem 2017, 10, 1460.
Contributor Information
Dr. Daan S. van Es, Email: daan.vanes@wur.nl
Prof. Dr. Johannes H. Bitter, Email: harry.bitter@wur.nl
References
- 1.
- 1a. de Jong E., Dam M. A., Sipos L., Gruter G.-J. M., ACS Symp. Ser. 2012, 1105, 1–13; [Google Scholar]
- 1b. Knoop R. J. I., Vogelzang W., van Haveren J., van Es D. S., J. Polym. Sci. Part A 2013, 51, 4191–4199; [Google Scholar]
- 1c. Matsuda K., Horie H., Ito K. (Canon Kabushiki-Gaisha, Tokyo), US20120258299, 2012;
- 1d. Moore J. A., Bunting W. W., Polyesters and Polyamides Containing Isomeric Furan Dicarboxylic Acids, in Advances in Polymer Synthesis (Eds.: B. M. Culbertson, J. E. McGrath), Plenum Press, New York, 1985, pp. 51–91; [Google Scholar]
- 1e. Sousa A. F., Vilela C., Fonseca A. C., Matos M., Freire C. S. R., Gruter G.-J. M., Coelho J. F. J., Silvestre A. J. D., Polym. Chem. 2015, 6, 5961–5983; [Google Scholar]
- 1f. Thiyagarajan S., Vogelzang W., Knoop R. J. I., Frissen A. E., van Haveren J., van Es D. S., Green Chem. 2014, 16, 1957–1966; [Google Scholar]
- 1g. Van Es D. S., Van Haveren J., Frissen A. E., Van De Kolk J. C. (Stichting Dienst Landbouwkundig Onderzoek, Wageningen; Archer Daniels Midland Company, Chicago, IL), WO2013184661, 2013;
- 1h. Vannini M., Marchese P., Celli A., Lorenzetti C., Green Chem. 2015, 17, 4162–4166; [Google Scholar]
- 1i. Wu J., Eduard P., Thiyagarajan S., Noordover B. A. J., van Es D. S., Koning C. E., ChemSusChem 2015, 8, 67–72. [DOI] [PubMed] [Google Scholar]
- 2. van Putten R.-J., van der Waal J. C., de Jong E., Rasrendra C. B., Heeres H. J., de Vries J. G., Chem. Rev. 2013, 113, 1499–1597. [DOI] [PubMed] [Google Scholar]
- 3. Rackemann D. W., Doherty W. O. S., Biofuels Bioprod. Biorefin. 2011, 5, 198–214. [Google Scholar]
- 4. van Zandvoort I., Wang Y., Rasrendra C. B., van Eck E. R. H., Bruijnincx P. C. A., Heeres H. J., Weckhuysen B. M., ChemSusChem 2013, 6, 1745–1758. [DOI] [PubMed] [Google Scholar]
- 5. Eerhart A. J. J. E., Faaij A. P. C., Patel M. K., Energy Environ. Sci. 2012, 5, 6407–6422. [Google Scholar]
- 6. Gruter G. J. M., Dautzenberg F. (Avantium International B.V., Amsterdam), EP1834950A1, 2007.
- 7.
- 7a. Elias H.-G., Macromolecules, Vol. 2 (Industrial Polymers and Synthesis), Wiley-VCH, Weinheim, 2005; [Google Scholar]
- 7b. Weissermel K. A., Arpe H.-J., Industrial Organic Chemistry, 4th ed., Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 8.
- 8a. http://faostat.fao.org/site/339/default.aspx;
- 8b.A. W. M. Huijbregts, in 1st joint II, RB-ASSBT Congress, San Antonio, Texas, USA, 2003, pp. 451–459;
- 8c. Oosterveld A., Beldman G., Schols H. A., Carbohydr. Res. 1996, 288, 143–153. [DOI] [PubMed] [Google Scholar]
- 9. Ramasamy U. S., Gruppen H., Schols H. A., J. Agric. Food Chem. 2013, 61, 6077–6085. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Kuivanen J., Dantas H., Mojzita D., Mallmann E., Biz A., Krieger N., Mitchell D., Richard P., AMB Express 2014, 4, 33; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Torrado A. M., Cortes S., Salgado J. M., Max B., Rodriguez N., Bibbins B. P., Converti A., Dominguez J. M., Braz. J. Microbiol. 2011, 42, 394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rommi K., Rahikainen J., Vartiainen J., Holopainen U., Lahtinen P., Honkapää K., Lantto R., J. Appl. Polym. Sci. 2016, 133, 42862. [Google Scholar]
- 12. Pfaltzgraff L. A., De Bruyn M., Cooper E. C., Budarin V., Clark J. H., Green Chem. 2013, 15, 307–314. [Google Scholar]
- 13.
- 13a. Otalora A. F. Belalcazar (PectCof B.V., Heelsum), WO2014083032, 2014;
- 13b. Coffee Pulp: Composition, Technology, and Utilization (Eds.: J. E. Braham, R. Bressani), IDRC, 1979; [Google Scholar]
- 13c. Padmapriya R., Tharian J. A., Thirunalasundari T., Int. J. Curr. Sci. 2013, 9, 83–91. [Google Scholar]
- 14. Leijdekkers A. G. M., Bink J. P. M., Geutjes S., Schols H. A., Gruppen H., Bioresour. Technol. 2013, 128, 518–525. [DOI] [PubMed] [Google Scholar]
- 15. Harding S. E., Smith I. H., Lawson C. J., Gahler R. J., Wood S., Carbohydr. Polym. 2011, 83, 329–338. [Google Scholar]
- 16. Nie S.-P., Wang C., Cui S. W., Wang Q., Xie M.-Y., Phillips G. O., Food Hydrocolloids 2013, 32, 221–227. [Google Scholar]
- 17. Adams G. A., Can. J. Chem. 1954, 32, 186–194. [Google Scholar]
- 18.
- 18a. Gírio F. M., Fonseca C., Carvalheiro F., Duarte L. C., Marques S., Bogel-Łukasik R., Bioresour. Technol. 2010, 101, 4775–4800; [DOI] [PubMed] [Google Scholar]
- 18b. Moriana R., Vilaplana F., Ek M., Cellulose 2015, 22, 3409–3423. [Google Scholar]
- 19.
- 19a. Raedts J., Kengen S. W. M., van der Oost J., Glycoconjugate J. 2011, 28, 57–66; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Haug A., Larsen B., Acta Chem. Scand. 1963, 17, 1653–1662. [Google Scholar]
- 20.
- 20a. Lu J., Yang H., Hao J., Wu C., Liu L., Xu N., Linhardt R. J., Zhang Z., Carbohydr. Polym. 2015, 122, 180–188; [DOI] [PubMed] [Google Scholar]
- 20b. Sánchez-Machado D. I., López-Cervantes J., López-Hernández J., Paseiro-Losada P., Simal-Lozano J., Biomed. Chromatogr. 2004, 18, 90–97. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Nguyen T. T., Brown S., Fedorov A. A., Fedorov E. V., Babbitt P. C., Almo S. C., Raushel F. M., Biochemistry 2008, 47, 1194–1206; [DOI] [PubMed] [Google Scholar]
- 21b. Nguyen T. T., Fedorov A. A., Williams L., Fedorov E. V., Li Y., Xu C., Almo S. C., Raushel F. M., Biochemistry 2009, 48, 8879–8890; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Takagi Y., Kanda M., Nakata Y., Biochim. Biophys. Acta 1959, 31, 264–265; [DOI] [PubMed] [Google Scholar]
- 21d. Wahba A. J., Hickman J. W., Ashwell G., J. Am. Chem. Soc. 1958, 80, 2594–2595. [Google Scholar]
- 22. McNaught A. D., Pure Appl. Chem. 2009, 68, 1919–2008. [Google Scholar]
- 23.
- 23a. Carlsson B., Samuelso O., Popoff T., Theander O., Acta Chem. Scand. 1969, 23, 261–267; [Google Scholar]
- 23b. Nelson E. R., Nelson P. F., Aust. J. Chem. 1968, 21, 2323–2325; [Google Scholar]
- 23c. Prihar H. S., Feingold D. S., Carbohydr. Res. 1980, 86, 302–304. [Google Scholar]
- 24. Angyal S. J. in Glycoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates (Ed.: A. E. Stütz), Springer, Berlin Heidelberg, 2001, pp 1–14. [Google Scholar]
- 25. Ehrlich F., Guttmann R., Ber. Dtsch. Chem. Ges. 1934, 67B, 573–589. [Google Scholar]
- 26.
- 26a. Keliang G., Dongzhi W., Appl. Microbiol. Biotechnol. 2006, 70, 135–139; [DOI] [PubMed] [Google Scholar]
- 26b. Hameršak Z., Pavlović N., Delić V., Šunjić V., Carbohydr. Res. 1997, 302, 245–249; [Google Scholar]
- 26c. Kambourakis S., Griffin B. M., Martin K. V. (Synthetic Genomics, Inc., La Jolla, CA), WO2015143381A3, 2015.
- 27. Votoček E., Malachta S., Collect. Czech. Chem. Commun. 1934, 6, 241–250. [Google Scholar]
- 28.
- 28a. Stutz E., Deuel H., Helv. Chim. Acta 1956, 39, 2126–2130; [Google Scholar]
- 28b. Stutz E., Deuel H., Helv. Chim. Acta 1958, 41, 1722–1730. [Google Scholar]
- 29. Luisa M., Ramos D., Madalena M., Caldeira M., Gil V. M. S., Carbohydr. Res. 1996, 286, 1–15. [Google Scholar]
- 30.
- 30a. Emaga T. H., Rabetafika N., Blecker C. S., Paquot M., Biotechnol. Agron. Soc. Environ. 2012, 16, 139–147; [Google Scholar]
- 30b. Leitão M. C. A., Alarcão Silva M. L., Januário M. I. N., Azinheira H. G., Carbohydr. Polym. 1995, 26, 165–169; [Google Scholar]
- 30c. Shorey E. C., Martin J. B., J. Am. Chem. Soc. 1930, 52, 4907–4915. [Google Scholar]
- 31. Eyrisch O., Sinerius G., Fessner W.-D., Carbohydr. Res. 1993, 238, 287–306. [Google Scholar]
- 32. Crawford T. C., Andrews G. C., Faubl H., Chmurny G. N., J. Am. Chem. Soc. 1980, 102, 2220–2225. [Google Scholar]
- 33. Chen C., Yamamoto H., Kwan T., Chem. Pharm. Bull. 1970, 18, 815–819. [Google Scholar]
- 34.
- 34a. Kosaka A., Aida M., Katsumoto Y., J. Mol. Struct. 2015, 1093, 195–200; [Google Scholar]
- 34b. Sinnott M., Carbohydrate Chemistry and Biochemistry: Structure and Mechanism, 2nd ed., RSC, Cambridge, 2013. [Google Scholar]
- 35. Rosatella A. A., Simeonov S. P., Frade R. F. M., Afonso C. A. M., Green Chem. 2011, 13, 754–793. [Google Scholar]
- 36.
- 36a. Binder J. B., Cefali A. V., Blank J. J., Raines R. T., Energy Environ. Sci. 2010, 3, 765–771; [Google Scholar]
- 36b. van Putten R.-J., Soetedjo J. N. M., Pidko E. A., van der Waal J. C., Hensen E. J. M., de Jong E., Heeres H. J., ChemSusChem 2013, 6, 1681–1687; [DOI] [PubMed] [Google Scholar]
- 36c. Yang G., Pidko E. A., Hensen E. J. M., J. Catal. 2012, 295, 122–132. [Google Scholar]
- 37. Matsuhiro B., Zanlungo A. B., Dutton G. G. S., Carbohydr. Res. 1981, 97, 11–18. [Google Scholar]
- 38. van der Klis F., Frissen A. E., van Haveren J., van Es D. S., ChemSusChem 2013, 6, 1640–1645. [DOI] [PubMed] [Google Scholar]
- 39.K. Iffland, J. Sherwood, M. Carus, A. Raschka, T. Farmer, J. Clark, nova paper #8 on bio-based economy 2015–11 2015, http://www.bio-based.eu/nova-papers.
- 40. Taarning E., Nielsen I. S., Egeblad K., Madsen R., Christensen C. H., ChemSusChem 2008, 1, 75–78. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Deng J., Song H.-J., Cui M.-S., Du Y.-P., Fu Y., ChemSusChem 2014, 7, 3334–3340; [DOI] [PubMed] [Google Scholar]
- 41b. Menegazzo F., Fantinel T., Signoretto M., Pinna F., Manzoli M., J. Catal. 2014, 319, 61–70; [Google Scholar]
- 41c. Shaikh A. S., Parker K. R., Partin L. R., Janka M. E. (Eastman Chemical Company, Kingsport, TN), WO2013191942, 2013.
- 42. Khusnutdinov R. I., Bayguzina A. R., Mukminov R. R., Russ. Chem. Bull. 2013, 62, 93–97. [Google Scholar]
- 43. Shackelford S. A., Anderson M. B., Christie L. C., Goetzen T., Guzman M. C., Hananel M. A., Kornreich W. D., Li H., Pathak V. P., Rabinovich A. K., Rajapakse R. J., Truesdale L. K., Tsank S. M., Vazir H. N., J. Org. Chem. 2003, 68, 267–275. [DOI] [PubMed] [Google Scholar]
- 44. Dawson M. I., Chan R., Hobbs P. D., Chao W., Schiff L. J., J. Med. Chem. 1983, 26, 1282–1293. [DOI] [PubMed] [Google Scholar]
- 45. Morandi B., Wickens Z. K., Grubbs R. H., Angew. Chem. Int. Ed. 2013, 52, 9751–9754; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9933–9936. [Google Scholar]
- 46. Hengne A. M., Kamble S. B., Rode C. V., Green Chem. 2013, 15, 2540–2547. [Google Scholar]
- 47.
- 47a. W. R. Mason III , Gray H. B., J. Am. Chem. Soc. 1968, 90, 5721–5729; [Google Scholar]
- 47b. Skibsted L. H., Bjerrum J., Acta Chem. Scand. Ser. A 1974, 28, 740–746. [Google Scholar]
- 48. Pyryaev P. A., Moroz B. L., Zyuzin D. A., Nartova A. V., Bukhtiyarov V. I., Kinet. Catal. 2010, 51, 885–892. [Google Scholar]
- 49.
- 49a. Thiyagarajan S., Pukin A., van Haveren J., Lutz M., van Es D. S., RSC Adv. 2013, 3, 15678–15686; [Google Scholar]
- 49b. Tsanaktsis V., Papageorgiou G. Z., Bikiaris D. N., J. Polym. Sci. Part A 2015, 53, 2617–2632. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary