

Abstract
During the last years there was a substantial increase in the use of antibodies and related proteins as therapeutics. The emphasis of the pharmaceutical industry is on IgG1, IgG2, and IgG4 antibodies, which are therefore in the focus of this article. In order to ensure appropriate quality control of such biopharmaceuticals, deep understanding of their chemical degradation pathways and the resulting impact on potency, pharmacokinetics, and safety is required. Criticality of modifications may be specific for individual antibodies and has to be assessed for each molecule. However, some modifications of conserved structure elements occur in all or at least most IgGs. In these cases, criticality assessment may be applicable to related molecules or molecule formats. The relatively low dissociation energy of disulfide bonds and the high flexibility of the hinge region frequently lead to modifications and cleavages. Therefore, the hinge region and disulfide bonds require specific consideration during quality assessment of mAbs. In this review, available literature knowledge on underlying chemical reaction pathways of modifications, analytical methods for quantification and criticality are discussed. The hinge region is prone to cleavage and is involved in pathways that lead to thioether bond formation, cysteine racemization, and iso‐Asp (Asp, aspartic acid) formation. Disulfide or sulfhydryl groups were found to be prone to reductive cleavage, trisulfide formation, cysteinylation, glutathionylation, disulfide bridging to further light chains, and disulfide scrambling. With regard to potency, disulfide cleavage, hinge cleavage, disulfide bridging to further light chains, and cysteinylation were found to influence antigen binding and fragment crystallizable (Fc) effector functionalities. Renal clearance of small fragments may be faster, whereas clearance of larger fragments appears to depend on their neonatal Fc receptor (FcRn) functionality, which in turn may be impeded by disulfide bond cleavage. Certain modifications such as disulfide induced aggregation and heterodimers from different antibodies are generally regarded critical with respect to safety. However, the detection of some modifications in endogenous antibodies isolated from human blood and the possibility of in vivo repair mechanisms may reduce some safety concerns.
Keywords: Antibody, Critical quality attribute, Disulfide, Fragmentation, Modification
Abbreviations
- Asp
aspartic acid
- CDR
complementarity determining region
- CE‐SDS‐NGS
CE‐SDS nongel sieving
- CH
constant part of heavy chain
- CL
constant part of light chain
- Cys
cysteine
- Fab
fragment antigen binding
- Fab/c
antibody fragment that results from loss of one Fab
- Fc
fragment crystallizable
- FcRn
neonatal Fc receptor
- FDA
Food and Drug Administration
- FLC
free light chain
- HC
heavy chain
- HCCF
harvested cell culture fluid
- LC
light chain or liquid chromatography
- PK
pharmacokinetic
- VH
variable part of heavy chain
1. Introduction
Based on differences in the constant region, antibodies are grouped into IgA, IgD, IgE, IgG, and IgM 1. Clear focus of pharmaceutical development is on the IgG subtype whereby IgG1, IgG2, and IgG4 are most relevant. In contrast to IgG1 and IgG2, IgG4 does not exhibit complement activation. In addition, IgG2 and IgG4 have lower FcγRI, FcγRII, FcγRIIIa/b receptor affinities than IgG1 1. The focus of this article is especially on disulfide bonds and the hinge between the epitope binding part of the antibody (fragment antigen binding, Fab) and the effector function bearing Fc due to the low dissociation energy and flexibility of these structural elements, respectively. The relatively low chemical stability of disulfide bonds, the extraordinary flexibility and reactivity of the hinge region, and the strong involvement of disulfide bonds in complex reaction mechanisms make these structures especially prone to changes and thus very important for quality assessment (Table 1). In dependence on receptor allotype and/or antibody glycosylation, IgG4 and IgG2 were found to activate FcγRI, FcγRIIIa 2, 3, 4, or FcγRIIa 2 receptors, respectively. IgG3 is not used as therapeutic 1, 5 due to its short half‐life (approximately 7 days for IgG3 versus approximately 21 days for IgG1 6) and its long hinge region that is prone to cleavage and allotypic polymorphisms. Therefore, this review on the quality assessment of disulfide and hinge modifications focusses on IgG1, IgG2, and IgG4 antibodies. In case of conserved structure elements, discussed literature information for endogenous standard antibody formats 1 may also by applicable to new format molecules 7, 8.
Table 1.
Overview of disulfide bond‐related modifications
| disulfiderelated modifications | Investigated IgG types | References |
|---|---|---|
| Cleavage of inter‐ and intrachain disulfide bonds (Sections 2.1 and 3.1) | IgG1 | [34, 49, 50, 51, 57] |
| IgG2 | 10, 34 | |
| IgG4 | 76 | |
| Hinge fragmentation (Sections 2.2 and 3.2) | IgG1 | 19, 23 |
| IgG3 | 1, 5 | |
| Thioether bond formation (Sections 2.2 and 3.3) | IgG1 | 21, 23, 24 |
| Cys racemization (Sections 2.2 and 3.4) | IgG1 | 22 |
| IgG2 | 22 | |
| Disulfide Scrambling (Sections 2.3 and 3.6) | IgG2 | 67, 68, 69 |
| Dimerization (Sections 2.3 and 3.6) | IgG2 | 66 |
| Half antibodies and Fab arm exchange (Sections 2.3 and 3.6) | IgG4 | 1, 73, 74, 75 |
| Trisulfide formation (Sections 2.4 and 3.7) | IgG1, IgG2, IgG3, and IgG4 | 47 |
| Cysteinylation (Sections 2.5 and 3.8) | IgG1 | 14, 90 |
| Disulfide bridging to additional LCs (Sections 2.5 and 3.8) | IgG1 | 30 |
| Aggregation (Sections 2.6 and 3) | IgG1 | 99 |
| IgG2 | 10 |
Criticality, chemical reaction pathways, and analytical methods are discussed in the respective sections.
To ensure a comprehensive quality assessment for such molecules, in depth understanding of all modification and cleavage processes is inevitable. They may arise from different chemical processes that result in fragmentation 9 and/or structural perturbation 10. Extend and occurrence may for example depend on the solvent exposure of an affected protein structure element 9, its amino acid sequence 11, its molecular flexibility 12, and its dynamic steady state in vivo (i.e. dynamic in vivo concentration that results from generation rate and clearance) 13. Subsequent aggregation, exposure of epitopes, or fragmentation can detrimentally affect potency 14, safety (i.e. immunogenicity 15, 16) or pharmacokinetic (PK) properties 13. Eventually, all critical quality attributes have to be identified and kept within acceptance criteria. A complete criticality assessment has to include all relevant efficacy, safety, and PK information (refer to FDA Guidance for Industry Q8‐Pharmaceutical Development).
Fragmentation is often observed in the solvent exposed, flexible loops of a protein 11. Water‐mediated fragmentation of peptide bonds is accelerated under acidic and basic conditions and has been observed for Gly‐Gly motives in the lower hinge and in the constant part of heavy chain 1 (CH1) domain of antibodies. Such cleavage can be attributed to the low steric hindrance by the small side chain. Other fragmentation processes are facilitated by different amino acid side chains like for aspartic acid (Asp), Ser, Thr, Cys, or Asn and also by the occurrence of free radicals 17, 18, 19. A total of 47 potential fragmentation sites have been summarized previously 9.
Chemical modification processes may not necessarily result in cleavage of the peptide backbone or of disulfide bonds. Examples are deamidation 20, iso‐Asp formation 20, 21, racemization 21, 22, thioether bond formation 23, 24, pyroglutamate formation 25, and oxidation 26. However, some of these modifications may still impede structural integrity or functionality.
The number and position of disulfide bonds depend on the IgG subtype. IgG1 and IgG4 antibodies contain 12 intra‐ and four interchain disulfide bonds. The intrachain bonds are located in the variable part of light chain (VL), constant part of light chain (CL), variable part of heavy chain (VH), CH1, CH2, and CH3 domains, whereas two interchain disulfide bonds connect the two heavy chains (HCs) and one links each of the LCs to the HCs. IgG2 antibodies contain 18 disulfide bonds (similar distribution but two additional disulfide bonds between the two HCs). The following disulfide‐related modifications are observed for IgG disulfide bonds:
-
(1)
Partial cleavage of inter‐ and intrachain disulfide bonds.
-
(2)
Disulfide scrambling 1.
-
(3)
Cysteinylation 14/glutathionylation 29/addition of LCs 30 in presence of cysteine (Cys), glutathione or free light chains (FLCs).
Open disulfide bonds can be a result of incomplete processing within the host cells or of reductive or radical cleavage of the secreted antibody. Especially under cellular stress conditions it can already occur within the endoplasmaic reticulum by an incomplete protein disulfide isomerase oxidation 28, 31, 32, 33. It is also caused by cellular redox‐systems such as glutathione, Cys, glutathione reductase, thioredoxin, and glutaredoxin 27, whereby the extend of reduction is influenced by the concentration of these redox systems and metal ions for example copper 34. Expression of redox systems or reduced nicotinamide adenine dinucleotide phosphate production by pentose phosphate pathway can be reduced by adaptation of cell culture conditions or choice of cell line 35, 36. A decrease of IgG1 disulfide bond reduction by knocking down expression of thioredoxin 1 (TXN1) was demonstrated during the room temperature hold step of harvested cell culture fluid 37. Approximately, 0.02–0.1 mol sulfhydryl per mol of protein was found to be reduced in recombinant antibodies secreted into the culture medium 28.
This reductive cleavage and related modifications can occur in the cell culture fluid, during harvest operations and subsequent purification steps, or during subsequent storage of the final drug until usage. Especially during large‐scale fermentations the surface to volume ratio and thus oxygen content becomes smaller and can lead to disulfide reduction 34, 38. By cell rupture during harvest operations, redox systems can be released into the harvested cell culture fluid (e.g. thioredoxin reductase and reduced nicotinamide adenine dinucleotide phosphate) 37, 38, 39. These redox systems are acting on the antibody for a relatively long time and are able to reduce disulfide bonds 39. Additives such as EDTA, CuSO4, and l‐Cys, usage of hermetic centrifuges and ensuring the presence of oxygen during harvest operations are proposed to decrease levels of reductive cleavage 34, 38, 39. Besides reductive cleavage, radical cleavage into two CysS• radicals has been described 40, 41. Cys thiyl radical (CysS•) pairs may be derived from homolytic dissociation of disulfide bonds during photostressing (8–12) or from oxygen in combination with metal ions 17, 19. A CysS• radical pair can disproportionate into thiol and thioaldehyde or may capture hydrogen atoms from surrounding amino acids 40. This may induce protein hydroperoxide, protein aggregates and/or protein fragmentation 40. Besides cleavage, formation of dithiohemiacetal and thioether cross‐links are described for intrachain disulfide bonds 41.
The flexibility of the hinge region was confirmed by crystallographic studies 42, 43 and can also be related to its length that increases in the following order: IgG3 > IgG1 > IgG4 > IgG2 44. This region is involved in different degradation processes: Upper hinge cleavage 45, 46, thioether bond formation 45, 46, Cys racemization 45, 46, and iso Asp formation 21. They result for example from β‐elimination 23, Michael‐like addition 23, or radical‐mediated processes 18, 19. Presence of H2S or Cys in the fermentation medium was found to be responsible for trisulfide formation at the light chain (LC) and heavy chain (HC) disulfide bond 47.
In summary, disulfide bonds and hinge region have to be considered for a thorough quality assessment of therapeutic antibodies. These structures require a comprehensive evaluation in order to obtain a complete picture about the quality of the respective biopharmaceutical product.
2. Disulfide‐ and hinge‐related modifications
At a first glance, the huge variety of reaction mechanisms seems to be confusing (see Section 2.2). However, most reaction pathways follow common rules. They depend on chemical characteristics of involved structure elements, such as flexibility and the conditions under which the reaction takes place:
-
(1)
Radical‐induced homolytic dissociation of covalent bonds strongly depends on their dissociation energy. Typical protein bonds dissociation energies decrease in the following order 48: C = O (743 kJ/mol) > C = N (613 kJ/mol)1 > C‐H (412 kJ/mol) > C‐O (360 kJ/mol) > C‐C (348 kJ/mol) > C‐N (305 kJ/mol) > S‐S (264 kJ/mol) > C‐S (259 kJ/mol). Due to mesomerie effects CN or CO bonds often have a partial double bond character (e.g. peptide bond). In conclusion, involvement of sulfur atoms makes bonds relatively weak and prone to cleavage. That explains preferred homolytic C‐S or S‐S cleavage upon radical attack and common observation of open disulfide bonds and disulfide‐related modifications 1, 49, 50, 51.
-
(2)
Different electronegativities of elements result in partial positive and partial negative charges. Electronegativity according to Alled‐Rochow decreases in the following order 52: O (3.50) > N (3.07) > C (2.50) > S (2.44) > H (2.20). Due to inductive effects oxygen and nitrogen are partially negatively charged, whereas carbon and hydrogen bear partial positive charges. Especially, carbon is very often the target of nucleophilic attacks by nitrogen or oxygen that often results in cleavage or isomerization.
-
(3)
Geometry of reaction counterparts, transient states and products, as well as steric hindrance of adjacent groups and overall flexibility of the whole molecule has an important influence on the course of reaction pathways 53, 54. Typical bond angles (120° for hybridization between one s and two p orbitals (sp2) or 109.5° for hybridization between one s and three p orbitals (sp3) have to be approached as closely as possible by the reacting counterparts. A commonly observed intermediate of many reaction pathways is the energetically favored five‐ring structure (angle of the regular five‐ring is closest to the standard sp3 tetraeder angle of 109.5°). However, for relaxation of bond angles rearrangement and adaptation of the overall molecular structure is required. This occurs faster if there are high molecular flexibility and few steric constraints.
-
(4)
Acidity and basicity of functional groups and their influence on different reaction pathways makes the pH value an important parameter for degradation reactions 53, 54. Proton‐free electron pairs of oxygen or nitrogen are frequently involved in nucleophilic attacks on partially positive carbon atoms. On the other hand, protonation of carbonyl oxygen often enables nucleophilic addition to adjacent carbon.
2.1. Reduction of inter‐ and intrachain disulfide bonds
Each LC consists of two and each HC of four immunoglobulin domains. Besides antiparallel β‐strands and β‐sheets, an intrachain disulfide bond is characteristic for each domain. Depending on the number of IgG domains, there are two or four intrachain disulfide bonds in each light and heavy chain, respectively. Depending on the antibody subtype, there are also two or four interchain disulfide bonds between the light and heavy chains 1.
Since disulfide bonds are relatively weak, some free sulfhydryl is generally found for each disulfide bond (1.5–5.6% for four investigated MAbs 50). Intrachain disulfide reduction in the variable region has been investigated in detail 49, 55, 56, 57. In one of these studies the ratio of free thiol versus intact disulfide was found to be around 1:5 49. Open disulfide levels in endogenous antibodies were found to be comparable to that of recombinant antibodies, demonstrating that open disulfide bonds are an inherent property of antibodies 10. Susceptibility of IgG1 disulfide bonds to reduction was investigated in detail and decreased in the following order 51: interchain > intrachain; LC‐HC intrachain > upper HC‐HC intrachain > lower HC‐HC‐intrachain; CH2 interchain > VL, CL, VH, CH1 interchain > CH3 interchain and LC‐HC interchain > HC‐HC interchain > Fab intrachain 51. Some structure variation may explain the higher abundance of disulfide reduction in the constant domains of serum derived IgG1 and IgG2 antibodies in comparison to the variable Ig domain that was found in another study 10. Aggregation upon agitation stress reduced the overall amount of open disulfide bonds which indicated their involvement in aggregate formation 10. The susceptibility of interchain disulfide bonds to reduction followed the order IgG1λ > IgG1κ > IgG2λ > IgG2κ 34. The reduced susceptibility of the κ IgGs to reduction may be attributed to hindrance by the proximity of the C‐terminal carboxyl group that is formed by the C‐terminal κ LC Cys itself (C‐terminal Serine of the λ LC is missing). Reduction of the interchain disulfide bond between the light and the heavy chain is more elaborately discussed in Section 2.2 since it is strongly involved in the complex reaction mechanisms that occur in the upper hinge.
There are different analytical methods for the identification and relative quantification of open disulfide bonds. Reduction of interchain disulfide bonds results in partially reduced MAbs (HC/HC/LC, HC/HC, HC/LC, HC, and LC). This can be monitored under denaturing conditions by nonreduced CE‐SDS‐NGS (CE‐SDS‐non‐gel sieving, in house data; Fig. 1) or by orthogonal SDS‐PAGE since the noncovalently bound chains dissociate from the molecule upon treatment with SDS.
Figure 1.
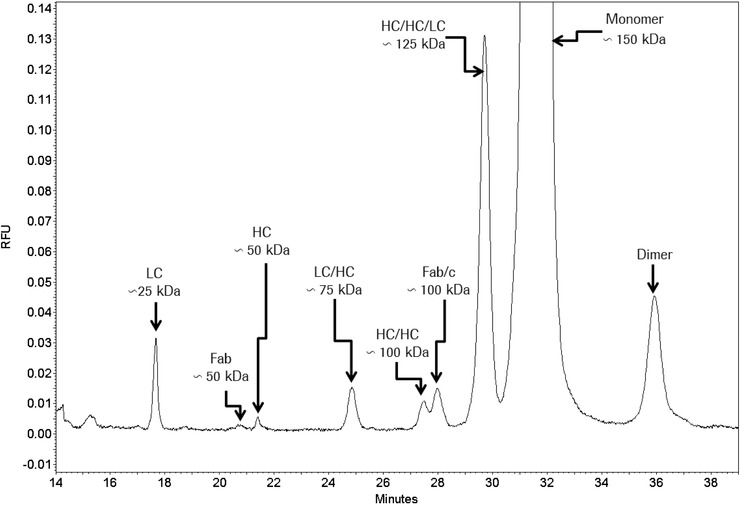
CE‐SDS NGS separation of a non‐reduced IgG1. Attribution to underlying structures shows its high resolution for hinge cleavage and partially reduced forms.
Many Cys residues involved in intrachain disulfide bonds are buried in the interior of the antibody and are not access‐ible for labeling reagents. Their reduction does not lead to molecule disintegration and causes only insignificant change in molecular weight. This is a challenge for size‐based separation technologies 49 and thus requires special analytical methodologies that involve labeling of free Cys residues. They are preferably applied under denaturing conditions in order to enable access to buried parts of the MAb structure:
-
(1)
Ellman assay 49, 57 5,5'‐dithiobis‐(2‐nitrobenzoic acid) reacts with free thiols. By this reaction 2‐nitro‐5‐thiobenzoate is released. The two‐fold negatively charged form of 2‐nitro‐5‐thiobenzoate is measured by absorbance at 412 nm.
-
(2)
Fluorescent assay [28, 58– 60] Free sulfhydryl reacts with fluorogenic N‐(1‐pyrenyl)maleimide (NPM) that results in fluorescent derivatives, allowing for detection without the need to purify or isolate the NPM derivative.
-
(3)
Ratio of reduced and oxidized SH groups can be determined by labeling with 12C and 13C iodoacetic acid and subsequent MS 50, 51. For the investigated MAbs, a portion of free sulfhydryl was found for every Cys 50.
-
(4)
Ratio of reduced and oxidized SH groups can also be determined by alkylation experiments using N‐ethylmaleimide 57 and deuterated N‐ethylmaleimide (in house data).
-
(5)
The reduced and nonreduced Cys‐22 and Cys‐96 in the variable domain of the HC could also be separated by ion exchange chromatography with a WCX‐10 column 49, 57, RP‐HPLC 49, or hydrophilic interaction chromatography after Papain cleavage 55.
-
(6)
LC‐MS peptide mapping can identify the free thiols at Cys‐22 and Cys‐96 in the HC 49.
2.2. Disulfide‐related modifications in the hinge region
In general, hinge modifications are induced by exposure to heat [21, 22, 23, 42, 46], H2O2 18, acidic/basic pH 22, 23, or UV light 41. Disulfide bonds are strongly involved in these processes that also comprise different cleavage processes 11, 17, 23, thioether bond formation 21, 23, 24 and Cys racemization 21, 22. Also iso‐Asp formation is an often observed variant of these modification processes 21, 61. Sometimes, predominant peptide bond cleavage sites are adjacent to upper hinge His and Asp residues, which facilitate cleavage due to the acidic and basic characteristics of their side chains 11. β‐elimination of Cys with subsequent thioether bond formation or hinge cleavage is prominent under basic conditions, whereas C‐terminal Asp cleavage is increased under acidic conditions 11. Radical‐induced hinge cleavage occurs in presence of oxygen and thermal stress or in presence of UV light 17. Enzymatic processes do not seem to play a role since host cell proteins did not enhance fragmentation, and protease inhibitors did not reduce it 42. IgG1 antibodies were found to be more susceptible to cleavage processes than IgG4 antibodies 44, 62. That may be caused by the higher length and flexibility of the IgG1 hinge.
A common method for the analysis of hinge fragmentation is CE‐SDS NGS (Fig. 1) or SDS‐PAGE 23. With nonreduced SDS‐PAGE, bands at 23 Da (Fab HC fragment and LC) and a band at 50 kDa (disulfide linked Fab) were detected 23. The underlying cleavage sites at several positions in the upper hinge (ladder formation) were detected by LC/MS and MALDI‐TOF‐MS 23. Fab fragments were also prefractionated by SEC and subsequently analyzed by RP‐HPLC‐TOF/MS and LC‐MS/MS peptide mapping 17.
Reduced CE‐SDS NGS or reduced SDS‐PAGE are also excellent methods for thioether quantification (in house data, not shown). After peak assignment by MS, the thioether level was also determined by RP‐HPLC separation with UV detection (complete protein without digestion) or by comparing the quantities of thioether peptides with that of the sum of thioether and disulfide linked peptides (after LysC digestion; based on extracted ion current or UV absorbance) 24. Further characterization of underlying reaction mechanisms was supported by forced degradation at elevated temperature and pH in D2O 24.
d‐Cys and iso Asp were detected by LC/MS and LC/MS/MS of tryptic peptides after forced degradation in D2O. In addition, amino acid analysis with chiral separation was applied 21. Since LysC digestion is blocked after iso‐Asp formation, the extend of iso Asp formation could be determined by analyzing the uncleaved peptides by RP LC/MS 61.
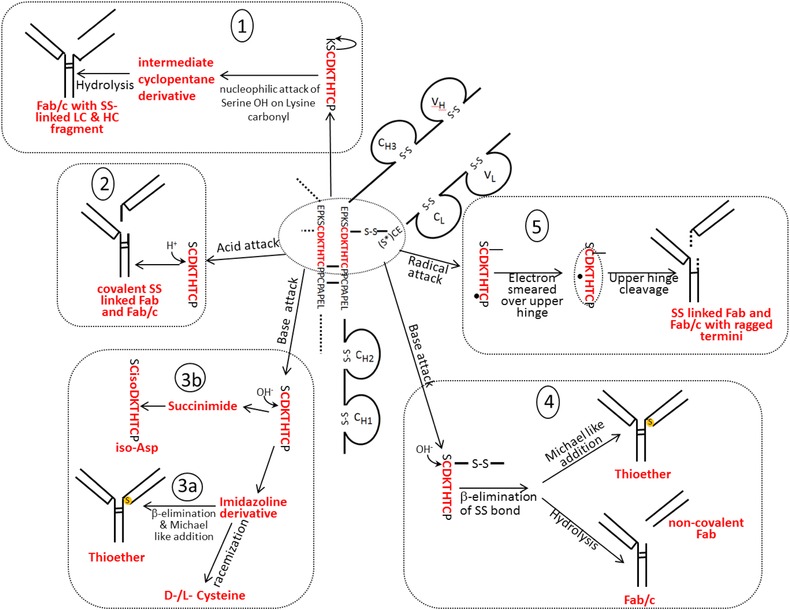
These investigations elucidated the underlying reaction mechanisms that lead to diverse hinge modification and fragmentation (see also Fig. 2):
-
(1)
Disulfide‐linked Fab fragments 17, 19 were described as a result of radical transfer between the LC‐HC disulfide bond and the first HC‐HC disulfide bond including the upper hinge His as radical center. This resulted in cleavage of any one peptide bond between these two disulfide bonds (formation of ragged termini or ladder formation as denoted above). Disulfide‐linked Fab fragments did also result from C‐terminal Asp cleavage under acidic conditions (mechanisms 2 and 5 in Fig. 2).
-
(2)
Noncovalent Fab (Fab part of the cleaved HC without disulfide bridge to adjacent LC) and Fc (Fc part of the cleaved HC) were described as a result of β‐elimination at the LC‐HC disulfide bond with subsequent hydrolysis under basic conditions (mechanism 4 in Fig. 2) 23 or by radical induced disulfide cleavage at the C‐S bonds 17. In the occurring fragments, sulfurized cysteins were also observed 17.
-
(3)
Formation of antibody fragment that results from loss of one Fab (Fab/c) with disulfide‐linked LC and Fab fragment of the HC was attributed to a nucleophilic attack of the serine hydroxyl on the lysine carbonyl with subsequent N‐terminal cleavage 11 (see mechanism 1 in Fig. 2).
-
(4)
In case of IgG1, thioether bond (lanthionine) formation was observed between the C‐terminal Cys of the LC and its counterpart in the HC 21, 23, 24, 46 (mechanisms 3 and 4 Fig. 2). Thioether bond formation was exclusively found in this disulfide bond, which was attributed to the exposed and flexible hinge region and the C‐terminal position of the LC Cys 46. In reduced CE‐SDS the thioether was found to be usually present at the 0.5–2% level of total protein 46.
Different mechanisms were proposed for thioether bond formation:
-
(i)
Formation of dehydroalanine by β‐elimination at the LC‐HC disulfide bond with subsequent Michael‐like addition (mechanism 4 in Fig. 2 23).
-
(ii)
Formation of a cyclic imidazoline intermediate (mechanism 3 in Fig. 2 21).
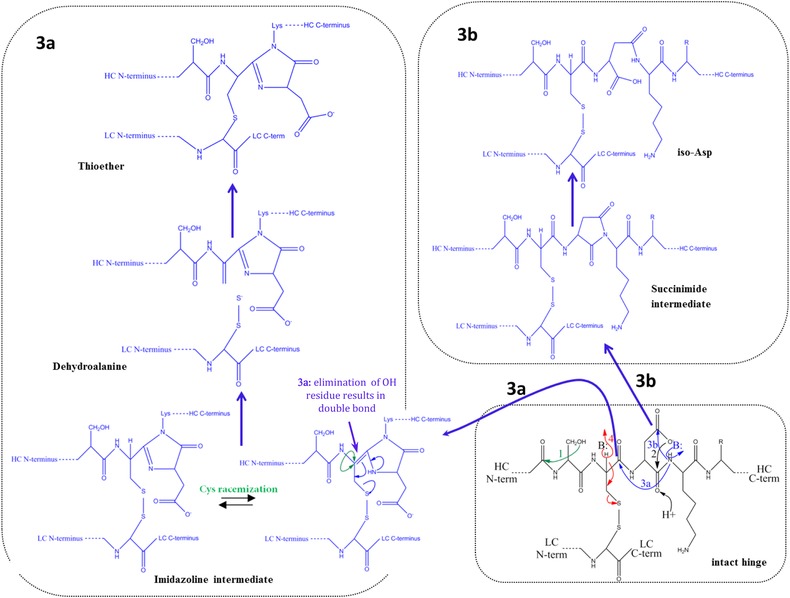
Figure 2.
Main fragmentation and modification processes in the upper hinge region. Mechanism 1 (A and B): Nucleophilic attack of serine OH on lysine carboxyl at pH 3–8 with subsequent hydrolysis (according to Vlasak et al. 11). Mechanism 2 (A and B): C‐terminal Asp cleavage is induced by protonation of the carbonyl of the peptide bond between Asp and Lys. Ring closure between the carbonyl carbon and the oxygen of the Asp carboxylgroup is succeeded by hydrolysis of the peptide bond (according to Vlasak et al. 11). This reaction is preferred under acidic conditions (below pH 5). Mechanism 3a (A and C): Abstraction of a proton from the nitrogen of the peptide bond between the upper hinge Asp and Lys and ring closure with the carbonyl of the Asp‐Cys peptide bond leads to an imidazoline derivative whose racemization leads to d‐Cys. Further β‐elimination of the disulfide bond and subsequent Michael addition leads to a thioether linkage. Mechanism 3b: Abstraction of a proton from the nitrogen of the peptide bond between the upper hinge Asp and Lys and ring closure with the carbonyl of the Asp side chain leads to a succinimide intermediate and subsequent hydrolysis to iso‐aspartate (according to Amano et al. 21). Mechanism 4 (A and B): Base attack leads to β‐elimination of the disulfide bond and formation of dehydroalanine. Subsequent hydrolysis leads to a noncovalent Fab amide and an Fab/c (pyruvoyl at the N‐terminus of the cleaved HC), whereas Michael‐like addition leads to Thioether formation (according to Vlasak et al. and Cohen et al. 11, 23). This reaction is preferred under basic conditions. Mechanism 5 (A): Radical attack of the C‐terminal upper hinge disulfide bond leads to sulfenic acid (Cys‐SOH) on one Cys and a thiyl radical (Cys‐S●) on the other 18. The thiyl radical transfers its electron to the upper hinge and induces backbone cleavage somewhere within the upper hinge. This forms Fab and Fab/c fragments with ragged termini 18, 23. The upper hinge histidine is assumed to be an important radical center during this process 19.
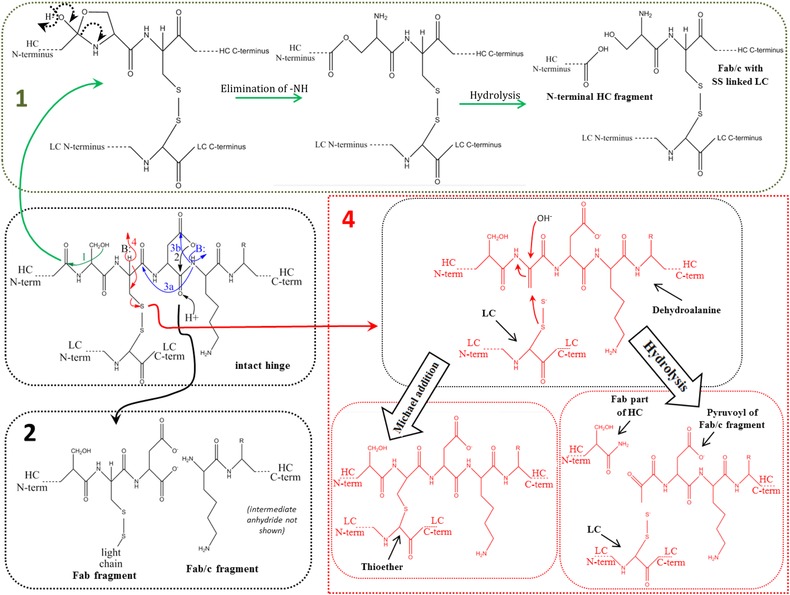
The portion of the respective thioether‐linked peptide was found to be increased after temperature stressing 46. Thioether bond formation was also increased after high pH stressing 24, which suggests a β‐elimination mechanism. Interestingly, λ LCs were more prone to thioether bond formation than κ LCs 24. It was assumed that the proximity of the carboxylic charge group at the C terminus inhibits the racemization on the κLC Cys 24.
Other thioether linkages between Cys and Asp 63, glutamic acid 63, histidine 64, or tyrosine 65 were also reported.
-
(1)
Cys racemization 21, 22 is a result of a base‐catalyzed attack of the N‐terminal peptide bond nitrogen of the upper hinge lysine on the C‐terminal peptide bond carbonyl carbon of the N‐terminal upper hinge Cys and subsequent formation of a transient imidazolone ring (mechanism 3 in Fig. 2). It was also described as a result of base‐catalyzed Cys racemization at the LC‐HC disulfide bond with dehydroalanine as intermediate 22 (mechanism 4 in Fig. 2). It occurred on both Cys in presence of lambda LCs (IgG1λ), but mainly on the HC Cys in case of kappa LCs (IgG1κ). A repulsion of nucleophiles by negatively charged carboxylic group at the C terminus was assumed to be the reason 22. After high pH stress, d‐Cys levels were higher for λLC of an IgG1 antibody in comparison to the levels in an IgG2 antibody. This is in agreement with the respective levels in endogenous antibodies, whose sensitivity to chemical reduction and racemization followed a similar order: IgG1λ > IgG1κ > IgG2λ > IgG2κ 34.
-
(2)
Also Asp isomerization was described as a result of this complex reaction mechanism. Isomerization of Asp is assumed to be induced by nucleophilic attack of the N‐terminal peptide bond nitrogen of the upper hinge lysine on the side chain carbonyl of the upper hinge aspartate. This then results in a cyclic succinimide intermediate 21 (mechanism 3 in Fig. 2). The intermediate can hydrolyze back to the original Asp form or to the iso Asp form. Partial positive charge of the side chain carbonyl carbon is higher in case of protonated carboxyl. Therefore, Asp isomerization is supported by acidic pH. Limited LysC proteolysis generated Fab and Fc fragments by C‐terminal cleavage at upper hinge lysine. After isomerization to d‐aspartate this cleavage was prevented, which explained partial resistance to this cleavage after IgG1 incubation under mildly acidic conditions at elevated temperatures. No relevant Asp isomerization occurred at 2–8 °C. However, it became important at elevated temperatures 61.
2.3. Disulfide scrambling
Serum and/or intracellular compounds such as albumin, Cys, glutathione, and homocysteine have an influence on the secondary and tertiary structure of antibodies. They were found to rearrange disulfide bonds, especially in the case of IgG2 and IgG4 antibodies 1.
2.3.1. Dimerization and disulfide bond shuffling of IgG2 antibodies
Typical characteristics of IgG2 MAbs are already described in Section 1. It was shown that the arrangement of disulfide bonds in this antibody subtype supports dimerization and disulfide bond shuffling:
-
(i)
IgG2 MAbs frequently exhibit low affinity to their antigens and were found to occur as binding‐active dimers (66 and Fig. 3). However, the higher number of binding sites that arise from dimerization is expected to increase binding by avidity effects 5, 66. IgG2 MAbs often bind to carbohydrates on bacterial surfaces with relatively low affinity. Dimerization then increases binding and thus protection. IgG2 dimers were detected by nonreduced SDS‐PAGE. In case of reduced CE‐SDS of the same sample, only LC and HC bands were visible 66. Cyanogen bromide cleavage analysis indicated peptides that are linked by cystin bridges in the hinge region 66. These results demonstrated that dimers are covalently linked by disulfide bonds of the hinge. Replacing one of the IgG2 hinge Cys with an inert amino acid may prevent dimerization 5, 66, which is expected to be advantageous for IgG2 therapeutics since in these cases cross‐functionalities may not be desired (see Section 3.6).
-
(ii)
IgG2 disulfide bond shuffling in serum leads to IgG2‐A, IgG2‐B, and IgG2‐A/B isoforms [67, 68, 69 and Fig. 4]. By using a flow‐through dialysis system, conversion kinetics of the IgG2‐A, IgG2‐A/B, and IgG2‐B isoforms were studied in vitro under physiological‐like conditions 70. The three isoforms were separated by CEX‐HPLC and RP‐HPLC 68, 69 and the obtained fractions were further characterized by nonreduced Lys‐C peptide mapping with subsequent LC/ESI‐MS 68. For IgG2κ antibodies, in vivo and in vitro shuffling kinetics matched well, which shows that it is not significantly influenced by clearance. If there had been preferred clearance of one isoform over the others, the steady state ratio between IgG2‐A, IgG2‐B, and IgG2‐A/B would have been shifted in comparison to the in vitro ratio. However, the isoform distribution is influenced by the different C‐termini of the κ and λ LCs 70. In IgG2κ antibodies, formation of IgG2‐B from IgG2‐A/B was faster than the same conversion of IgG2λ antibodies 70. By nonreducing peptide mapping and Edman sequencing even more disulfide subvariants of the IgG2‐A/B and IgG2‐B structures could be identified 71. IgG2‐A1 and IgG2‐A2 isoforms with slight differences in disulfide linkages are also described elsewhere 72. A2 was more resistant to reduction and had a lower Fc glycan flexibility.
Figure 3.
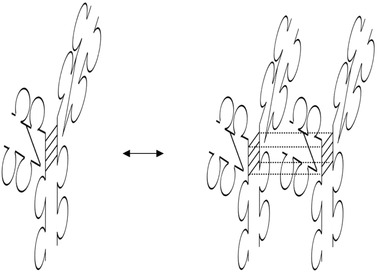
Formation of functional covalent dimers between identical as well as different IgG2 molecules.
Figure 4.
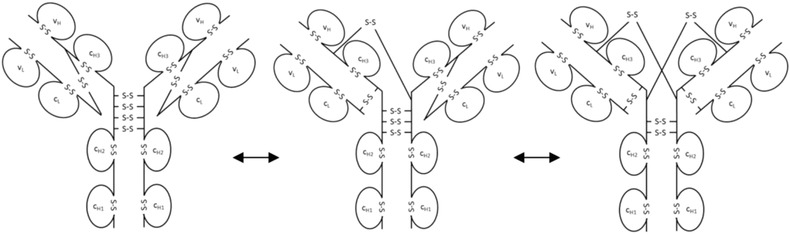
From left to right: IgG2‐A, IgG2‐A/B, and IgG2‐B isoforms.
2.3.2. Formation of half IgG4 antibodies and Fab arm exchanges
Endogenous and therapeutic IgG4 antibodies can exchange HC/LC parts in circulation that results in undesired bispecific antibodies 1, 73, 74, 75 (Fig. 5). This can be attributed to an equilibrium between inter‐ and intrachain cystines of the two HCs that are not always linked covalently 76. In vivo, this mechanism requires reducing conditions in blood or at cell surfaces. It was also described that interactions between the CH3 domains play a role in Fab arm exchange between two MAbs 73, 77. From a faster exchange of IgG4 Fc fragments it was concluded that Fab arms stabilize the covalent interactions between the HCs 78. Hinge mutations (e.g. from CPSC to CPPC) have been performed in order to prevent this undesired exchange in therapeutic IgG4 molecules 1, 76, 79, 80, 81.
Figure 5.
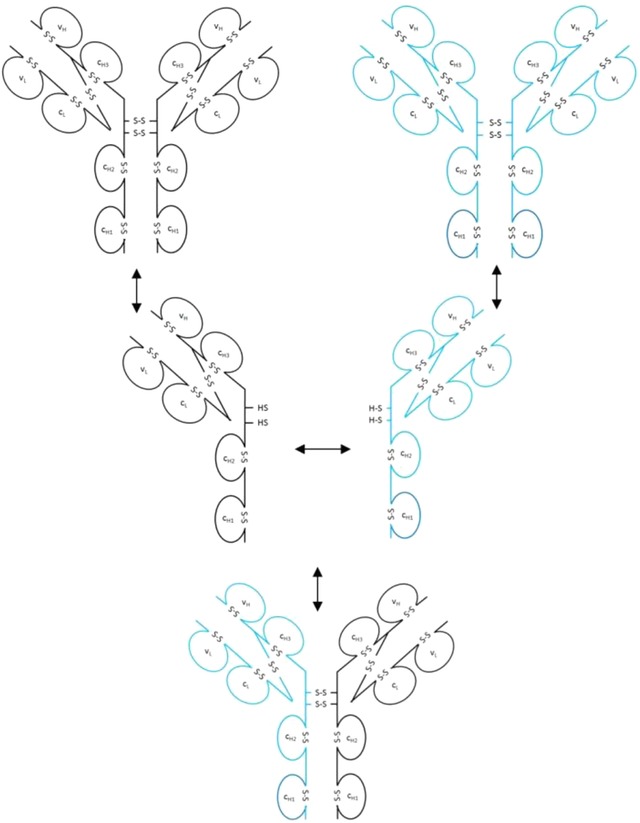
Exchange of Fab arms between endogenous and therapeutic IgG4 molecules.
Antibodies are normally directed against foreign antigens and help the body to defend itself against exogenous matter. However, in case of autoantibodies, these react with self‐antigens and protection mechanisms are needed. Due to their weak Fc effector functions (see Section 1), IgG4 antibodies are not harmful themselves in this respect but instead block harmful IgG1 autoantibodies from binding 5. If an IgG1 autoantibody binds to self‐epitopes, harmful inflammatory Fc effector cascades may be the result. This is prevented by IgG4 epitope occupancy and explains why therapeutic IgG4 are preferred for immunotherapies. The weak inter HC disulfide bonds and the HC/LC exchange are assumed to be a common anti‐inflammatory protection mechanism in vivo 5, 73, 82, since they prevent IgG4 molecules with two different specificities from cross‐linking self‐antigens 83, 84.
Due to their noncovalent interaction, half IgG4 antibodies are not detected by non‐denaturing assays. That makes denaturing SDS–PAGE the method of choice 85. Half IgG4 antibodies were also analyzed with nonreduced CE‐SDS after 35S methionine labeling and immune precipitation 76. Formation of bispecific IgG4 MAbs was shown by ELISA 73, 81 and by measurement of antibody mediated cross‐linking of a bound antigen with a radioactively labeled second antigen 74. The exchange rates of half IgG4 or fragments thereof were also monitored by SEC, SDS‐PAGE, mixed mode chromatography, LC–MS, and Foerster resonance energy transfer (FRET) assays 77, 78, 81. The latter method did not require an often incomplete quenching of disulfide exchange reactions by iodacetamide and therefore was preferred 78.
2.4. Trisulfide formation
Trisulfides result from insertion of an additional sulfur atom into a disulfide bond by the following mechanism 47:
They were found to occur in the heavy‐light and heavy‐heavy interchain linkages but not in the intrachain disulfides of IgG1, IgG2, IgG3, and IgG4 47. The highest levels were observed in the heavy‐LC linkage 47. Production of H2S by mammalian cells 86 and high Cys concentration in the cell culture medium 87 were described as sources for trisulfide formation during cell fermentation. Culture duration, cell density, and feeding strategies were also shown to influence the trisulfide level 47. Depending on the culture conditions, the trisulfide levels can be in the range of 1 to 40% 47, 87, 88. In endogenous antibodies, trisulfide levels in the LC‐HC disulfide linkage were found to be less than 0.1% 24. In case of antibody drug conjugates that use disulfides for linking the drug to the antibody, it was found that the trisulfide levels influence the requested TCEP:antibody molar ratio that is needed to achieve the intended drug‐to‐antibody ratio in the product 89.
The relative content of trisulfides at the LC‐HC disulfide linkage can be determined by peptide mass fingerprinting after alkylation with 4‐vinylpyridine and Lys‐C digestion 47. This method provided the relative content of the trisulfide‐linked peptides. Random localization is assumed and binominal distribution can be applied (47 and Table 2).
Table 2.
Binominal distribution can be applied for calculation of the relative amount of the expected species (x = relative content of the trisulfide linked LC‐HC peptides)
| Species | Relative content |
|---|---|
| No trisulfide at both LC‐HC disulfide linkages | (1 − x)2 × 100% |
| Trisulfide at one LC‐HC disulfide linkage | x(1 − x) × 100% |
| Trisulfide at both LC‐HC disulfide linkages | x 2 × 100% |
2.5. Cystein modification by cysteinylation, glutathionylation, or binding of further LCs
Cysteinylation has been observed in the variable region of a recombinant monoclonal IgG1 antibody 14, 90. S‐Gluthathionylation sites were predicted in silico and were shown to depend on the surrounding structure 29. Cysteinylated and noncysteinylated forms could be separated by cation exchange (CEX) and SEC 14 or by limited Lys‐C digestion with subsequent LC/MS analyses 90. In another study glutathionylation was detected by MALDI‐TOF/TOF, nano LC‐MS/MS analysis (for peptides), or nano ESI‐Q‐TOF‐MS (for intact molecules) 91.
Open disulfide bonds in the complementarity determining regions (CDRs) of IgG1 LCs were also found to form disulfide bonds with an additional FLC resulting in an IgG with three LCs 30. The free Cys at the C‐terminus of this third LC was found to form a disulfide bridge to the C‐terminal Cys of a fourth LC that then results in an IgG1 variant with four LCs. Binding of additional LCs was detected by SEC or CE‐SDS in these studies 30.
2.6. Aggregation
Despite of open disulfide bonds, hydrophobic and electrostatic interactions seem to be sufficient for the formation of the correct tertiary structure of antibodies. Nevertheless, cleavage of disulfide bonds results in lower protein stability (e.g. in case of heat or agitation stress), mispairing of disulfide bonds, and may thereby lead to aggregation 10, 92, 93, 94, 95, 96, 97. The stabilizing effect of intrachain disulfide bonds is in the range of 19 kJ/mol 98. Correlation of free –SH content with circular dichroism (CD) showed that a higher level of free –SH lowers thermal (T m) stability 58. That explains why an IgG2 antibody was found to form protein particles during agitation stress 10. In SEC, more than 40% of this particle protein appeared as high molecular weight species. Denatured SEC in the presence of 2% SDS and 6 M urea (dSEC) did not lead to dissociation of these aggregates and confirmed the covalent nature of the interactions. In contrast, DTT was able to reduce the aggregates to light and HCs, which showed that disulfide bonds were responsible for this particle formation 10. Also for an Fc‐fusion protein, SDS‐PAGE was applied to show that aggregate species purified from SEC were covalently linked by disulfide bonds. In this case treatment with l‐Cys was sufficient to initiate covalent aggregation of purified monomer via disulfide‐bond shuffling 31. Aggregation or fragmentation can be caused by light induced CysS• radicals and by subsequent carbon‐centered radicals as well 40. It was found that in case of IgG1, aggregation is favored at alkaline pH and fragmentation is favored at acidic pH 99. Due to intermediary carbon‐centered radicals and resulting carbon involvement in subsequent covalent bonds some of these aggregates cannot be dissociated under reducing conditions. Besides dityrosine formation 100 also other carbon‐carbon bonds or carbon‐sulfur bonds may form this type of aggregates 40. IgG4 antibodies were found to be more prone to aggregation than IgG1 antibodies 44, 62. That correlated with a higher conformational stability of IgG1 during temperature stress.
3. Assessment of criticality of disulfide‐related modifications influence on safety, potency, and pharmacokinetics
A criticality assessment of modifications in therapeutic antibodies should comprise a detailed evaluation of the impact on safety, potency and PK properties. While safety is mainly related to adverse events such as formation of anti‐drug antibodies 15, 16, loss of potency, and altered clearance impede the effectiveness of the drug.
For safety evaluations, in vitro drug stability can be complemented by assessment of in vivo levels of the respective modifications, i.e. investigating the steady state between formation and depletion 1, 13. In addition, in vivo occurrence in endogenous antibodies and in vivo repair of modifications may lead to positive safety assessments (see also subsequent sections for more details). However, some modifications may result in critical structural perturbations that lead to for example:
-
(1)
Aggregation after oxidation of open disulfide bonds, i.e. formation of wrong intermolecular disulfide bonds (Section 2.6)
-
(2)
Structural changes after cysteinylation (Section 3.8)
-
(3)
Cross‐specificities between therapeutic and endogenous antibodies (IgG2 and IgG4) (Section 3.6)
The extent of potentially harmful modifications depends on the antibody and requires MAb‐specific assessments.
The influence of modifications or cleavage processes on the potency of a therapeutic generally depends on the mode of action (e.g. monovalent or bivalent binding, requirement of Fc effector functionalities, or detailed mechanism of CDR‐epitope interaction). The relevance of modifications may be directly derived from the involvement of affected functionalities in the overall mode of action.
PK properties such as clearance are influenced by size and neonatal Fc receptor (FcRn) binding properties. Small fragments such as the LC (FLC, Bence Jones protein) are cleared by the kidney. They can be found in the urine and in serum 101, 102. However, loss of larger serum proteins is prevented by a size selective barrier in the kidney 103. Whereas Fabs are still partially segregated by the kidney 104, clearance of larger fragments such as Fab/c or Fc appears to depend on FcRn‐binding properties that in turn can themselves be affected by modifications or cleavage processes. In case of decreased binding to the FcRn receptor recycling of antibodies through endosomes is impeded, which then correlates with reduced serum half‐life 1, 105, 106, 107, 108. However, slow clearance is not always desired. In order to limit functionality on single organs FcRn‐binding activity is sometimes even removed from therapeutic antibodies. Reduction of FcRn‐binding properties is also discussed for antibodies that are used for acute therapies 109.
3.1. Open inter‐ and intrachain disulfide‐bonds
As already mentioned, safety concerns may be reduced by the presence of endogenous repair mechanisms and by the detection of some modifications in endogenous antibodies.
Due to the high number of hydrogen bonds, salt bridges, and hydrophobic interactions between Ab chains, cleaved Cysteines are still assumed to be structurally adjacent. This structural integrity supports reformation of correct cystines and explains why the presence of “redox” compounds such as albumin, Cys, cystine, glutathione, or homocysteine in serum enabled closure of open Cys‐22 Cys‐96 disulfide bonds of IgG1 VH domains in vivo 1, 57. In addition, the CE‐SDS peak levels of LC, HC, HC/HC, HC/LC, and HC/HC/LC in cell culture fluid were found to decrease with time 34. These observations indicated that disulfide bonds can reform by endogenous or cell‐based redox‐repair systems.
Free Cys residues were found similarly in endogenous IgG1 and IgG2 10, 110 as well as in recombinant IgG1 and IgG2 antibodies 10. To a small extent all endogenous IgG1 CH and CL domains contain open disulfide bonds. For IgG2 they were more pronounced in CH domains 10. Free Cys levels were in the range of 0.17–0.59 mol per mole of antibody and were slightly lower for IgG1 antibodies than for IgG2 antibodies 10. The content of open disulfide bonds in recombinant and endogenous antibodies was comparable.
Also FLCs are present (free light chain, Bence Jones protein) in urine and serum 101, 102. The serum concentration of FLC arises from the steady state between FLC production and renal clearance. Typical FLC half‐lives are in the range of 2 to 5 h and typical serum concentrations were found to be in the range of 3.3–19.4 mg/L for κFLC and 5.7–26.3 mg/L for λFLC 101, 111.
In general, knowledge about in vivo abundance in combination with potential in vivo repair may influence how criticality of open disulfide bonds or respective cleavage products in administered biopharmaceuticals is regarded. However, reduced structural stability in absence of natural disulfide bonds leading to the formation of aggregates has to be considered. Contrary to widely held opinion, findings using a particular mAb (mAb1) demonstrated that noncovalently modified aggregates are not immunogenic 16. Nevertheless, it has been claimed using animal models 16, 112, 113 that aggregates or misfolding may induce immunogenic responses.
In conclusion, open disulfide bonds are a common endogenous phenomenon that can apparently be repaired by in vivo redox processes. However, open disulfides may also induce structural perturbation and aggregation that may impact patient safety.
As discussed before, disulfide bonds are expected to be partly reduced in therapeutic antibody preparations. Therefore, it is important to gain understanding of the effect of disulfide bond cleavage on different antibody functionalities.
For example, the binding activity may be affected by open disulfide bonds in the CDR. In a study about an IgG1, unpaired cysteins (Cys‐22 and Cys‐96) were found within the variable domain of the HC. Binding and complement‐dependent cytotoxicity activities of reduced and intact Fab could be compared 49. In this case no influence on the activity was found 49. This can be explained by other interactions that in this case seem to be sufficient for maintaining the original conformation and binding activity 94.
However, for Omalizumab 57 and other MAbs 55, 56 incomplete disulfide bond formation has been shown to impede binding activity and to reduce potency of the molecule 1, 3, 55, 110. This shows that the effect of unpaired cysteins strongly depends on the detailed structure of the antibody and its interaction with the antigen.
In case of disulfide cleavage between the two HC immune effector functionality may be reduced. This assumption is based upon investigations that have shown that a single proteolytic cleavage within the lower hinge of trastuzumab reduces immune effector function and in vivo efficacy 114.
Beside safety and potency also the influence of open disulfide bonds on the PK properties, i.e. FcRn‐binding properties of the regarded MAb and increasing renal clearance of smaller cleavage products should be discussed.
Influence of disulfide cleavage on the molecular size and renal clearance strongly depends on noncovalent interactions that are still present after disulfide cleavage and may prevent complete dissociation. In this case, disulfide cleavage may not influence renal clearance.
There are hints that one HC is sufficient for full FcRn functionality of an antibody. Two studies confirmed that oxidation of methionine residues in the FcRn‐binding region decreased the affinity for the FcRn receptor 1, 105, 115. However, the oxidation of only one chain at this position did not lead to faster clearance 26 and the same may apply to cleavage of HC‐HC interchain disulfide bonds.
3.2. Hinge fragmentation
Fab fragments are often used therapeutics. LUCENTIS® (ranibizumab) is a Fab fragment that binds to the vascular epidermal growth factor and thus prevents ingression of chroidal capillaries into the retina 8. Other therapeutic Fabs are directed toward poisons such as Digoxin (Digibind), colchicine, and other compounds such as tricyclic antidepressants 116. Ongoing treatment and respective clinical studies have not shown any notable side effects or allergic reactions 117 and antibody fragments were tolerated 116, 118, 119. Transferability of these findings to randomly cleaved Fab may not be possible and requires an individual assessment. The same applies to other hinge cleavage products such as Fab/c.
Upper hinge cleavage can have an influence on epitope binding (loss of bivalent binding properties), Fc effector (cleavage in the lower hinge) 114, and FcRn functionalities 120. Therefore, impact of hinge cleavage should always be discussed on the basis of the mode of action requirements that may include not only antigen binding but also Fc effector (e.g. antibody‐dependent cell cytotoxicity) and FcRn binding (PK) capabilities.
Lower hinge fragmentation was found in vivo and had a detrimental effect on Fc effector functions. Metalloproteinases secreted by tumors cleave human IgG1 in the lower hinge which in nonreduced SDS‐PAGE resulted in respective C‐terminal Fc part of the HC and its disulfide‐linked Fab/c counterpart 114. In this study, cleaved trastuzumab was found in human breast cancer tumor tissue. In spite of still having one complete HC the occurring Fab/c fragments had reduced immune effector function (especially FcγRIIA and FcγRIIIA). Tumor tissues that were treated with this fragment showed reduced immune cell infiltration compared with trastuzumab‐treated tissues 114.
Due to loss of Fc functionality a faster clearance is expected for covalent Fab (see Section 2.2). Typical half‐life of Fab fragments is 12–20 h 116. However, in renal failure the half‐life of Fab fragments is prolonged. Fab fragments then remain detectable in plasma for 2–3 wks after administration 116, 121. In case of noncovalent Fab fragmentation (see Section 2.2), the resulting LC and the respective HC fragment should be rapidly cleared by the kidney 122. However, that is only in case of complete dissociation which may be impeded by noncovalent interactions. Clearance of larger fragments such as Fab/c strongly depends on FcRn functionalities. After endosomal intake larger fragments with impeded FcRn‐binding properties are not recycled to the cell surface and are then cleared faster. For 36 IgG molecules FcRn binding was found to be comparable and it was concluded that the antibody‐specific Fab domain does not significantly change FcRn binding 123. However, in another study with briakinumab, ustekinumab and engineered variants thereof, the Fab domain was found to be involved in FcRn binding and thereby influence FcRn‐dependent terminal half‐lives 120. The effects observed in the latter study may be reduced by hinge cleavage.
3.3. Formation of thioether bonds
Safety concerns are again assumed to be tempered by the natural occurrence of thioethers at the LC‐HC linkage 24. This was tested for endogenous IgG1 from healthy persons and for myeloma patients. The lower thioether level of the myeloma patients was attributed to the shorter circulating half‐life of the myeloma IgG. After intravenous injection of a therapeutic IgG1κ antibody a thioether conversion rate of about 0.1 %/day was found 24. This agrees very well with in vitro conversion rates (0.09%/day) 24. Higher in vitro degradation rates were found for IgG1λ (0.16%/day) that again agreed with the higher in vivo thioether levels of IgG1λ 24.
Thioether bond formation could have an influence on the flexibility of the Fab. Therefore, especially bivalent binding may be affected 24.
3.4. d‐Cys formation
Therapeutic antibodies can form d‐Cys at the HC‐LC disulfide bond 22. d‐amino acid residues have been detected in long‐lived proteins and play a critical role in aggregation 124, 125, 126. However, as for thioethers, the criticality of d‐Cys should also be assessed in the context of its natural appearance and its location within a conserved region. Like endogenous thioethers, also endogenous d‐Cys were found in IgG1 and IgG2 antibodies of myeloma patients and healthy volunteers 22. That was determined by RP‐HPLC‐MS separation of respective Lys‐C peptides under reducing conditions.
3.5. Asparagine isomerization
Asp isomerization is no disulfide modification. But since it is directly linked to thioether bond formation and Cys racemization (see reaction mechanism 3a and b in Fig. 2) it has been added to this discussion.
Asp isomerization was negligible under recommended storage conditions (5°C) 61. That could be shown by limited LysC proteolysis at K222, which results in Fab and Fab‐Fc fragments instead of Fc and Fab (one cleavage site blocked by isomerization 61). Iso‐Asp does also occurs in endogenous MAbs 61, 90, 127) and is involved in the determination of protein age 61. In addition, endogenous l‐isoaspartyl methyltransferase (PIMT) converts l‐isoaspartyl back to normal l‐aspartyl 126, which means that in vivo repair systems are available. In summary, low abundance, in vivo abundance, involvement in natural processes, and natural repair systems support a positive safety evaluation for this modification.
3.6. Disulfide‐bond scrambling
Endogenous human myeloma IgG2 showed the same disulfide bond heterogeneity as a human monoclonal IgG2 antibody 68. The same applies to IgG2 dimers that were also detected in pooled human γ globulin (Miles IgG) and in serum.
IgG2 is important for recognition of carbohydrate antigens in humans 128, 129 and for defense against encapsulated bacterial pathogens 130, 131, 132, 133. Dimerization increases affinity of human IgG2 to bacterial surfaces. These special characteristics of IgG2 isotypes are deemed to be important for their role against low affinity carbohydrate antigens 66. On the other hand, formation of bispecific IgG4 antibodies is assumed to be a common anti‐inflammatory protection mechanism in vivo 5, 73, 82.
Effect of disulfide bond shuffling on biological binding activity depends on individual molecular properties of regarded IgG2 MAbs 69. The same IgG2A, IgG2A/B, IgG2B conversion pattern was found for all IgG2 MAbs. In some cases adjustment of isoform ratios allows a modulation of binding activity. However, in dependence on individual molecular characteristics in the CDR, IgG2A, IgG2A/B, and IgG2B forms do not always have different binding activities 68, 69.
In conclusion, the same disulfide bond re‐shuffling is also observed for endogenous antibodies, which supports the assumption that an analog disulfide bond re‐shuffling in therapeutic products is safe.
However, heterodimers of therapeutic IgG2 with endogenous IgG2 molecules (cross‐functionalities), homodimers of therapeutic IgG2 molecules (higher affinity), bispecificity after Fab arm exchanges of therapeutic IgG4 with endogenous IgG4 MAbs 76 or IgG2 isoform conversion (especially in combination with altered affinities) may be critical or could influence functionality 5, 73. Therefore, modifications that disable these special features are considered (1 and Section 2.3). The FDA requested some mitigation action for IgG4 antibodies 134. So far, preclinical or clinical safety data did not show any safety concerns with therapeutic IgG2 and IgG4 antibodies 5.
3.7. Trisulfide formation
Trisulfides were observed in endogenous human IgG1, IgG2, IgG3, and IgG4 antibodies. Their ubiquitous presence can be attributed to H2S in human tissue 47, 135, 136. After in vivo circulation in rat trisulfides converted back into disulfides 47, which demonstrated the presence of physiological repair systems. A high trisulfide level did not show a significant effect on the function or stability of the antibody 47. In summary, these findings support a positive safety evaluation.
3.8. Cysteinylation and additional LCs
Criticality of cysteinylation has been addressed by Banks et al. 14. Cysteinylation was shown to impede the structural integrity of antibodies (i.e. alteration of the tertiary or quaternary structure). This decreased the melting point and increased the aggregation rate. Also, the bioactivity was shown to be impeded by cysteinylation in the Fab region. In this study, a direct correlation between cysteinylation and reduced biological activity was found 14.
Potency was also reduced after binding of additional LCs to the CDR, which was attributed to a reduced accessibility of the antigen‐binding site 30.
4. Conclusions
Due to their relatively low dissociation energy, disulfides are involved in many different modification and fragmentation processes such as hinge cleavage, disulfide cleavage, thioether bond formation, racemization, aggregation, dimerization, trisulfide formation, cysteinylation, glutathionylation, and disulfide bond shuffling. The high flexibility of the hinge region is responsible for its strong involvement in these processes, which are induced by chemical stress (e.g. pH or redox stress), by light stress (radical formation) or by incomplete processing during in vivo production. Often these modifications are an inherent characteristic of the regarded IgG subtype (e.g. disulfide bond shuffling of IgG2 and IgG4 antibodies). A high number of different methods such as MS (mass spectrometry), CE‐SDS, hydrophilic interaction chromatography, or RP‐HPLC are available for characterization or quantification of these modifications.
Formation of thioethers, iso‐Asp, open disulfide bonds, d‐Cys, and trisulfides often occurs located in conserved regions and is also commonly observed in endogenous antibodies. Its appearance in endogenous antibodies may temper safety concerns. However, other sulfhydryl induced modifications such as aggregation or formation of heterodimers (cross‐specificities) are considered to be more critical.
Influence on potency strongly depends on the mode of action, i.e. monovalent or bivalent binding and requirement of Fc receptor functionalities. With respect to potency, hinge cleavage may be less critical in case of monovalent binding or nonrequired Fc receptor functionality. Influence of open intrachain disulfide bonds on binding activity depends on individual properties of the CDR. Cysteinylation of sulfhydryl groups was shown to impede functionality.
Influence of modifications on serum half‐life has always to be regarded in the context of desired PK profile and desired location of bioactivity. Cleavage may result in smaller fragments that are faster cleared by the kidney. Disulfide and hinge related fragmentation or modification processes may influence FcRn functionality. But a long serum half‐life is not always required.
Disulfide bonds and the hinge region represent an important excerpt of the whole set of potential critical quality attributes that have to be controlled for ensuring product quality. Many of the observed modifications are also present in endogenous antibodies. In conclusion, focus should be on critical modifications that have to be part of the control system of therapeutic antibodies.
The authors have declared no conflict of interest.
Acknowledgements
The authors thank Dietmar Reusch for careful review of the manuscript. They also acknowledge Christof Finkler and Harald Wegele for enabling this work.
Colour Online: See the article online to view Fig. 2 in colour.
5 References
- 1. Correia, I. R. , MAbs. 2010, 2, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Sorge, N. M. , van der Pol, W. L. , van de Winkel, J. G. , Tissue Antigens. 2003, 61, 189–202. [DOI] [PubMed] [Google Scholar]
- 3. Jefferis, R. , Biotechnol. Prog. 2005, 21, 11–16. [DOI] [PubMed] [Google Scholar]
- 4. Niwa, R. , Natsume, A. , Uehara, A. , Wakitani, M. , Iida, S. , Uchida, K. , Satoh, M. , Shitara, K. , J. Immunol. Methods. 2005, 306, 151–160. [DOI] [PubMed] [Google Scholar]
- 5. Salfeld, J. G. , Nat. Biotechnol. 2007, 25, 1369–1372. [DOI] [PubMed] [Google Scholar]
- 6. Jefferis, R. , Expert Opin. Biol. Ther. 2007, 7, 1401–1413. [DOI] [PubMed] [Google Scholar]
- 7. Williams, A. J. , Giese, G. , Persson, J. , Biotechnol. Prog. 2015, 31, 1315–1322. [DOI] [PubMed] [Google Scholar]
- 8. Mohr‐Andra, M. , Mohr, K. , Pharm. Unserer Zeit. 2007, 36, 437–440. [DOI] [PubMed] [Google Scholar]
- 9. Liu, H. , Gaza‐Bulseco, G. , Lundell, E. , J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 876, 13–23. [DOI] [PubMed] [Google Scholar]
- 10. Huh, J. H. , White, A. J. , Brych, S. R. , Franey, H. , Matsumura, M. , J. Pharm. Sci. 2013, 102, 1701–1711. [DOI] [PubMed] [Google Scholar]
- 11. Vlasak, J. , Ionescu, R. , MAbs. 2011, 3, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang, G. S. , Chen, Y. S. , Yeh, H. W. , Nano Lett. 2006, 6, 2467–2471. [DOI] [PubMed] [Google Scholar]
- 13. Goetze, A. M. , Schenauer, M. R. , Flynn, G. C. , MAbs. 2010, 2, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Banks, D. D. , Gadgil, H. S. , Pipes, G. D. , Bondarenko, P. V. , Hobbs, V. , Scavezze, J. L. , Kim, J. , Jiang, X. R. , Mukku, V. , Dillon, T. M. , J. Pharm. Sci. 2008, 97, 775–790. [DOI] [PubMed] [Google Scholar]
- 15. Wang, Y. M. , Jawa, V. , Ma, M. , Bioanalysis. 2014, 6, 79–87. [DOI] [PubMed] [Google Scholar]
- 16. Bessa, J. , Boeckle, S. , Beck, H. , Buckel, T. , Schlicht, S. , Ebeling, M. , Kiialainen, A. , Koulov, A. , Boll, B. , Weiser, T. , Singer, T. , Rolink, A. G. , Iglesias, A. , Pharm. Res. 2015, 32, 2344–2359. [DOI] [PubMed] [Google Scholar]
- 17. Yan, B. , Boyd, D. , J. Biol. Chem. 2011, 286, 24674–24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan, B. , Yates, Z. , Balland, A. , Kleemann, G. R. , J. Biol. Chem. 2009, 284, 35390–35402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yates, Z. , Gunasekaran, K. , Zhou, H. , Hu, Z. , Liu, Z. , Ketchem, R. R. , Yan, B. , J. Biol. Chem. 2010, 285, 18662–18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diepold, K. , Bomans, K. , Wiedmann, M. , Zimmermann, B. , Petzold, A. , Schlothauer, T. , Mueller, R. , Moritz, B. , Stracke, J. O. , Molhoj, M. , Reusch, D. , Bulau, P. , PLoS One 2012, 7, e30295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amano, M. , Hasegawa, J. , Kobayashi, N. , Kishi, N. , Nakazawa, T. , Uchiyama, S. , Fukui, K. , Anal. Chem. 2011, 83, 3857–3864. [DOI] [PubMed] [Google Scholar]
- 22. Zhang, Q. , Flynn, G. C. , J. Biol. Chem. 2013, 288, 34325–34335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen, S. L. , Price, C. , Vlasak, J. , J. Am. Chem. Soc. 2007, 129, 6976–6977. [DOI] [PubMed] [Google Scholar]
- 24. Zhang, Q. , Schenauer, M. R. , McCarter, J. D. , Flynn, G. C. , J. Biol. Chem. 2013, 288, 16371–16382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu, Y. D. , Goetze, A. M. , Bass, R. B. , Flynn, G. C. , J. Biol. Chem. 2011, 286, 11211–11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stracke, J. , Emrich, T. , Rueger, P. , Schlothauer, T. , Kling, L. , Knaupp, A. , Hertenberger, H. , Wolfert, A. , Spick, C. , Lau, W. , Drabner, G. , Reiff, U. , Koll, H. , Papadimitriou, A. , MAbs 2014, 6, 1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qi, Y. , Grishin, N. V. , Proteins. 2005, 58, 376–388. [DOI] [PubMed] [Google Scholar]
- 28. Zhang, W. , Czupryn, M. J. , Biotechnol. Prog. 2002, 18, 509–513. [DOI] [PubMed] [Google Scholar]
- 29. Zhao, X. , Ning, Q. , Ai, M. , Chai, H. , Yin, M. , Mol. Biosyst. 2015, 11, 923–929. [DOI] [PubMed] [Google Scholar]
- 30. Lu, C. , Liu, D. , Liu, H. , Motchnik, P. , MAbs. 2013, 5, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strand, J. , Huang, C. T. , Xu, J. , J. Pharm. Sci. 2013, 102, 441–453. [DOI] [PubMed] [Google Scholar]
- 32. Frand, A. R. , Cuozzo, J. W. , Kaiser, C. A. , Trends Cell Biol. 2000, 10, 203–210. [DOI] [PubMed] [Google Scholar]
- 33. Jessop, C. E. , Chakravarthi, S. , Watkins, R. H. , Bulleid, N. J. , Biochem. Soc. Trans. 2004, 32, 655–658. [DOI] [PubMed] [Google Scholar]
- 34. Hutterer, K. M. , Hong, R. W. , Lull, J. , Zhao, X. , Wang, T. , Pei, R. , Le, M. E. , Borisov, O. , Piper, R. , Liu, Y. D. , Petty, K. , Apostol, I. , Flynn, G. C. , MAbs 2013, 5, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moritz, B. , Striegel, K. , de Graaf, A. A. , Sahm, H. , Metab. Eng. 2002, 4, 295–305. [DOI] [PubMed] [Google Scholar]
- 36. Moritz, B. , Striegel, K. , De Graaf, A. A. , Sahm, H. , Eur. J. Biochem. 2000, 267, 3442–3452. [DOI] [PubMed] [Google Scholar]
- 37. Koterba, K. L. , Borgschulte, T. , Laird, M. W. , J. Biotechnol. 2012, 157, 261–267. [DOI] [PubMed] [Google Scholar]
- 38. Trexler‐Schmidt, M. , Sargis, S. , Chiu, J. , Sze‐Khoo, S. , Mun, M. , Kao, Y. H. , Laird, M. W. , Biotechnol. Bioeng. 2010, 106, 452–461. [DOI] [PubMed] [Google Scholar]
- 39. Kao, Y. H. , Hewitt, D. P. , Trexler‐Schmidt, M. , Laird, M. W. , Biotechnol. Bioeng. 2010, 107, 622–632. [DOI] [PubMed] [Google Scholar]
- 40. Zhou, S. , Mozziconacci, O. , Kerwin, B. A. , Schoneich, C. , Pharm. Res. 2013, 30, 1291–1299. [DOI] [PubMed] [Google Scholar]
- 41. Mozziconacci, O. , Kerwin, B. A. , Schoneich, C. , Chem. Res. Toxicol. 2010, 23, 1310–1312. [DOI] [PubMed] [Google Scholar]
- 42. Cordoba, A. J. , Shyong, B. J. , Breen, D. , Harris, R. J. , J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 818, 115–121. [DOI] [PubMed] [Google Scholar]
- 43. Amzel, L. M. , Poljak, R. J. , Annu. Rev. Biochem. 1979, 48, 961–997. [DOI] [PubMed] [Google Scholar]
- 44. Neergaard, M. S. , Nielsen, A. D. , Parshad, H. , Van De Weert, M. , J. Pharm. Sci. 2014, 103, 115–127. [DOI] [PubMed] [Google Scholar]
- 45. Rustandi, R. R. , Wang, Y. , Electrophoresis 2011, 32, 3078–3084. [DOI] [PubMed] [Google Scholar]
- 46. Tous, G. I. , Wei, Z. , Feng, J. , Bilbulian, S. , Bowen, S. , Smith, J. , Strouse, R. , McGeehan, P. , Casas‐Finet, J. , Schenerman, M. A. , Anal. Chem. 2005, 77, 2675–2682. [DOI] [PubMed] [Google Scholar]
- 47. Gu, S. , Wen, D. , Weinreb, P. H. , Sun, Y. , Zhang, L. , Foley, S. F. , Kshirsagar, R. , Evans, D. , Mi, S. , Meier, W. , Pepinsky, R. B. , Anal. Biochem. 2010, 400, 89–98. [DOI] [PubMed] [Google Scholar]
- 48. Atkins, P. W. , Physical Chemistry, Oxford University Press, Oxford: 1978. [Google Scholar]
- 49. Zhang, T. , Zhang, J. , Hewitt, D. , Tran, B. , Gao, X. , Qiu, Z. J. , Tejada, M. , Gazzano‐Santoro, H. , Kao, Y. H. , Anal. Chem. 2012, 84, 7112–7123. [DOI] [PubMed] [Google Scholar]
- 50. Xiang, T. , Chumsae, C. , Liu, H. , Anal. Chem. 2009, 81, 8101–8108. [DOI] [PubMed] [Google Scholar]
- 51. Liu, H. , Chumsae, C. , Gaza‐Bulseco, G. , Hurkmans, K. , Radziejewski, C. H. , Anal. Chem. 2010, 82, 5219–5226. [DOI] [PubMed] [Google Scholar]
- 52. Wiberg, E. , Holleman, A. F. , Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin: 1971. [Google Scholar]
- 53. Morrison, R. T. , Boyd, R. N. , Organic Chemistry, Allyn and Bacon, Boston: 1983. [Google Scholar]
- 54. Carey, F. A. , Sundberg, R. J. , Advanced Organic Chemistry, Springer, New York: 2007. [Google Scholar]
- 55. Chaderjian, W. B. , Chin, E. T. , Harris, R. J. , Etcheverry, T. M. , Biotechnol. Prog. 2005, 21, 550–553. [DOI] [PubMed] [Google Scholar]
- 56. Harris, R. J. , Dev. Biol. 2005, 122, 117–127. [PubMed] [Google Scholar]
- 57. Ouellette, D. , Alessandri, L. , Chin, A. , Grinnell, C. , Tarcsa, E. , Radziejewski, C. , Correia, I. , Anal. Biochem. 2010, 397, 37–47. [DOI] [PubMed] [Google Scholar]
- 58. Lacy, E. R. , Baker, M. , Brigham‐Burke, M. , Anal. Biochem. 2008, 382, 66–68. [DOI] [PubMed] [Google Scholar]
- 59. Woodward, J. , Tate, J. , Herrmann, P. C. , Evans, B. R. , J. Biochem. Biophys. Methods. 1993, 26, 121–129. [DOI] [PubMed] [Google Scholar]
- 60. Winters, R. A. , Zukowski, J. , Ercal, N. , Matthews, R. H. , Spitz, D. R. , Anal. Biochem. 1995, 227, 14–21. [DOI] [PubMed] [Google Scholar]
- 61. Hambly, D. M. , Banks, D. D. , Scavezze, J. L. , Siska, C. C. , Gadgil, H. S. , Anal. Chem. 2009, 81, 7454–7459. [DOI] [PubMed] [Google Scholar]
- 62. Ishikawa, T. , Ito, T. , Endo, R. , Nakagawa, K. , Sawa, E. , Wakamatsu, K. , Biol. Pharm. Bull. 2010, 33, 1413–1417. [DOI] [PubMed] [Google Scholar]
- 63. Vandenberghe, I. , Kim, J. K. , Devreese, B. , Hacisalihoglu, A. , Iwabuki, H. , Okajima, T. , Kuroda, S. , Adachi, O. , Jongejan, J. A. , Duine, J. A. , Tanizawa, K. , Van Beeumen, J. , J. Biol. Chem. 2001, 276, 42923–42931. [DOI] [PubMed] [Google Scholar]
- 64. Gielens, C. , De Geest, N. , Xin, X. Q. , Devreese, B. , Van Beeumen, J. , Preaux, G. , Eur. J. Biochem. 1997, 248, 879–888. [DOI] [PubMed] [Google Scholar]
- 65. Diaz, A. , Horjales, E. , Rudino‐Pinera, E. , Arreola, R. , Hansberg, W. , J. Mol. Biol. 2004, 342, 971–985. [DOI] [PubMed] [Google Scholar]
- 66. Yoo, E. M. , Wims, L. A. , Chan, L. A. , Morrison, S. L. , J. Immunol. 2003, 170, 3134–3138. [DOI] [PubMed] [Google Scholar]
- 67. Liu, Y. D. , Chen, X. , Enk, J. Z. , Plant, M. , Dillon, T. M. , Flynn, G. C. , J. Biol. Chem. 2008, 283, 29266–29272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wypych, J. , Li, M. , Guo, A. , Zhang, Z. , Martinez, T. , Allen, M. J. , Fodor, S. , Kelner, D. N. , Flynn, G. C. , Liu, Y. D. , Bondarenko, P. V. , Ricci, M. S. , Dillon, T. M. , Balland, A. , J. Biol. Chem. 2008, 283, 16194–16205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dillon, T. M. , Ricci, M. S. , Vezina, C. , Flynn, G. C. , Liu, Y. D. , Rehder, D. S. , Plant, M. , Henkle, B. , Li, Y. , Deechongkit, S. , Varnum, B. , Wypych, J. , Balland, A. , Bondarenko, P. V. , J. Biol. Chem. 2008, 283, 16206–16215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu, Y. D. , Wang, T. , Chou, R. , Chen, L. , Kannan, G. , Stevenson, R. , Goetze, A. M. , Jiang, X. G. , Huang, G. , Dillon, T. M. , Flynn, G. C. , Mol. Immunol. 2013, 54, 217–226. [DOI] [PubMed] [Google Scholar]
- 71. Zhang, B. , Harder, A. G. , Connelly, H. M. , Maheu, L. L. , Cockrill, S. L. , Anal. Chem. 2010, 82, 1090–1099. [DOI] [PubMed] [Google Scholar]
- 72. Liu, Y. D. , Chou, R. Y. , Dillon, T. M. , Poppe, L. , Spahr, C. , Shi, S. D. , Flynn, G. C. , Protein Sci. 2014, 23, 1753–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. van der Neut Kolfschoten, M. , Schuurman, J. , Losen, M. , Bleeker, W. K. , Martinez‐Martinez, P. , Vermeulen, E. , den Bleker, T. H. , Wiegman, L. , Vink, T. , Aarden, L. A. , De Baets, M. H. , van de Winkel, J. G. , Aalberse, R. C. , Parren, P. W. , Science 2007, 317, 1554–1557. [DOI] [PubMed] [Google Scholar]
- 74. Schuurman, J. , Van Ree, R. , Perdok, G. J. , Van Doorn, H. R. , Tan, K. Y. , Aalberse, R. C. , Immunology. 1999, 97, 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Labrijn, A. F. , Buijsse, A. O. , van den Bremer, E. T. , Verwilligen, A. Y. , Bleeker, W. K. , Thorpe, S. J. , Killestein, J. , Polman, C. H. , Aalberse, R. C. , Schuurman, J. , van de Winkel, J. G. , Parren, P. W. , Nat. Biotechnol. 2009, 27, 767–771. [DOI] [PubMed] [Google Scholar]
- 76. Schuurman, J. , Perdok, G. J. , Gorter, A. D. , Aalberse, R. C. , Mol. Immunol. 2001, 38, 1–8. [DOI] [PubMed] [Google Scholar]
- 77. Rispens, T. , Davies, A. M. , Ooijevaar‐de Heer, P. , Absalah, S. , Bende, O. , Sutton, B. J. , Vidarsson, G. , Aalberse, R. C. , J. Biol. Chem. 2014, 289, 6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rispens, T. , Ooijevaar‐de Heer, P. , Bende, O. , Aalberse, R. C. , J. Am. Chem. Soc. 2011, 133, 10302–10311. [DOI] [PubMed] [Google Scholar]
- 79. Bloom, J. W. , Madanat, M. S. , Marriott, D. , Wong, T. , Chan, S. Y. , Protein Sci. 1997, 6, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Angal, S. , King, D. J. , Bodmer, M. W. , Turner, A. , Lawson, A. D. , Roberts, G. , Pedley, B. , Adair, J. R. , Mol. Immunol. 1993, 30, 105–108. [DOI] [PubMed] [Google Scholar]
- 81. Yang, X. , Wang, F. , Zhang, Y. , Wang, L. , Antonenko, S. , Zhang, S. , Zhang, Y. W. , Tabrizifard, M. , Ermakov, G. , Wiswell, D. , Beaumont, M. , Liu, L. , Richardson, D. , Shameem, M. , Ambrogelly, A. , J. Pharm. Sci. 2015, 104, 4002–4014. [DOI] [PubMed] [Google Scholar]
- 82. Aalberse, R. C. , Schuurman, J. , Immunology. 2002, 105, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. van der Zee, J. S. , van Swieten, P. , Aalberse, R. C. , J. Immunol. 1986, 137, 3566–3571. [PubMed] [Google Scholar]
- 84. Margni, R. A. , Binaghi, R. A. , Annu. Rev. Immunol. 1988, 6, 535–554. [DOI] [PubMed] [Google Scholar]
- 85. Forrer, K. , Hammer, S. , Helk, B. , Anal. Biochem. 2004, 334, 81–88. [DOI] [PubMed] [Google Scholar]
- 86. Singh, S. , Padovani, D. , Leslie, R. A. , Chiku, T. , Banerjee, R. , J. Biol. Chem. 2009, 284, 22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kshirsagar, R. , McElearney, K. , Gilbert, A. , Sinacore, M. , Ryll, T. , Biotechnol. Bioeng. 2012, 109, 2523–2532. [DOI] [PubMed] [Google Scholar]
- 88. Pristatsky, P. , Cohen, S. L. , Krantz, D. , Acevedo, J. , Ionescu, R. , Vlasak, J. , Anal. Chem. 2009, 81, 6148–6155. [DOI] [PubMed] [Google Scholar]
- 89. Cumnock, K. , Tully, T. , Cornell, C. , Hutchinson, M. , Gorrell, J. , Skidmore, K. , Chen, Y. , Jacobson, F. , Bioconjug. Chem. 2013, 24, 1154–1160. [DOI] [PubMed] [Google Scholar]
- 90. Gadgil, H. S. , Bondarenko, P. V. , Pipes, G. D. , Dillon, T. M. , Banks, D. , Abel, J. , Kleemann, G. R. , Treuheit, M. J. , Anal. Biochem. 2006, 355, 165–174. [DOI] [PubMed] [Google Scholar]
- 91. Chou, C. C. , Chiang, B. Y. , Lin, J. C. , Pan, K. T. , Lin, C. H. , Khoo, K. H. , J. Am. Soc. Mass Spectrom. 2015, 26, 120–132. [DOI] [PubMed] [Google Scholar]
- 92. Proba, K. , Honegger, A. , Pluckthun, A. , J. Mol. Biol. 1997, 265, 161–172. [DOI] [PubMed] [Google Scholar]
- 93. Worn, A. , Pluckthun, A. , J. Mol. Biol. 2001, 305, 989–1010. [DOI] [PubMed] [Google Scholar]
- 94. Uson, I. , Bes, M. T. , Sheldrick, G. M. , Schneider, T. R. , Hartsch, T. , Fritz, H. J. , Fold Des. 1997, 2, 357–361. [DOI] [PubMed] [Google Scholar]
- 95. Proba, K. , Worn, A. , Honegger, A. , Pluckthun, A. , J. Mol. Biol. 1998, 275, 245–253. [DOI] [PubMed] [Google Scholar]
- 96. Anfinsen, C. B. , Science 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- 97. Kikuchi, H. , Goto, Y. , Hamaguchi, K. , Biochemistry 1986, 25, 2009–2013. [DOI] [PubMed] [Google Scholar]
- 98. Frisch, C. , Kolmar, H. , Schmidt, A. , Kleemann, G. , Reinhardt, A. , Pohl, E. , Uson, I. , Schneider, T. R. , Fritz, H. J. , Fold Des. 1996, 1, 431–440. [DOI] [PubMed] [Google Scholar]
- 99. Mason, B. D. , Schoneich, C. , Kerwin, B. A. , Mol. Pharm. 2012, 9, 774–790. [DOI] [PubMed] [Google Scholar]
- 100. Kato, Y. , Kitamoto, N. , Kawai, Y. , Osawa, T. , Free Radic. Biol. Med. 2001, 31, 624–632. [DOI] [PubMed] [Google Scholar]
- 101. Altinier, S. , Seguso, M. , Zaninotto, M. , Varagnolo, M. , Adami, F. , Angeli, P. , Plebani, M. , Clin. Biochem. 2013, 46, 691–693. [DOI] [PubMed] [Google Scholar]
- 102. Jenner, E. , Clin. Chim. Acta 2014, 427, 15–20. [DOI] [PubMed] [Google Scholar]
- 103. Roopenian, D. C. , Akilesh, S. , Nat. Rev. Immunol. 2007, 7, 715–725. [DOI] [PubMed] [Google Scholar]
- 104. Wochner, R. D. , Strober, W. , Waldmann, T. A. , J. Exp. Med. 1967, 126, 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bertolotti‐Ciarlet, A. , Wang, W. , Lownes, R. , Pristatsky, P. , Fang, Y. , McKelvey, T. , Li, Y. , Drummond, J. , Prueksaritanont, T. , Vlasak, J. , Mol. Immunol. 2009, 46, 1878–1882. [DOI] [PubMed] [Google Scholar]
- 106. Medesan, C. , Matesoi, D. , Radu, C. , Ghetie, V. , Ward, E. S. , J. Immunol. 1997, 158, 2211–2217. [PubMed] [Google Scholar]
- 107. Kim, J. K. , Tsen, M. F. , Ghetie, V. , Ward, E. S. , Eur. J. Immunol. 1994, 24, 2429–2434. [DOI] [PubMed] [Google Scholar]
- 108. Ghetie, V. , Ward, E. S. , Immunol. Today 1997, 18, 592–598. [DOI] [PubMed] [Google Scholar]
- 109. Stern, M. , Herrmann, R. , Crit. Rev. Oncol. Hematol. 2005, 54, 11–29. [DOI] [PubMed] [Google Scholar]
- 110. Gevondyan, N. M. , Volynskaia, A. M. , Gevondyan, V. S. , Biochemistry 2006, 71, 279–284. [DOI] [PubMed] [Google Scholar]
- 111. Katzmann, J. A. , Clark, R. J. , Abraham, R. S. , Bryant, S. , Lymp, J. F. , Bradwell, A. R. , Kyle, R. A. , Clin. Chem. 2002, 48, 1437–1444. [PubMed] [Google Scholar]
- 112. Braun, A. , Kwee, L. , Labow, M. A. , Alsenz, J. , Pharm. Res. 1997, 14, 1472–1478. [DOI] [PubMed] [Google Scholar]
- 113. Maas, C. , Hermeling, S. , Bouma, B. , Jiskoot, W. , Gebbink, M. F. , J. Biol. Chem. 2007, 282, 2229–2236. [DOI] [PubMed] [Google Scholar]
- 114. Fan, X. , Brezski, R. J. , Fa, M. , Deng, H. , Oberholtzer, A. , Gonzalez, A. , Dubinsky, W. P. , Strohl, W. R. , Jordan, R. E. , Zhang, N. , An, Z. , Breast Cancer Res. 2012, 14, R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pan, H. , Chen, K. , Chu, L. , Kinderman, F. , Apostol, I. , Huang, G. , Protein Sci. 2009, 18, 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Flanagan, R. J. , Jones, A. L. , Drug Saf. 2004, 27, 1115–1133. [DOI] [PubMed] [Google Scholar]
- 117. Smolarz, A. , Roesch, E. , Lenz, E. , Neubert, H. , Abshagen, P. , J. Toxicol. Clin. Toxicol. 1985, 23, 327–340. [DOI] [PubMed] [Google Scholar]
- 118. Hickey, A. R. , Wenger, T. L. , Carpenter, V. P. , Tilson, H. H. , Hlatky, M. A. , Furberg, C. D. , Kirkpatrick, C. H. , Strauss, H. C. , Smith, T. W. , J. Am. Coll. Cardiol. 1991, 17, 590–598. [DOI] [PubMed] [Google Scholar]
- 119. Smith, T. W. , Am. J. Emerg. Med. 1991, 9, 1–6; discussion 33–34. [DOI] [PubMed] [Google Scholar]
- 120. Schoch, A. , Kettenberger, H. , Mundigl, O. , Winter, G. , Engert, J. , Heinrich, J. , Emrich, T. , Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ujhelyi, M. R. , Robert, S. , Clin. Pharmacokinet. 1995, 28, 483–493. [DOI] [PubMed] [Google Scholar]
- 122. Levinson, S. S. , Clin. Chem. Lab. Med. 2015, 54 6, 1039–1043. [DOI] [PubMed] [Google Scholar]
- 123. Neuber, T. , Frese, K. , Jaehrling, J. , Jager, S. , Daubert, D. , Felderer, K. , Linnemann, M. , Hohne, A. , Kaden, S. , Kolln, J. , Tiller, T. , Brocks, B. , Ostendorp, R. , Pabst, S. , MAbs 2014, 6, 928–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hipkiss, A. R. , Exp. Gerontol. 2006, 41, 464–473. [DOI] [PubMed] [Google Scholar]
- 125. Shapira, R. , Austin, G. E. , Mirra, S. S. , J. Neurochem. 1988, 50, 69–74. [DOI] [PubMed] [Google Scholar]
- 126. Shimizu, T. , Matsuoka, Y. , Shirasawa, T. , Biol. Pharm. Bull. 2005, 28, 1590–1596. [DOI] [PubMed] [Google Scholar]
- 127. Kleemann, G. R. , Beierle, J. , Nichols, A. C. , Dillon, T. M. , Pipes, G. D. , Bondarenko, P. V. , Anal. Chem. 2008, 80, 2001–2009. [DOI] [PubMed] [Google Scholar]
- 128. Yount, W. J. , Dorner, M. M. , Kunkel, H. G. , Kabat, E. A. , J. Exp. Med. 1968, 127, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Riesen, W. F. , Skvaril, F. , Braun, D. G. , Scand. J. Immunol. 1976, 5, 383–390. [DOI] [PubMed] [Google Scholar]
- 130. Dhooge, I. J. , van Kempen, M. J. , Sanders, L. A. , Rijkers, G. T. , Int. J. Pediatr. Otorhinolaryngol. 2002, 64, 133–141. [DOI] [PubMed] [Google Scholar]
- 131. Ekdahl, K. , Braconier, J. H. , Svanborg, C. , Scand. J. Infect. Dis. 1997, 29, 401–407. [DOI] [PubMed] [Google Scholar]
- 132. Ambrosino, D. M. , Bone Marrow Transplant. 1991, 7 (Suppl 3), 48–51. [PubMed] [Google Scholar]
- 133. Kuijpers, T. W. , Weening, R. S. , Out, T. A. , Allergol. Immunopathol. 1992, 20, 28–34. [PubMed] [Google Scholar]
- 134. Weinberg, W. C. , Frazier‐Jessen, M. R. , Wu, W. J. , Weir, A. , Hartsough, M. , Keegan, P. , Fuchs, C. , Cancer Metastasis Rev. 2005, 24, 569–584. [DOI] [PubMed] [Google Scholar]
- 135. Goodwin, L. R. , Francom, D. , Dieken, F. P. , Taylor, J. D. , Warenycia, M. W. , Reiffenstein, R. J. , Dowling, G. , J. Anal. Toxicol. 1989, 13, 105–109. [DOI] [PubMed] [Google Scholar]
- 136. Zhao, W. , Zhang, J. , Lu, Y. , Wang, R. , EMBO J. 2001, 20, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]