

Abstract
Purpose
X-linked agammaglobulinemia is a primary humoral immunodeficiency characterized by hypogammaglobulinemia and increased susceptibility to infection. Although there is increased awareness of autoimmune and inflammatory complications in XLA, the spectrum of gastrointestinal manifestations has not previously been fully explored.
Methods
We present a case report of a family with two affected patients with XLA. Given the gastrointestinal involvement of the grandfather in this family, we performed a retrospective descriptive analysis of XLA patients with reported diagnoses of GI manifestations and inflammatory bowel disease (IBD) or enteritis registered at the USIDNet, a national registry of primary immunodeficiencies.
Results
In this cohort of patients with XLA, we found that up to 35% had concurrent gastrointestinal manifestations, and 10% had reported diagnoses of IBD or enteritis. The most commonly reported mutations were missense, which have been associated with a less severe XLA phenotype in the literature. The severity of symptoms were wide-ranging, and management strategies were diverse and mainly experimental.
Conclusions
Patients with XLA may require close monitoring with particular attention for GI manifestations including IBD and infectious enteritis. Further studies are needed to improve diagnosis and management of GI conditions in XLA patients.
Introduction
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency (PID) affecting approximately three to six cases per one million males[1]. In XLA, mutations in Bruton’s tyrosine kinase (BTK) result in defective tonic pre-BCR signaling, impaired B-cell survival, and arrest in maturation at the pre-B-cell stage. CD19+ B cells are typically <2% and the classical clinical presentation is agammaglobulinemia with recurrent bacterial infections. Hypomorphic variants with low, but not absent, immunoglobulin levels and decreased, but not absent, B cells also exist. Recently, autoimmune and inflammatory manifestations have been highlighted as a feature of XLA[2]. Among the inflammatory complications, the prevalence of inflammatory bowel disease (IBD)/enteritis in the USIDNet XLA patient cohort was found to be 3.4%, which is higher than the reported prevalence of 0.4% in the general population[2].
We present a case of an XLA family in which inflammatory bowel disease (IBD) was the presenting feature of the primary case, leading to a significant delay in diagnosis. A literature review did not reveal any publications regarding best practices for gastrointestinal manifestations such as IBD/enteritis in XLA patients. Therefore, we searched the United States Immunodeficiency Network (USIDNet) database for XLA patients with IBD/enteritis to improve our understanding of the natural history and management strategies, and present a descriptive analysis of 19 XLA patients with diagnoses of IBD/enteritis.
Case
The index case was a 23-month-old boy (Patient A) with a history of recurrent infections (including acute otitis media, conjunctivitis, Staphylococcus perianal cellulitis) since 7 months of age and a history of chronic diarrhea attributed to soy protein intolerance. Although closely followed by his pediatrician, the clinical history of infections had not been alarming and therefore no immune evaluation had been pursued prior to his visit with the Immunology service. A week before evaluation, he developed a febrile seizure and was admitted to an outside hospital. He was treated with intravenous ceftriaxone for a presumed infection and was found to have neutropenia. Because of the maternal grandfather’s history of hypogammaglobulinemia, the family requested quantitative immunoglobulin testing for the child, and he was found unexpectedly to have an undetectable immunoglobulin G (IgG) level. Therefore, he was referred to the Immunodeficiency Program at our institution for evaluation of absolute neutropenia and agammaglobulinemia. At the time of initial immune evaluation, the neutropenia appeared to be resolving with an absolute neutrophil count of 1170k/μL. His Ig levels were significant for an undetectable IgG, IgA, IgE and low IgM of 20mg/dL (Table 1). His lymphocyte panel was notable for a near absence of B cells (4 cells/mm3, < 1%), complete absence of switched memory B cells, and increased double negative T cells (3%), but otherwise preserved numbers of T and NK cell compartments with age-appropriate ratio of naive and memory T cells. BTK gene sequencing revealed a missense mutation c.41C>A, p.Ser14Tyr, suggesting a diagnosis of XLA. Two patients with a mild phenotype of XLA were reported to have the same mutation in a cohort study [3].
Table 1.
Laboratory Characteristics of XLA Family
| Laboratory Test | Patient A | Patient B |
|---|---|---|
| Immunoglobulins (mg/dL) | ||
| IgG | Undetectable (normal age-adjusted range: 313–1,170 mg/dL) | 788 (on IVIG) (normal range: 767–1,590 mg/dL) |
| IgA | Undetectable (normal age-adjusted range: 36–79 mg/dL) | Undetectable (normal range: 61–356 mg/dL) |
| IgM | 20 (normal age-adjusted range: 46–152 mg/dL) | 6 (normal range: 37–286 mg/dL) |
| IgE | Undetectable (normal age-adjusted range: < or =97kU/L) | 5 (normal range: < or =214kU/L) |
| Flow cytometry (cells/mm3, percent) | ||
| CD19+ (total B cells) | 4, <1% (normal range: 2–76%) | <1, <1% (normal range: 6–25%) |
| CD19+ CD27+ IgM−IgD−(switched memory B cells) | 1, <1% (normal range: 1.5–4.1%) | <1, <0.01% (normal range: 9.2–18.9%) |
| CD4+CD3 T cells | 2049, 46% (normal range: 28–47%) | 426, 32% (normal range: 31–60%) |
| CD16+56+ NK cells | 671, 15% (normal range:3–30%) | 223, 17% (normal range: 2–27%) |
| Gene analysis | Btk mutation missense 41C>A, pSer14Tyr | |
Laboratory evaluation was notable for undetectable IgG, IgA, IgE, low IgM, extremely low B cell counts, and absence of switched memory B cells.
The index patient’s family history was significant for a 71-year-old maternal grandfather (Patient B) diagnosed with presumed common variable immunodeficiency (CVID) at 48 years of age given low immunoglobulin levels and prior history of multiple ear infections, bronchitis, and pneumonias as a child, and frequent walking pneumonias as an adult. Prior to his “CVID” diagnosis, he had been diagnosed with Crohn’s disease at 35 years of age for symptoms including severe decrease in appetite, weight loss, bloating, loose non-bloody stools, nausea, vomiting, and abdominal pain requiring recurrent hospitalizations. His gastrointestinal (GI) disease was initially treated with prednisone and 5-amino salicylates. Interestingly, although he continued to have mild GI complaints, he did not require active treatment or hospitalization after starting intravenous immunoglobulin (IVIG) replacement for “CVID” at 48 years of age. To our knowledge, the patient did not have B cell quantitation performed at the time of the “CVID” diagnosis.
In light of his grandson being diagnosed with a BTK missense mutation c.41C>A, p.Ser14Tyr and immunological phenotype of near absence of B cells, the maternal grandfather was also evaluated for BTK mutation. Laboratory evaluation was notable for a low-normal absolute lymphocyte count of 1440k/μL, undetectable B cell count (<1cell/mm3, <1%) with preserved T and NK cell count, IgG 788mg/dL (on IVIG replacement), low IgM of 6mg/dL, low IgE of 5mg/dL, and undetectable IgA (Table 1). Flow cytometry revealed reduced but not absent BTK protein expression in monocytes (CD14+) compared to control (Patient B: 66.31%, mean fluorescence intensity (MFI) 0.98 versus control 97.48%, MFI 4.01, corrected for isotype). Genetic testing of the BTK gene revealed the same missense mutation c.41C>A, p.Ser14Tyr, confirming the diagnosis of partial (hypomorphic) XLA, similar to his grandchild.
Family history was significant for a maternal first male cousin who died over 30 years ago at 18 months of age (Patient C)(Figure 1). The family reports that the cousin was diagnosed with a sudden flare of acute otitis media that progressed to systemic illness requiring treatment in an intensive care unit, where he died from complications of myocarditis and/or pericarditis. Reportedly the child had frequent ear infections before this sudden fatal decline.
Figure 1.
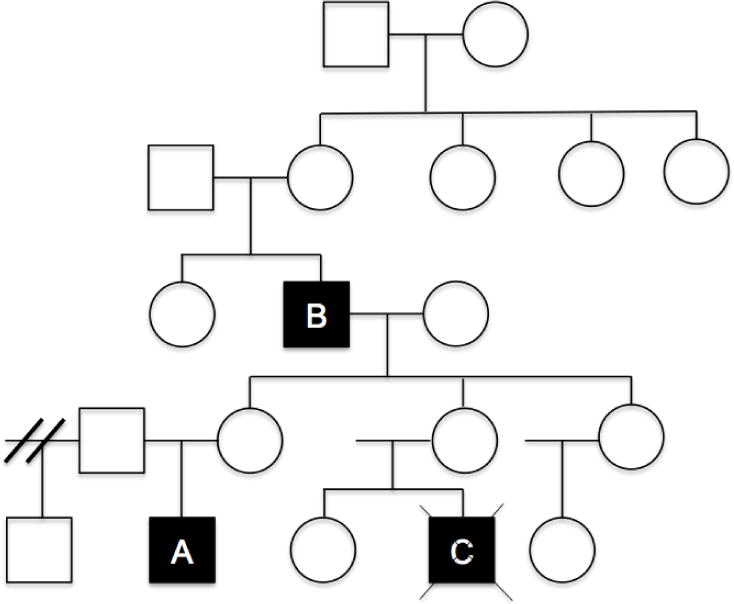
Patient A: Index case — 23 month old boy. Patient B: 71 year old maternal grandfather. Patient C: Maternal first cousin who died at 18 months of age from infectious complications over 30 years ago. He was diagnosed with bilateral ear infections that progressed to sepsis requiring admission to the intensive care unit, where he suffered from fatal cardiac failure due to myocarditis/pericarditis.
Methods
Given the paucity of published data regarding the natural history of GI manifestations, disease progression, and treatment strategies in XLA patients, we performed a retrospective analysis of XLA patients with reported GI manifestations, and specifically those with IBD/enteritis, registered at the USIDNet (www.usidnet.org), a national registry with over 5786 patients of primary immunodeficiencies in the United States of America. Data received from USIDNet included diagnosis, age of onset of symptom, age of diagnosis, year of birth, race, ethnicity, gender, patient height and weight and body mass index, immunophenotyping including T cell subsets and immunoglobulin levels, previous infectious and species of organisms (if known), details of immunoglobulin replacement therapy (age at initiation of therapy, route, frequency of adverse reactions, and dosing), immunomodulatory therapy (including tumor necrosis factor-α inhibitors, aminosalicylates, corticosteroids, azathioprine/mercaptopurine or methotrexate), history of gastrointestinal surgeries, and non-surgical treatments.
We reviewed BTKbase (http://databases.lovd.nl/shared/genes/BTK), the RAPID database (http://web16.kazusa.or.jp/rapid/), and published cases in the literature for genotype and clinical phenotype correlations. For mutations where a phenotype has not been reported, phenotype predictions were made based on prior genotype-phenotype association study predictions [3,4].
Statistical analyses were performed using Stata/IC 14.9 (Stat-Corp, College Station, Tex). Wilcoxon rank sum test was used to compare T cell subset numbers and immunoglobulin levels between XLA patients with a diagnosis of IBD/enteritis and XLA patients with complete absence of any GI manifestations.
Results
In the USIDNet database, 200 cases of XLA were identified, 71 (35%) with concurrent gastrointestinal manifestations (Table 2). The year of birth ranged from 1945 to 2010. The average age of diagnosis was 3.45 years old (range 0–16 years). All 71 patients (100%) were male, as expected. Of these patients, the most common GI manifestations included diarrhea (intermittent = 38%, chronic = 26%), abdominal pain (22%), gastroesophageal reflux disease (18%), and gastroenteritis (14%).
Table 2.
Gastrointestinal conditions reported in USIDNet cohort of XLA patients
| GI Disorders/Symptoms | Number of Patients (#) | Percent of total patients with GI manifestations (%) |
|---|---|---|
| Diarrhea (intermittent) | 27 | 38.0 |
| Diarrhea (chronic) | 19 | 26.8 |
| Abdominal pain | 16 | 22.5 |
| GE reflux | 13 | 18.3 |
| Gastroenteritis | 10 | 14.1 |
| Colitis/enteritis | 8 | 11.3 |
| Vomiting | 8 | 11.3 |
| Constipation | 7 | 9.9 |
| Malabsorption | 4 | 5.6 |
| Appendicitis | 3 | 4.2 |
| Hepatosplenomegaly | 3 | 4.2 |
| Viral Gastroenteritis | 3 | 4.2 |
| Obstruction | 2 | 2.8 |
| IBD-not otherwise spec | 2 | 2.8 |
| Lactose intolerance | 2 | 2.8 |
| Protein losing gastroenteropathy | 2 | 2.8 |
| Dysphagia | 2 | 2.8 |
| Cirrhosis | 2 | 2.8 |
| Celiac disease | 2 | 2.8 |
| Abdominal bloating | 1 | 1.4 |
| Aphthous ulcers | 1 | 1.4 |
| Cholecysitis | 1 | 1.4 |
| Diverticulitis | 1 | 1.4 |
| Nausea | 1 | 1.4 |
| Pancreatitis | 1 | 1.4 |
| Biliary dyskinesia | 1 | 1.4 |
| Fistula | 1 | 1.4 |
| Esophageal Varices | 1 | 1.4 |
| Jaundice | 1 | 1.4 |
| Liver function abnormalities | 1 | 1.4 |
| Nodular regenerative hyperplasia (liver) | 1 | 1.4 |
| Portal Hypertension | 1 | 1.4 |
| Gall bladder disease | 1 | 1.4 |
| Perianal ulceration | 1 | 1.4 |
| Bloody stool | 1 | 1.4 |
| Liver failure | 1 | 1.4 |
| Gastric varices | 1 | 1.4 |
| Esophagitis | 1 | 1.4 |
| Hemorrhoids | 1 | 1.4 |
| Hematochezia | 1 | 1.4 |
There were 19 patients (9.5% of total cohort) who had a specific diagnoses of IBD and/or enteritis. The majority of these cases were Caucasian (63%), six (32%) were registered as unknown, and one (5%) was African-American. At least thirteen (68%) patients had a family member who had XLA or who was an XLA carrier: brother (n=5), maternal uncle (n=3), maternal cousin (n=2), mother (n=3). The majority of the patients had received immunoglobulin replacement therapy (17/19, 89%) (Table 3). There were no significant differences between the T cell subsets and immunoglobulin A, M, and E levels of XLA patients with IBD/enteritis compared to the XLA patients without GI manifestations (data not shown). Of note, T cell subset data was limited and there was likely insufficient power to detect significant differences.
Table 3.
Patient Demographics and Treatment Characteristics of Patients Diagnosed with IBD or enteritis (n= 19)
| Demographics | n (%) |
|---|---|
| Gender: | |
| Male | 19 (100) |
| Female | 0 (0) |
| Ethnicity/Race: | |
| Caucasian/white | 12 (63) |
| African American | 1 (5) |
| Unknown | 6 (32) |
| Family History of XLA | 13 (68) |
| Treatment Characteristics | n (%) |
| IVIG | 17 (89) |
| Corticosteroids | 2 (11) |
| Parenteral nutrition | 14 (74) |
| Gastrointestinal Surgery | 0 (0) |
The spectrum of GI manifestations observed in the subset of patients specifically diagnosed with IBD/enteritis is listed in Table 4. Enteritis was the most common GI manifestation, occurring in 17 of the 19 XLA patients (90%). Diarrhea was reported as a dominant symptom in the majority of patients (11/19, 58%). Five patients were identified as having infectious diarrhea, and a causative organism was identified in four of these five patients. These organisms included Giardia lamblia, rotavirus, adenovirus, and Clostridium difficile. Two of the 19 patients (11%) were diagnosed with IBD. The date of IBD diagnosis was not reported and could not be compared to the date of XLA diagnosis. Of note, although a specific infectious agent was not identified in several of the patients classified as having enteritis, we can not fully exclude the possibility of an infection-associated enteritis.
Table 4.
Gastrointestinal conditions reported for patients with IBD and Enteritis from the USIDNet cohort of XLA patients
| Patient | GI Disorders/Symptoms | GI Infections | Organism |
|---|---|---|---|
| 1 | Abdominal pain Colitis/enteritis |
Diarrhea (infectious) | |
| 2 | Gastroenteritis | Diarrhea (infectious) | Giardia |
| 3 | Diarrhea (intermittent) Gastroenteritis | ||
| 4 | Colitis/enteritis Diarrhea (chronic) Gastroenteritis |
Gastroenteritis | |
| 5 | Gastroenteritis | ||
| 6 | Gastroenteritis | ||
| 7 | Gastroenteritis | Gastroenteritis | |
| 8 | Gastroenteritis | ||
| 9 | Gastroenteritis | ||
| 10 | Gastroenteritis | ||
| 11 | Gastroenteritis | Diarrhea (infectious) | Giardia |
| 12 | Protein losing gastroenteropathy | Viral gastroenteritis | |
| 13 | Abdominal pain IBD-not otherwise specified |
||
| 14 | Gastroenteritis IBD-not otherwise specified |
||
| 15 | Colitis/enteritis Diarrhea (chronic) Vomiting |
Diarrhea (infectious) | Giardia |
| 16 | Colitis | ||
| 17 | Diarrhea (intermittent) Viral gastroenteritis |
||
| 18 | Colitis/enteritis | ||
| 19 | Abdominal pain Colitis/enteritis Diarrhea (chronic) Protein losing gastroenteropathy |
Diarrhea | Adenovirus Clostridium difficile Giardia Rotavirus |
GI manifestations appeared to be severe enough to require parenteral nutrition in many of the XLA patients with GI manifestations (14/19, 74%). Two patients (2/19, 11%) required immunomodulatory therapy with corticosteroids, one for IBD and one for protein-losing gastroenteropathy (this patient also required parenteral nutrition). None of the patients required gastrointestinal surgery for complications of inflammatory bowel disease.
The BTK mutations reported in the subset of XLA patients with IBD/enteritis are listed in Table 5 (n=7). In this subgroup of XLA patients with IBD/enteritis, the reported mutations were missense (n=4), nonsense (n=1), and frameshift (n=1). Missense mutations were the most commonly reported mutations. These mutations have been associated with a less severe XLA phenotype, with low but detectable levels of immunoglobulin and B cells[5,6]. This is consistent with our finding that our XLA family with GI manifestations had a missense mutation and a phenotype with intrafamilial variability in severity.
Table 5.
We referred to BTKbase (http://databases.lovd.nl/shared/genes/BTK) and published literature for clinical associations between genotype and phenotype. Available reports of observed clinical phenotypes were identified in the literature. For mutations where a phenotype had not been reported, phenotype predictions were made based on prior genotype-phenotype association study predictions.
| (3) Patient | DNA Change | Protein Change | Mutation Type | Phenotype* | Reference [PMID] |
|---|---|---|---|---|---|
| 2 | c.653del | p.(Lys218Argfs*11) | Frameshift | Severe, R | Conley et al. [20] [7849697] |
| 12 | c.1567-14 T>A | – | – | – | – |
| 14 | – | p.(Arg600*) | Nonsense | Severe, P | – |
| 15 | c.1589 T>C | p.(Leu486Pro) | Missense | Less severe, P | Futatani et al. [14] [11472359] |
| 17 | c.43 G>A | p.(Arg28His) | Missense | Less severe, P | – |
| 19 | c.1565 T>C | p.(Leu522Pro) | Missense | Less severe, R | Toth et al. [22] [19419768] |
| Patients A and B | c.41 C>A | p.(Ser14Tyr) | Missense | Less severe, R | Lee et al. [3] [19904586] |
P = predicted in literature, R = reported in literature
Discussion
The classical clinical presentation of XLA is agammaglobulinemia in a patient with recurrent bacterial infections. Immunologically, the notable feature is the lack or decrease in total B cell count in blood. The case presented herein of the grandfather (Patient B), highlights the importance of recognizing GI manifestations as a presenting feature in XLA. The patient had several features to suggest an underlying humoral immunodeficiency and was even started on replacement immunoglobulin, however, the correct diagnosis was missed because of an incomplete immune work-up. Diagnosis of XLA in the index case (Patient A) led to a subsequent diagnosis of the primary case of XLA in the grandfather, who had previously been misdiagnosed with Crohn’s Disease and then CVID. The presence of a BTK missense mutation c.41C>A, p.Ser14Tyr, resulted in decreased but not absent BTK protein as shown by flow cytometry (Figure 2), and near-absent but detectable levels of IgM confirm a diagnosis of a hypomorphic variant of XLA (Table 1).
Figure 2.
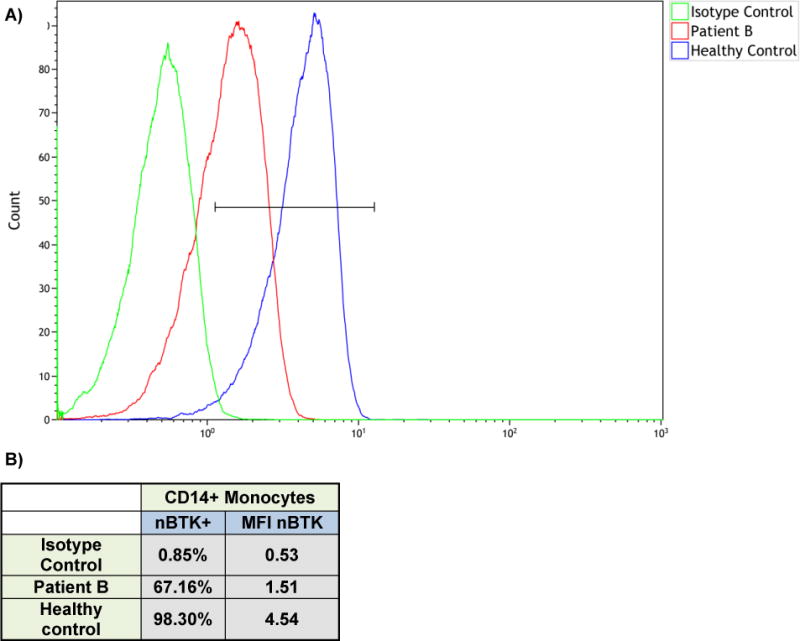
A) Flow cytometry for Patient B showing decreased (but not absent) bruton tyrosine kinase (btk) expression in monocytes (CD14+). B) Table showing flow cytometric analysis for btk.
Multiple studies have evaluated the correlation of disease severity with specific types of BTK mutations. Individuals with amino acid substitutions or splice site defects tend to be older at time of diagnosis with higher serum concentrations of IgM and relatively higher levels of peripheral B cells [5,7]. Additionally, previous studies evaluating genotype-phenotype correlations have found significant correlations between genotype and disease severity, with a later age of onset of symptoms in patients with missense mutations as compared to those with other types of mutations [3]. However, the correlation between specific BTK mutations and the clinical course of patients remains unknown. Interestingly, this case, when taken together with data from the USIDNet cases, suggests that patients with a hypomorphic variant of XLA are more prone to developing IBD/enteritis. This group may present with symptoms of abdominal pain, diarrhea, weight loss, nausea, vomiting, and varying degrees of enteric inflammation prior to their diagnosis of XLA. In some of these instances, there may be an underlying infectious trigger that has not been identified, rather than true autoimmune enteropathy or IBD. Commonly implicated culprit organisms for diarrhea in XLA include Giardia lamblia, Salmonella, Campylobacter, Cryptosporidium, mycoplasma, and enterovirus infections [2,8]. Additionally, patients who present with XLA may require vigilance for immune dysregulation such as monitoring for the development of IBD/enteritis.
A recent publication by Hernandez-Tujillo and colleagues highlighted the high prevalence of IBD/enteritis in the USIDNet XLA patient cohort (3.4% versus 0.4% in the general population) [2]. We delved further into this phenomenon by performing a detailed retrospective review of the clinical manifestations and management strategies employed for XLA patients with any GI manifestation in the USIDNet database. When including any GI manifestation (not limited to IBD and enteritis alone), we found that GI manifestations are present in up to 35% of XLA patients. The spectrum of GI manifestations ranged from diarrhea and infectious gastroenteritis to IBD. Of importance, we found that these GI manifestations can be severe, requiring parenteral nutrition and immunomodulatory therapy. Autoimmune GI manifestations are thought to be less common in XLA than in other primary immunodeficiencies, presumably because it is T cell dysfunction that drives intestinal disease in other immunodeficiency syndromes [8]. For example, GI complications are reported in 20–50% of patients with CVID [9], with GI infections occurring at a reported incidence of 27% [10]. It is possible that these hypomorphic XLA mutations are leading to increased autoimmunity. Interestingly, Meyers et al. recently reported decreased frequency of regulatory T cells in patients with XLA, possibly suggesting an effect of B cells on regulatory T cells, and suggesting a possible mechanism of action [11]. IBD is considered to be a group of multifactorial disorders, and interestingly, there have been several monogenic disorders causing primary immunodeficiency described that can present with clinical and histopathological features similar to IBD [12,13]. Our findings suggest that it is important for clinicians to be aware of the potential GI complications that can cause significant morbidity in patients with XLA. In patients presenting with primary GI symptoms in addition to clinically significant infections, it may be reasonable to assess quantitative immunoglobulins and lymphocyte subsets to determine if there is an underlying immunodeficiency, such as XLA. To improve diagnosis and management of GI conditions in XLA patients, longitudinal studies that characterize the progression of GI manifestations and investigate successful management strategies are needed.
Finally, this case report and retrospective case series raises questions regarding best practices for managing GI manifestations in XLA patients. The most commonly administered therapy was IVIG for the primary disease process (XLA). Interestingly, IVIG has recently been gaining recognition as a potentially beneficial treatment for Crohn’s disease [10,11]. It is possible that IVIG use in the XLA patient population is preventing IBD progression. In these cases, there was a broad range of IVIG dosing, however, it was largely dosed at higher doses than traditional replacement level dosing (400mg/kg), which may have contributed to the anti-inflammatory and immunomodulatory effect [10,11]. The therapeutic benefits of IVIG may involve not just the passive transfer of antibodies, but also possibly an active role in aiding the immune cellular compartment and reducing inflammation and autoimmunity [12]. There are a number of emerging biologics that have shown promise in IBD [17], and interestingly, oral immunoglobulins have recently been suggested as a potential therapy for certain types of gastrointestinal disease [18,19]. Investigating the physiologic effects of immunoglobulin therapy in PID may provide further insight into immune pathways.
In the USIDNet cohort, therapies utilized purely for GI manifestations in XLA patients were limited to corticosteroids and supportive rescue therapy with parenteral nutrition. Longitudinal studies are needed to investigate the progression of IBD in XLA patients and to identify what treatment strategies for IBD are most successful in XLA patients.
Our case report is also notable for wide intrafamilial variability with a fatal outcome for Patient C (his disease was not confirmed, but consistent with XLA, likely related to the BTK mutation carried in the family). It is yet to be determined how the clinical outcome of young Patient A will be in comparison to his grandfather (Patient B), and if GI symptoms will be partly alleviated secondary to early use of immunoglobulin replacement therapy.
Conclusions
We present a case of a family where diagnosis of XLA in the index case led to a subsequent diagnosis of XLA in the grandfather, who had previously been diagnosed with Crohn’s Disease and then misdiagnosed with CVID. The predominance of the grandfather’s GI manifestations likely accounted for the delay in diagnosis and highlights the need for improved recognition of GI manifestations as complications of XLA.
GI manifestations are present in up to 35% of XLA patients, with IBD/enteritis diagnosed in up to 10% of patients. To improve diagnosis and management of GI conditions in XLA patients, longitudinal studies that characterize the progression of GI manifestations and investigate successful management strategies are needed.
Investigating the physiologic effects of IVIG therapy in PID may provide further insight into immune pathways. IVIG therapy has recently been gaining recognition as a potentially beneficial treatment for Crohn’s disease. It has been suggested that IVIG, in addition to passive replacement, may provide some protection against autoimmunity and inflammation.
Data from this case and the USIDNet case series suggest that hypomorphic BTK mutations in the XLA patients are associated with intrafamilial variability. These patients may be predisposed to immune dysregulation, and possibly have a tendency to develop gastrointestinal manifestations. Patients with XLA may require monitoring with particular attention for GI manifestations.
Footnotes
The authors declare that they have no conflict of interest.
References
- 1.Conley MEHV. X-Linked Agammaglobulinemia [Internet] GeneReviews® [Internet] 2011 Available from: http://www-ncbi-nlm-nih-gov.ezp-prod1.hul.harvard.edu/books/NBK1453/
- 2.Hernandez-Trujillo VP, Scalchunes C, Cunningham-Rundles C, Ochs HD, Bonilla Fa, Paris K, et al. Autoimmunity and inflammation in X-linked agammaglobulinemia. J Clin Immunol. 2014 Aug;34(6):627–32. doi: 10.1007/s10875-014-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee PPW, Chen T-X, Jiang L-P, Chan K-W, Yang W, Lee B-W, et al. Clinical characteristics and genotype-phenotype correlation in 62 patients with X-linked agammaglobulinemia. J Clin Immunol. 2010 Jan;30(1):121–31. doi: 10.1007/s10875-009-9341-5. [DOI] [PubMed] [Google Scholar]
- 4.Conley ME. Genetics of Hypogammaglobulinemia : What do we really know? Curr Opionion Immunol. 2010;21(5):466–71. doi: 10.1016/j.coi.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broides A, Yang W, Conley ME. Genotype/phenotype correlations in X-linked agammaglobulinemia. Clin Immunol Orlando Fla. 2006;118(2–3):195–200. doi: 10.1016/j.clim.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Jones A, Bradley L, Alterman L, Tarlow M, Thompson R, Kinnon C, et al. X linked agammaglobulinaemia with a “leaky” phenotype. Arch Dis Child. 1996 Jun;74(6):548–9. doi: 10.1136/adc.74.6.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.López-Granados E, Pérez de Diego R, Ferreira Cerdán A, Fontán Casariego G, García Rodríguez MC. A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol. 2005 Sep;116(3):690–7. doi: 10.1016/j.jaci.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal S, Mayer L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc. 2013 Sep;11(9):1050–63. doi: 10.1016/j.cgh.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Muhsen SZ. Gastrointestinal and hepatic manifestations of primary immune deficiency diseases. Saudi J Gastroenterol Off J Saudi Gastroenterol Assoc. 2010 Jun;16(2):66–74. doi: 10.4103/1319-3767.61230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis Off Publ Infect Dis Soc Am. 2008 May 15;46(10):1547–54. doi: 10.1086/587669. [DOI] [PubMed] [Google Scholar]
- 11.Meyers G, Ng Y-S, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, et al. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc Natl Acad Sci U S A. 2011 Jul 12;108(28):11554–9. doi: 10.1073/pnas.1102600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014 Nov;147(5):990–1007e3. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bianco AM, Girardelli M, Tommasini A. Genetics of inflammatory bowel disease from multifactorial to monogenic forms. World J Gastroenterol. 2015 Nov 21;21(43):12296–310. doi: 10.3748/wjg.v21.i43.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah S, Terdiman J, Gundling K, Mahadevan U. Immunoglobulin therapy for refractory Crohn’s disease. Ther Adv Gastroenterol. 2014 Mar;7(2):99–102. doi: 10.1177/1756283X13504728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogosnitzky M, Danks R, Holt D. Intravenous immunoglobulin for the treatment of Crohn’s disease. Autoimmun Rev. 2012 Dec;12(2):275–80. doi: 10.1016/j.autrev.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Kaveri SV, Maddur MS, Hegde P, Lacroix-Desmazes S, Bayry J. Intravenous immunoglobulins in immunodeficiencies: more than mere replacement therapy. Clin Exp Immunol. 2011 Jun;164(Suppl):2–5. doi: 10.1111/j.1365-2249.2011.04387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan HC-H, Ng SC. Emerging biologics in inflammatory bowel disease. J Gastroenterol. 2016 Nov;:10. doi: 10.1007/s00535-016-1283-0. [DOI] [PubMed] [Google Scholar]
- 18.Jasion VS, Burnett BP. Survival and digestibility of orally-administered immunoglobulin preparations containing IgG through the gastrointestinal tract in humans. Nutr J. 2015 Mar 7;14:22. doi: 10.1186/s12937-015-0010-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Arsdall M, Haque I, Liu Y, Rhoads JM. Is There a Role for the Enteral Administration of Serum-Derived Immunoglobulins in Human Gastrointestinal Disease and Pediatric Critical Care Nutrition? Adv Nutr Bethesda Md. 2016 May;7(3):535–43. doi: 10.3945/an.115.011924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conley ME, Fitch-Hilgenberg ME, Cleveland JL, Parolini O, Rohrer J. Screening of genomic DNA to identify mutations in the gene for Bruton’s tyrosine kinase. Hum Mol Genet. 1994 Oct;3(10):1751–6. doi: 10.1093/hmg/3.10.1751. [DOI] [PubMed] [Google Scholar]
- 21.Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton’s tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001 Jul;114(1):141–9. doi: 10.1046/j.1365-2141.2001.02905.x. [DOI] [PubMed] [Google Scholar]
- 22.Tóth B, Volokha A, Mihas A, Pac M, Bernatowska E, Kondratenko I, et al. Genetic and demographic features of X-linked agammaglobulinemia in Eastern and Central Europe: a cohort study. Mol Immunol. 2009 Jun;46(10):2140–6. doi: 10.1016/j.molimm.2009.03.012. [DOI] [PubMed] [Google Scholar]