

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder with cognitive deficits. Amyloidogenic processing of amyloid precursor protein (APP) produces amyloid β (Aβ), the major component of hallmark AD plaques. Synaptic activity stimulates APP cleavage, whereas APP promotes excitatory synaptic transmission, suggesting APP participates in neuronal homeostasis. However, mechanisms linking synaptic activity to APP processing are unclear. Here we show that Polo-like kinase 2 (Plk2), an activity-inducible regulator of homeostatic plasticity, directly binds and phosphorylates threonine-668 and serine-675 of APP in vitro and associates with APP in vivo. Plk2 accelerates APP amyloidogenic cleavage by β-secretase at synapses and is required for neuronal overactivity-stimulated Aβ secretion. These findings implicate Plk2 as a novel mediator of activity-dependent APP amyloidogenic processing.
Keywords: Alzheimer’s disease, amyloidogenic processing, polo-like kinase 2, synaptic plasticity, neuronal signaling, neurodegeneration, hyperexcitation
Graphical Abstract
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive cognitive deficits accompanied by hallmark neurofibrillary tangles and aggregated amyloid β (Aβ) plaques (Holtzman et al., 2011). Aberrant accumulation of Aβ may instigate pathogenesis (Hardy and Selkoe, 2002), based on dominant familial AD (FAD) mutations that increase production or aggregation of Aβ (Price and Sisodia, 1998). Soluble Aβ oligomers also block hippocampal long-term potentiation (LTP) and facilitate long-term depression (LTD), prominent models of learning and memory, and induce cognitive impairments in wild-type rodents (Li et al., 2009; Shankar et al., 2008; Walsh et al., 2002). Aβ is produced by amyloidogenic processing of amyloid precursor protein (APP) in which APP is initially cleaved by β-secretase (BACE-1), releasing secreted N-terminal sAPPβ and a C-terminal fragment (β-CTF) that is subsequently processed by γ-secretase to form Aβ and APP intracellular domain (AICD). A competing nonamyloidogenic pathway is initiated by α-secretases, producing sAPPα and α-CTF which is also processed by γ-secretase (Hardy and Selkoe, 2002). Despite intense interest in therapeutic manipulation of APP amyloidogenic processing, the physiological mechanism controlling Aβ production is still poorly understood.
Increasing evidence suggests that synaptic activity regulates APP processing and AD pathogenesis. Elevated incidence of seizures is observed in AD patients (Palop and Mucke, 2009), and brain regions with greatest basal metabolic activity, the “default-mode network”, exhibit highest amyloid burden in AD (Bero et al., 2011; Buckner et al., 2005). Furthermore, Aβ production is modulated by heightened neuronal activity in vitro and in vivo (Cirrito et al., 2008, 2005; Kamenetz et al., 2003), in a stimulus-dependent manner (Hoey et al., 2009; Verges et al., 2011). While multiple kinases may influence APP metabolism or trafficking, and hyperphosphorylation of APP is observed in human AD brain (Lee et al., 2003; Schettini et al., 2010), the precise role of APP phosphorylation and the signaling pathways linking synaptic activity to APP processing remain obscure.
Human Aβ soluble oligomers induce synaptic depression via AMPA receptor (AMPAR) removal and dendritic spine loss (Hsieh et al., 2006; Priller et al., 2006). The finding that Aβ production by chronic synaptic overexcitation causes synaptic depression suggests a homeostatic function for Aβ at excitatory synapses (Kamenetz et al., 2003), similar to the known role of Polo-like kinase 2/serum-inducible kinase (Plk2/SNK), an important regulator of homeostatic synaptic plasticity (Seeburg et al., 2005). Like Aβ, Plk2 is also upregulated by hyperexcitation (Kauselmann et al., 1999) to promote synaptic depression, AMPAR internalization, and shrinkage of dendritic spines, whereas Plk3 (the most closely related family member to Plk2 and the only other Plk family member detected in adult brain (Kauselmann et al., 1999)), is not required for these functions (Evers et al., 2010; Lee et al., 2011; Pak and Sheng, 2003; Seeburg et al., 2008). These observations suggest a preferential role for Plk2 in homeostatic plasticity and a potential relationship between Plk2 and Aβ generation.
Here, we show Plk2 directly binds to and phosphorylates threonine (668) and serine (675) of synaptic APP. Phosphorylation of the two sites is required for activity-inducible APP internalization and amyloidogenic processing, resulting in enhanced Aβ production in hippocampal neurons. These findings implicate Plk2 as an important physiological regulator of Aβ formation and possibly a target molecule for treatment of AD.
2. Material and Methods
2.1. Cell cultures
COS-7 cells were grown in DMEM with 10% fetal bovine serum and 0.1% gentamicin and transfected with 1 μg of DNA using Lipofectamine 2000 (Invitrogen) for 24 h. For cultured neurons, time pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA). Wild-type C57BL/6J and APP−/− (B6.129S7-Apptm1Dbo/J, stock #004133) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animal protocols were performed in accordance with guidelines of the Georgetown University Animal Care and Use Committee and the NIH. Hippocampal neurons were prepared from E18 rat embryos (Lee et al., 2011) and early postnatal (P0-P1) mice (Beaudoin et al., 2012). Neurons were plated on coverslips coated with poly-D-lysine and laminin and grown in Neurobasal media (Invitrogen) supplemented with B27, 0.5 mM glutamine and 12.5 μM glutamate. Between DIV 20–24, neurons were transfected with 2 μg DNA using Lipofectamine 2000 for 3 days, or infected with Sindbis virus expressing GFP, Plk2-KD, Plk2, APP-WT, or APP-2A for 18 hours at high titer to achieve complete infection of the culture. For synaptic overactivity modulation, neurons were treated with 25 μM picrotoxin for 18–20 h.
2.2. Antibodies
Rabbit Plk2 antibodies (#7382, ICC (immunocytochemistry) 1:200, IB (immunoblotting) 1:500) used for western blotting and immunoprecipitation and guinea pig SPAR (ICC 1:400) antibodies have been described (Lee et al., 2011). The following antibodies were purchased from the indicated suppliers and used at the indicated dilutions: APP-N (Sigma A8967, ICC 1:400, IB 1:1000); RB9023 (Thermo Scientific, ICC 1:5); M3.2 (Biolegend, ICC 1:500); 4G8 (Biolegend, ICC 1:500); APP-C (Sigma A8717, ICC 1:1000); Y188 (OriGene, ICC 1:500, IB:1:1000~1:7000); C1/6.1 (Biolegend, ICC 1:400); sAPPβ (Immuno-Biological Laboratories, IB 1:500); 6E10 (Covance, ICC 1:500, IB 1:1000); 22C11 (Millipore, MAB348, IB:1:1000); mouse anti-MAP2 (Sigma, HM-2, ICC 1:400); chicken anti-MAP2 (NeuroMAb, ICC 1:400); Plk2 C-terminal C-18 and N-terminal N-17 and H-90 (Santa Cruz, ICC 1:200, IB 1:400); GFP (Invitrogen and NeuroMAb, ICC 1:200, IB 1:1000); GluA2 (BD Pharmingen, ICC 1:50~1:100); mouse anti-PSD-95 (NeuroMAb, ICC 1:200); rabbit anti-PSD-95 (Cell Signaling, ICC 1:200); guinea pig anti-Synaptotagmin 1 (Synaptic Systems, ICC 1:500); mouse anti-VGlut1 (NeuroMAb, N28/9, ICC 1:200); rabbit anti-HA (Santa Cruz, ICC 1:100); and unlabeled donkey anti-mouse IgG (Sigma). AlexaFluor-488 and AlexaFluor-555 (Invitrogen, ICC 1:200~400) were secondary antibodies used for immunocytochemistry.
2.3. Inhibitors
The following inhibitors were used: β-secretase inhibitor II (BSI, Calbiochem, 1 μM), GM6001 (Calbiochem, 5 μM), TAPI-1 (Calbiochem, 1 μM), DAPT (Calbiochem, 1 μM), BI2536 (Axon MedChem, 50 nM), APV (Tocris Bioscience, 50 μM), CNQX (Tocris Bioscience, 40 μM) or phosphatase inhibitor I and II (Sigma). PTX was freshly prepared as a 10 mM stock in 0.1 M NaOH and used at 25 μM final concentration.
2.4. DNA constructs
Myc epitope-tagged Plk2, constitutively active Plk2 (Plk2-CA, T236E), kinase-dead Plk2 (Plk-KD, K108M), human APP770, human APP770 MV (M671V), and phosphorylation site mutants were expressed in pGW1-CMV (British Biotechnology). Oligonucleotides for Plk2 shRNA (Evers et al., 2010; Lee et al., 2011) (5′-GCA TAG GGA TCT CAA GCT A-3′), rescue constructs (Evers et al., 2010; Lee et al., 2011) (5′-GCA TAG AGA CCT CAA GCT-3′), and APP shRNA (Hoe et al., 2009) ( 5′-GCA CTA ACT TGC ACG ACT A-3′) were inserted into pLL3.7 vector which expresses RNAi-inducing shRNAs and GFP simultaneously under the U6 and CMV promoters. For purified protein production for in vitro binding and kinase assays, the APP C-terminal 47 amino acids were inserted in pMAL-c2x (New England Biolabs) containing maltose binding protein (MBP) with a 73 amino acid multiple cloning site tail. These cloning sites were substituted with the APP C-terminal, leading to a 3 kD decrease in molecular weight of MBP-APPc compared to MBP alone. Plk2 C-terminus including the polo box domain was inserted in pGEX. Site-directed mutagenesis was performed to generate all phosphosite mutants in the hAPP-770 backbone and correct mutations were verified by DNA sequencing (Genewiz). The following oligonucleotides are primer sequences for APP T668 and S675 mutagenesis: hAPP-2A; 5′-GAG GTT GAC GCC GCT GTC GCC CCA GAG GAG CGC CAC CTG GCC AAG ATG CAG CAG AAC GGC-3′ (sense) and 5′-GCC GTT CTG CTG CAT CTT GGC CAG GTG GCG CTC CTC TGG GGC GAC AGC GGC GTC AAC CTC-3′(antisense), hAPP-2E; 5′-GAG GTT GAC GCC GCT GTC GAA CCA GAG GAG CGC CAC CTG GAA AAG ATG CAG CAG AAC GGC-3′ (sense) and 5′-GCC GTT CTG CTG CAT CTT TTC CAG GTG GCG CTC CTC TGG TTC GAC AGC GGC GTC AAC CTC-3′ (antisense).
2.5. Immunoprecipitation and immunoblotting
For immunoprecipitation, cells or tissues were lysed in immunoprecipitation buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP40) or RIPA buffer (50 mM Tris pH 7.6, 150 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, 1% NP40, 0.1% SDS) with protease and phosphatase inhibitors (Sigma). After incubation for 20 h at 4°C with 1 μg of antibody and either protein A-Sepharose or protein G-Sepharose, bound proteins were eluted in Laemmli sample buffer and separated by Tris-glycine SDS-PAGE. Proteins were transferred to 0.45 μm pore size nitrocellulose and detected with enhanced chemiluminescence (Pierce) following incubation of HRP-linked secondary antibody. To obtain better resolution and retention of low molecular weight APP-CTF species, protein samples were separated on a 15% Tris-tricine SDS-PAGE gel and transferred to 0.2 μm pore size nitrocellulose.
2.6. Immunocytochemistry
For immunolabeling of APP and synaptic markers in primary hippocampal cultured neurons, an optimized fixation protocol for synaptic proteins was used (1% paraformaldehyde for 5 min followed by ice-cold methanol for 7 min), and incubated with primary antibody in GDB buffer (0.1% gelatin (wt/vol), 0.3% Triton X-100 (vol/vol), and 450 mM NaCl in phosphate buffered saline (PBS). Cells were treated with Alexa 488- and Alexa 555-tagged secondary antibody (Invitrogen) in GDB buffer. Transfected neurons were immunolabeled with GFP antibodies to visualize transfected cells. For Sindbis virus experiments, cells were immunolabeled with GFP or Plk2 to visualize infected cells. To detect surface proteins, live labeling was used (Lee et al., 2011). Primary hippocampal cultured neurons were incubated with primary antibody in conditioned media at 37°C for 10 min, and fixed with 4% paraformaldehyde for 4 minutes. Cells were immunolabeled with the fluorescence-tagged secondary antibody in non-permeable condition (ADB buffer, 3% normal goat serum, 0.1% bovine serum albumin in PBS). For further detection of transfected GFP signal, neurons were permeabilized with cold methanol and immunolabeled with GFP followed by the fluorescence-tagged secondary antibody in GDB buffer.
2.7. Surface biotinylation
Hippocampal neurons (DIV 20–24) were infected with Sindbis-APP-WT or −2A, and treated with PTX for 18 hours. Neurons were washed twice with PBS supplemented with 0.5 mM MgCl and 1 mM CaCl (sPBS), followed by biotinylation reaction (EZ-Link Sulfo-NHS-SS-Biotin, Pierce, 1 mg/ml in sPBS) for 12 min at 4°C with gentle agitation. After biotinylation, cells were washed three times with quenching solution (50 mM glycine and 0.5% BSA in sPBS) and two times with sPBS. Lysates were collected with RIPA buffer with protease inhibitors. A 10% aliquot of the lysates were reserved for total protein fraction, then avidin slurry (NeutrAvidin Agarose Resin, Pierce) was added and rotated for 2 hrs at 4°C. Beads were washed with RIPA buffer and then biotinylated proteins were eluted in Laemmli sample buffer and resolved by SDS-PAGE. All steps were carried out on ice with cold solutions to prevent trafficking of membrane proteins during biotinylation.
2.8. In vitro binding and kinase assay
Bacterially purified MBP-APPc was incubated with GST alone or GST-Plk2c and proteins pulled down with glutathione sepharose resin for immunoblotting with APP C-terminus and GST antibody. For in vitro kinase reactions, MBP-APPc was incubated in 50 mM Tris pH 7.5, 10 mM MgCl2, 5 mM DTT and 2 mM EGTA with calf intestine phosphatase (Promega) at 37°C. After 30 min of dephosphorylation, 200 ng of recombinant GST-tagged full-length wild-type Plk2 (Active Motif), 200 μM ATP and phosphatase inhibitor were added, incubated 30 min at 30°C, and then analyzed by immunoblotting with phosphothreonine and APP C-terminus antibodies. For some in vitro kinase assays, full-length myc-hAPP-WT or myc-hAPP-2A was immunoprecipitated from COS-7 cell lysates with antibody 6E10, or with nonimmune mouse IgG, and incubated with recombinant Plk2 as above except with the inclusion of 10 μCi 32P-γ-ATP (Perkin Elmer). Reaction products were examined by immunoblotting with myc antibodies and by autoradiography of SDS-PAGE gels.
2.9. Surface plasmon resonance
Assays were performed on a Biacore T-200 instrument in the Georgetown Biacore Molecular Interactions Shared Resource of Lombardi Comprehensive Cancer Center. Response unit (RU) was captured using 5842 RU of ligand. Bacterially purified MBP-APPc, GST-Plk2c and negative control proteins MBP and GST were immobilized on CM5 chips by amine coupling method. 510 RU of GST, 1,041 RU of GST-Plk2c, 956 RU of MBP-APPc and 989 RU of MBP were immobilized on Fc1, Fc2, Fc3 and Fc4 respectively. GST-Plk2c and GST proteins were immobilized in 10 mM acetate at pH 4.0; MBP and MBP-APPc were immobilized in 10 mM acetate at pH 4.5. For immobilization steps HBS-P (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% P-20) was used as the background running buffer at 10 μl/min flow rate. For binding experiments buffer was switched to a kinase reaction buffer (25 mM Tris pH 7.5, 5 mM MgCl2, 2.5 nM DTT, 1 mM EGTA) at 100 μl/min flow rate. MBP-APPc and MBP were injected on flow cells Fc1 and Fc2 with the concentrations 1 μM, 0.5 μM, 0.25 μM, 0.125 μM, 0.0625 μM, 0.0313 μM, and 0 μM (only data for 1 μM shown). The surface was regenerated with 1 M NaCl after each injection. Proteins were injected at 0 seconds, and 60 seconds after complex formation, injection was stopped and wash buffer applied. To control for nonspecific binding, the specific signal (Fc2-1) was calculated by subtracting empty buffer and the binding to GST alone (Fc1) from binding to target protein, Plk2c (Fc2). Injections were done in duplicates at least 4 different times, which had similar results.
2.10. Aβ ELISA
Culture media from hippocampal neurons infected with Sindbis virus expressing human APP were collected after PTX stimulation and BI2536 treatment. N2a cell media were collected 24h after vector or Plk2 transfection. Collected media were analyzed for human Aβ40 and Aβ42 using species-specific sandwich ELISA (Invitrogen) as described (Lee et al., 2010).
2.11. Measurement of cell viability
Hippocampal neurons were cultured on 12-well plates at a density of 3× 105 per well. Viability of cells was measured by incubation for 1 h at 37°C with 0.2 mg/ml of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma, M2128) dissolved in culture media. The formation of the formazan product, which is proportional to the number of viable cells, was extracted by DMSO and the absorbance was read by a spectrophotometer at 570 nm.
2.12. Fluorescence internalization assay
Hippocampal neurons at 21 DIV were co-transfected using Lipofectamine 2000 with hAPP-770 and either empty vector GW1 or HA-Plk2-GW1. Following transfection, live neurons were incubated for 16 hours with 6E10 antibody (which recognizes an extracellular epitope on human APP) to allow for surface labeling and internalization. Neurons were then lightly fixed in 2% paraformaldehyde/4% sucrose/PBS for 5 min at room temperature under nonpermeabilizing conditions, washed in PBS, and incubated overnight at 4 °C also under nonpermeabilizing conditions with saturating concentration of unlabeled secondary donkey anti-mouse IgG (100 μg/ml) to mask remaining surface 6E10 antibodies. Neurons were again lightly fixed in 2% paraformaldehyde/4% sucrose/PBS for 5 min at room temperature under nonpermeabilizing conditions, washed in PBS, permeabilized in 0.3% Triton X-100, and incubated overnight at 4°C in GDB buffer with rabbit anti-HA antibody to detect Plk2. Some samples were left unpermeabilized by withholding detergent in the buffer for all incubations. Lastly, neurons were incubated in anti-mouse Alexa Fluor 555 to detect internalized 6E10 antibody, and anti-rabbit Alexa Fluor 488 to detect cytoplasmic HA-Plk2.
2.13. Quantification and image analysis
Images were obtained using Axiovert 200M (Zeiss) epifluorescence for cultured neurons. Metamorph and ImageJ software was using for image analysis. From culture neuron immunocytochemistry, integrated intensity was measured in 20–30 μm dendritic segments from proximal regions (within 40 μm distance from soma) using Metamorph software.
2.14. Statistical analysis
All values were expressed as mean±SEM of at least duplicate experiments. Sample number represents the number of neurons, cells, or independent cultures as indicated in the figure legends. For analysis of immunofluorescent intensity in cultured hippocampal neurons, 3 dendritic segments from each neuron were measured and averaged per neuron. Two-tailed unpaired Student’s t-tests were used for comparisons between two independent groups, and one-way analysis of variance (ANOVA) was used for multiple group comparisons with Tukey’s post hoc test. Statistically significant differences were determined at P<0.05. All image quantification was performed blind with respect to experimental condition to avoid sampling bias.
3. Results
3.1. Synaptic APP displays distinct epitopes
We hypothesized that neuronal activity promotes amyloidogenic processing selectively of synaptic APP. However, APP localization at synapses is controversial, as some groups observed highly synaptic APP distribution (Hoe et al., 2009; Hoey et al., 2009) but others showed that several APP antibodies were apparently nonspecific for immunocytochemistry (Guo et al., 2012). To more thoroughly investigate immunostaining specificity of APP antibodies, we examined a panel of 7 anti-rodent APP antibodies directed against N-terminal (A8967 and RB9023), Aβ-site (M3.2 and 4G8), and C-terminal (Y188, A8717, and C1/6.1) epitopes (Fig. 1a). All antibodies were tested against primary hippocampal neurons cultured from wild-type (WT) mice, as well as from APP knockout (KO) mice as an essential specificity control.
Fig. 1. APP localized to excitatory synapses is detected exclusively by N-terminal directed antibodies.

(a) Schematic of APP sequence showing epitopes of antibodies used in immunostaining (epitope for N-terminal RB9023 has not been reported). (b–d) Immunocytochemistry of dendrites from cultured hippocampal neurons (14 days in vitro (DIV)) of wild-type (APP+/+, WT C57BL/6) or APP knockout (APP−/−, KO) mouse using N-terminal, Aβ site, and C-terminal directed antibodies as indicated (magenta), co-labeled with excitatory marker PSD-95 (green) (co-localization shows as white in merge) under permeabilizing conditions. Note complete absence of APP signal in knockout neurons. Scale bars, 10 μm. (e) Hypothetical interpretation of results. Synaptic APP (co-localizing with PSD-95, green), is recognized only by N-terminal antibodies (C-terminal and Aβ site antibodies are masked, possibly by APP scaffold proteins (gray)). APP in dendrites is recognized by C-terminal and Aβ site antibodies only (N-terminal antibodies are blocked, possibly by vesicular membranes). It is unknown if Aβ site and C-terminal antibodies recognize the same subpopulation of full-length APP; these antibodies may also visualize APP processing fragments. APP at other locations such as presynaptic terminals cannot be assessed with these antibodies.
Interestingly, we found that both N-terminal antibodies (A8967 and RB9023) reproducibly showed punctate APP staining that colocalized highly with the excitatory postsynaptic marker PSD-95, but not with the presynaptic markers Synaptotagmin 1 or VGlut1 (Fig. 1b and Supplementary Fig. 1a,b) (focusing specifically on distal dendrite synapses at high magnification and using fixation conditions and image acquisition settings optimized for detecting synaptic proteins; see Methods). These APP puncta also co-localized extensively with postsynaptic SPAR and AMPAR subunit GluA2 (Supplementary Fig. 1c). In striking contrast, the APP Aβ-site (Fig. 1c) and C-terminal (Fig. 1d) antibodies showed punctate and granular staining that was entirely excluded from excitatory synapses (i.e., completely non-overlapping with PSD-95). Furthermore, the APP-N (A8967) signals showed negligible co-localization with APP-C (C1/6.1) or with dendritic MAP2, while the APP-C signal was entirely colocalized with MAP2 within dendritic shafts (Supplementary Fig. 1b).
Importantly, dendritic and synaptic staining for all 7 antibodies was abolished in APP KO cultured neurons (Fig. 1b–d), indicating that these antibodies are indeed specific and recognize bona fide APP with differential localization patterns. We did observe some weak diffuse staining in KO neurons, but this background was largely restricted to the soma (data not shown). We further verified antibody specificity against APP KO brain lysates as well as cultured neurons in which APP expression was suppressed by previously characterized shRNA (Hoe et al., 2009) (Supplementary Fig. 2). We conclude that APP displays distinct epitopes at post-synapses compared to other subcellular locations (Fig. 1e).
3.2. Plk2 is required for activity-induced synaptic APP depletion
Having validated APP synaptic localization and immunocytochemistry specificity, we were able to examine the effect of heightened neuronal activity selectively on synaptic APP. We stimulated cultured hippocampal neurons with GABAA receptor antagonist picrotoxin (PTX), which increases Aβ levels in organotypic hippocampal slices (Kamenetz et al., 2003) and in vivo (Bero et al., 2011; Cirrito et al., 2008). Immunolabeling with APP-N antibodies revealed that APP was markedly downregulated after PTX stimulation when quantifying all APP signal (Fig. 2a,b) or only postsynaptic APP pixels overlapping with PSD-95 (Supplementary Fig. 3). This reduction began after 8 hours and reached steady-state level by 12 hours, with somatic APP decreasing in parallel (Supplementary Fig. 4a–d). APP loss was dose-dependent, plateauing at 20 μM PTX (Supplementary Fig. 4e). Activation of ionotropic glutamate receptors mediated PTX-induced Plk2 expression (Pak and Sheng, 2003). Indeed, a combination of APV and CNQX (NMDA and AMPA receptor antagonists, respectively) blocked APP reduction by PTX, but the inhibitors by themselves did not have an effect on APP levels. This result suggests that NMDA and AMPA receptors are involved in APP processing, possibly through Plk2 upregulation (Supplementary Fig. 5a–c). Thus, chronic neuronal hyperactivity removes synaptic APP with a slow time course suggestive of homeostatic plasticity mechanisms, and the extent of APP loss is related to the degree of overexcitation imposed.
Fig. 2. Activity-dependent synaptic APP loss requires β-secretase and Plk2.

(a, c) Hippocampal neurons (day in vitro (DIV) 20–24) were treated with picrotoxin (PTX, 25 μM, 20 h) or vehicle and co-treated with β-secretase inhibitor (BSI II, 1 μM), Plk inhibitor BI2536 (50 nM), or vehicle as indicated, then immunolabeled for endogenous rat APP with polyclonal APP-N antibodies. Representative dendrites shown below. (b) Quantification of a (n=44 neurons for NT, 38 for PTX, 16 for BSI, 18 for PTX+BSI). (d) Quantification of c (n=34 neurons for NT, 26 for PTX, 10 for BI2536, 9 for PTX+BI2536). (e) Neurons were transfected with empty vector (pLL3.7) or constructs as indicated for 3 days and treated with PTX (25 μM, 20 h) or vehicle (NT), then immunolabeled for endogenous APP and either transfected GFP or Plk2 as indicated. (f) Quantification of e (n=16 neurons for vector, 9 for vector+PTX, 10 for shRNA, 18 for shRNA+PTX, 7 for Plk2 shRNA+rescue, 7 for Plk2 shRNA+rescue+PTX, 11 for GFP NT, 9 for GFP+PTX, 7 for Plk2 KD, 7 for Plk2 KD+PTX). (g) Neurons were transfected with GFP or Plk2-WT, treated with BSI II (1 μM, 20 h) or vehicle (DMSO), and immunolabeled for GFP or Plk2 and APP N-terminus. (h) Quantification of g (n=32 neurons for GFP+vehicle, 16 for GFP+BSI, 11 for Plk2+vehicle, 9 for Plk2+BSI). ****P<0.0001; ***P<0.001; **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate. Scale bars, 10 μm.
To test whether synaptic APP loss was due to amyloidogenic processing, we applied β-secretase inhibitor (BSI) and found that this treatment blocked the removal of synaptic APP following PTX (Fig. 2a,b and Supplementary Fig. 3c), whereas two different α-secretase inhibitors (GM6001 and TAPI-1) or γ-secretase inhibitor (GSI) failed to restore APP with PTX stimulation (Supplementary Fig. 5d–l).
APP removal was also not due to excitotoxicity, as PTX did not affect dendritic morphology, MAP2 levels, cell viability (Supplementary Fig. 6), or β-actin levels (data not shown). Moreover, APP depletion was unlikely to be simply secondary to postsynapse elimination because co-treatment of PTX with either GSI or BSI blocked loss of PSD-95 that normally occurs with enhanced activity (Lee et al., 2011; Pak and Sheng, 2003) (Supplementary Fig. 7). BSI or GSI by themselves had no effect on PSD-95 levels. Thus, heightened neuronal activity decreases APP synaptic content specifically in a β-secretase-dependent manner, while loss of PSD-95 requires both β- and γ-secretase activities.
Since PTX strongly induces endogenous Plk2 expression (Evers et al., 2010; Lee et al., 2011; Pak and Sheng, 2003; Seeburg et al., 2008), we used BI2536, a Plk inhibitor (Steegmaier et al., 2007), to evaluate the role of Plk2 in the above APP loss. When co-applied with PTX stimulation, BI2536 abolished downregulation of synaptic APP levels by hyperactivity (Fig. 2c,d and Supplementary Fig. 3a,b). Because BI2536 antagonizes all Plk family members, we also specifically knocked down Plk2 using previously validated shRNA (Evers et al., 2010; Lee et al., 2011). This manipulation similarly prevented APP removal by PTX, whereas co-transfection of Plk2 shRNA with an shRNA-insensitive Plk2 rescue construct (Lee et al., 2011) reduced APP intensity by itself and occluded further effect of overactivity (Fig. 2e,f). Moreover, APP reduction by PTX was abrogated in neurons transfected with a kinase-dead (KD) Plk2 mutant (Fig. 2e,f) that functions in a dominant negative manner in vitro and in vivo (Lee et al., 2011). Conversely, overexpression of wild-type (WT) Plk2 was sufficient to remove APP without synaptic activity modulation, which was also prevented by BSI (Fig. 2g,h). Taken together, these results suggest that Plk2 kinase function links overactivity to β-secretase-mediated reduction in synaptic APP.
We further examined the Plk2-APP relationship in COS-7 cells. Co-transfection of WT or constitutively active (CA) Plk2 with human APP (hAPP) caused loss of hAPP protein, as visualized by immunostaining and immunoblotting, whereas Plk2-KD appeared to roughly colocalize with and “protect” hAPP, boosting its expression above control level (Supplementary Fig. 8a–d). These effects were blocked by BSI but not by α-secretase inhibitor GM6001 (Supplementary Fig. 8c,d). Accordingly, neither Plk2-CA nor Plk2-KD had any discernible effect on levels of hAPP-MV (Kamenetz et al., 2003), a mutant resistant to BACE-1 cleavage (Supplementary Fig. 8c,d). Plk2 had no impact on γ-processing of hAPP C-terminal fragments (α- and β-CTF); on the expression of other BACE-1 substrates ApoER2, APLP-1, or LRP (Li and Südhof, 2004; von Arnim et al., 2005); or on BACE-1 itself (Supplementary Fig. 8e–h). Thus, Plk2 specifically drives β-secretase cleavage of hAPP in mammalian cells.
3.3. Plk2 increases APP β-processing
To confirm that Plk2 promotes APP amyloidogenic processing, we analyzed endogenous APP cleavage products (De Strooper and Annaert, 2000) (Fig. 3a) in hippocampal neurons. Compared to control GFP-Sindbis virus-infected hippocampal neurons, cells expressing Plk2-WT-Sindbis virus showed decreased full-length APP and increased sAPPβ levels; sAPPβ level in cells expressing Plk2-WT was also significantly greater than in cells expressing Plk2-KD (Fig. 3b–d). Furthermore, the β-CTF/α-CTF ratio, an indicator of amyloidogenic APP processing by β-secretase, was enhanced by PTX treatment, while co-treatment of PTX with BI2536 blocked this increase and BI2536 by itself did not have any effect on this ratio (Fig. 3e,f). To assay human Aβ (hAβ) formation, we virally expressed Sindbis-hAPP in hippocampal neurons (Fig. 3g,h). PTX stimulation increased levels of hAβ40, although there was no significant change in hAβ42. BI2536 had no effect by itself, but importantly abolished PTX-induced hAβ generation (Fig. 3h).
Fig. 3. Plk2 stimulates APP β-processing.

(a) Schematic of APP processing pathways. (b) Hippocampal cultured neurons (21 DIV) were infected with Sindbis viruses for 18 hr as shown at bottom and lysates analyzed by immunoblotting for proteins indicated at left. (c,d) Quantification of (c) full-length APP and (d) sAPPβ from b, normalized to actin and GFP (for full-length APP: n=9 independent cultures for GFP, 11 for Plk2-KD, 18 for Plk2-WT; for sAPPβ: n=6 cultures for each group). (e) Hippocampal neurons (DIV 19–20) were treated with PTX or vehicle and co-treated with BI2536 (50 nM) as indicated, then immunolabeled with rabbit monoclonal APP-C antibodies (Y188). Neurons were pretreated with γ-secretase inhibitor (DAPT, 1 μm) 30 min prior to stimulation in order to prevent CTF degradation, which confirms the specificity of β- and α-CTF bands (note absence of CTFs without DAPT). (f) Quantification β-/α-CTF ratio from e (n=5 cultures). (g) Neurons were uninfected or infected with Sindbis-hAPP770 or GFP. Human APP was detected by immunoblotting with APP-N antibodies (exogenous hAPP770 appears larger than the dominant endogenous rat APP695 form). (h) Sindbis-hAPP-infected or uninfected neurons were treated with PTX (25 μM, 20 h) or vehicle (NT) and co-treated with BI2536 (50 nM) or vehicle (DMSO). Human Aβ40/42 (hAβ) were measured in conditioned media by species-specific ELISA and normalized against hAPP expression levels from immunoblotting (n=2 cultures for uninfected, 6 for NT, 6 for BI2536, 5 for PTX, 6 for PTX+BI2536). (i) N2a-myc-hAPP-Swe cells were transfected as indicated and lysates analyzed by immunoblotting. (j) Quantification of hAβ40 in conditioned media from i (n=7 cultures for vector, 8 for Plk2-KD, 8 for Plk2-WT, 9 for Plk2-CA). ***P<0.001, **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate. Molecular weights are kDa. Cropped blots are displayed for clear and concise presentation. Full western blots are shown in Supplementary Fig. 10.
To examine the effect of Plk2 on APP-Swe, an FAD mutant that is a superior substrate for BACE-1 versus APP-WT (Price and Sisodia, 1998), we used N2a cells stably expressing hAPP770-Swe (Thinakaran et al., 1996). Plk2-WT or Plk2-CA transfection, but not Plk2-KD, increased hAβ40 in culture media compared to vector control (Fig. 3i,j). Thus, Plk2 may act on a central regulatory step upstream of BACE-1 common to rodent and human APP, as well as WT and FAD forms.
3.4. Robust and direct interaction of Plk2 and APP
Based on the above findings, we tested whether Plk2 formed a physical complex with APP. First, we observed co-immunoprecipitation (IP) of Plk2-KD and hAPP from COS-7 cells using hAPP-specific 6E10 antibodies but not nonimmune IgG (Fig. 4a). Interaction experiments in heterologous cells required Plk2-KD to avoid active kinase-dependent loss of APP as shown above. In addition, endogenous APP was co-precipitated with Plk2 in cortical neurons infected with Sindbis-Plk2-KD (Fig. 4b). This interaction was also confirmed in hippocampal neurons in which the co-IP of endogenous Plk2 and APP was detected in both directions (Fig. 4c). Moreover, APP was recovered from brain lysates of APP-SwDI transgenic AD mice (Davis et al., 2004) using two independent Plk2 antibodies (Fig. 4d). Finally, we performed co-IPs from WT mouse brain to confirm endogenous APP and Plk2 association (Fig. 4e). Crucially, we observed no APP expression or interaction with Plk2 from APP-KO mouse brain (Fig. 4e), confirming that the in vivo association was specific and genuine. We noted that Plk2 antibody H-90 recovered a much greater fraction of Plk2 than of APP relative to total inputs (Fig. 4c). This finding suggests that a minor fraction of APP is associated with Plk2, similar to other Plk2 substrates (Lee et al., 2011). Interestingly, however, APP antibody Y188 pulled down roughly similar fractions of both APP and Plk2 relative to total inputs (Fig. 4c,e). Although these immunoblotting data are not linearly quantitative, we infer that a substantial proportion of Plk2 is associated with APP endogenously.
Fig. 4. Plk2 interacts directly with APP.

(a) COS-7 cells were transfected with myc-tagged Plk2-KD and either myc-tagged APP or empty vector. Lysates were immunoprecipitated with APP 6E10 antibody or IgG and immunoblotted with 6E10 and Plk2 H-90 antibodies as indicated. (b) Rat cortical neurons (20 DIV) were infected with Sindbis-Plk2-KD. Lysates were immunoprecipitated with Plk2 7382 antibody or IgG and immunoblotted with Plk2 7382 and APP N-terminal antibodies. Overexposure (bottom) is required to visualize endogenous full-length APP in input lane. Each blot was obtained from same gel with same exposure and spliced together for concise display. (c) Rat cultured hippocampal neurons (24 DIV) were harvested and immunoprecipitated with Plk2 antibody (H-90), APP antibody (Y188), or nonimmune rabbit IgG and immunoblotted with H-90 and Y188 to detect endogenous APP and Plk2 proteins. (d) APP-SwDI mouse (4-month-old) forebrain lysates were incubated with IgG or Plk2 antibody N-17 or H-90 and immunoblotted with Plk2 N-17 and human/rodent APP C-terminus antibodies. (e) WT and APP knockout (KO) forebrain lysates obtained from 2 month old animals were immunoprecipitated with Y188, H-90, or rabbit IgG and immunoblotted with 22C11 and N-17 for endogenous APP and Plk2 detection in vivo. (f) Coomassie blue staining of bacterially purified proteins as indicated. (g) Purified GST-Plk2c and MBP-APPc were incubated, pulled down with glutathione sepharose, and immunoblotted using GST and APP C-terminus antibodies. Inputs, 5% of lysates. (h) Surface plasmon resonance assay was performed with purified GST-Plk2c and MBP-APPc, or negative control MBP. Binding response (relative units, RU) showed robust interaction between immobilized GST-Plk2 and injected MBP-APPc but not MBP alone. Background was defined as MBP-APPc binding to GST alone, which was subtracted from the values shown. Bar indicates protein injection. Molecular weights in kDa. Experiments were performed in at least duplicate. Cropped blots are displayed for clear and concise presentation. Full blots are shown in Supplementary Fig. 10.
Because co-immunoprecipitation cannot determine whether Plk2 and APP interact directly, we performed in vitro binding assays using purified maltose binding protein (MBP)-tagged APP cytosolic C-terminus (APPc) mixed with GST-tagged Plk2 C-terminus (Plk2c) containing the conserved “polo box domain” important for substrate interactions (Elia et al., 2003) (Fig. 4f). GST pulldowns demonstrated that MBP-APPc co-purified with GST-Plk2c, but not with the negative control GST alone, indicating direct interaction of Plk2 and APP (Fig. 4g). Surface plasmon resonance, an independent method to assess direct binding, confirmed that MBP-APPc bound with GST-Plk2c (Fig. 4h). As controls for nonspecific binding, MBP-APPc failed to bind GST alone, and GST-Plk2 showed no binding to MBP alone (see Methods). Thus, the C-termini of APP and Plk2 interact directly in the absence of other proteins.
3.5. Phosphorylation of APP by Plk2
Direct binding implied that Plk2, a serine-threonine (S/T) kinase, may phosphorylate the intracellular tail of APP (Fig. 5a). Indeed, alanine replacement of all 5 S/T sites in the C-terminus of full-length hAPP (termed 5A mutant) conferred insensitivity to Plk2 co-expression in COS-7 cells as well as to PTX hyperexcitation in hippocampal neurons (Supplementary Fig. 9a–d). However, mutagenesis of any individual S/T site to alanine had no effect (Supplementary Fig. 9e,f). Thus, multiple putative S/T phosphosites seemed required for Plk2- and activity-induced APP β-processing.
Fig. 5. Direct phosphorylation of APP by Plk2.

(a) APP intracellular domain showing mutants in the study; amino acid numbering given for both hAPP-695 and hAPP-770 (in parentheses). Bold blue, serine/threonine residues. Underline, residues hyperphosphorylated in AD brain. Magenta, mutants insensitive to Plk2. (b) Myc-tagged APP double mutants T668/S675A, T668/T686A or S675/T686A were co-transfected with HA-vector, HA-Plk2-KD or HA-Plk2-WT in COS-7 cells and labeled with myc antibody for APP (green) and HA antibody for vector or Plk2 (magenta). (c) Quantification of b (T668/S675A: n=8 cells for vector, 9 for Plk2-KD, 6 for Plk2-WT; T668/T686A: n=8 cells for vector, 8 for Plk2-KD, 6 for Plk2-WT; S675/T686A: n=5 for vector, 5 for Plk2-KD, 7 for Plk2-WT); **P<0.01, ANOVA with Tukey’s post hoc test; data are means±SEM. (d) In vitro kinase assay using purified MBP, MBP-APPc, or MBP-APPc-2A with or without purified recombinant full-length GST-Plk2 protein. Reactions were immunoblotted using phosphothreonine and human/rodent-specific APP C-terminus antibodies. (e) (Top) Autoradiogram of in vitro kinase assays using full-length (FL) myc-hAPP-WT or myc-hAPP-2A immunopurified from COS-7 cells with antibody 6E10, or nonimmune IgG, and incubated in the presence or absence of GST-Plk2. (Bottom) Representative myc western blots of immunopurified myc-hAPP-WT and myc-hAPP-2A from COS-7 for in vitro kinase assay. Recovery of hAPP-2A was consistently greater than hAPP-WT with 6E10 antibody, but no recovery was obtained with nonimmune mouse IgG. Molecular weights in kDa. Experiments were performed in at least duplicate. Cropped blots are displayed for clear and concise presentation. Full blots are shown in Supplementary Fig. 10.
Narrowing candidate residues further, a 4A mutant with alanine replacement of 4 known human AD-associated S/T phosphoresidues (Lee et al., 2003) was found to be resistant to Plk2, as was triple mutant 3A-2 (T668A/S675A/T686A), whereas a different triple mutant 3A-1 remained sensitive (Supplementary Fig. 9g,h). Among the possible combinations of double mutants derived from 3A-2, only one (T668A/S675A; termed 2A mutant) was blocked from Plk2-mediated processing in COS-7 cells (Fig. 5b,c).
To test if T668/S675 were phosphorylated by Plk2, we conducted in vitro kinase reactions with purified APPc domain and full-length Plk2. Immunoblotting of reaction samples with phosphothreonine antibody revealed autophosphorylated Plk2 as shown previously (Lee et al., 2011) as well as phosphorylated APPc-WT, but not APPc-2A (Fig. 5d), indicating direct phosphorylation of APP by Plk2 at T668. Plk2 also phosphorylated full-length immunopurified hAPP in vitro, and this phosphorylation was reduced with the hAPP-2A mutant (34.49±7.6% of hAPP-WT measured by quantitative autoradiography, normalized to total input protein), consistent with phosphorylation of the 2A sites as well as an additional site by Plk2 (Fig. 5e). These results suggest that Plk2 directly phosphorylates T668/S675 to control APP amyloidogenic processing.
3.6. Plk2 phosphosites regulate surface APP expression
Because neuronal activity-dependent APP β-processing occurs mainly in endosomes (Cirrito et al., 2008), we examined whether Plk2 mediated-APP phosphorylation regulates its surface expression and internalization. Hippocampal neurons were transfected with hAPP-WT, hAPP-2A, or hAPP-2E (phosphomimic mutant with dual glutamate replacement of T668/S675). Under basal conditions, surface levels of hAPP-2A and hAPP-2E were greater and lower, respectively, than hAPP-WT (Fig. 6a–d), despite similar total expression (Fig. 6e–h). Moreover, PTX stimulation induced loss of surface and total hAPP-WT, similar to endogenous rat APP, but not hAPP-2A (Fig. 6a,c,e,g). Surface biotinylation assays independently confirmed that total and surface hAPP-WT, but not hAPP-2A, were diminished by PTX (Fig. 6i,j). Finally, we demonstrated that Plk2 enhanced the endocytosis of hAPP using an antibody-feeding fluorescence internalization assay (Fig. 6k,l). These findings suggest that phosphorylation status of T668/S675 bidirectionally regulates APP surface residence.
Fig. 6. APP-T668/S675 regulate surface expression and activity-dependent internalization.

(a, b) Hippocampal neurons (21–24 DIV) were co-transfected with GFP and hAPP-WT, −2A, or −2E, and treated with vehicle (NT) or PTX (25 μM, 20 h) as indicated. Surface/extracellular hAPP (APPec) was live-labeled with 6E10 antibody, followed by permeabilization and GFP labeling. (c) Quantification of a (n=15 neurons for APP-WT+NT, 21 for APP-WT+PTX, 9 for APP-2A+NT, 18 for APP-2A+PTX) (d) Quantification of b (n=8 neurons for APP-WT, 9 for APP-2A, 13 for APP-2E). (e, f) Total levels (tAPP) of transfected APP-WT, −2A, or −2E were labeled with human-specific 6E10 antibody along with GFP antibody under permeabilizing conditions. (g) Quantification of e (n=15 neurons for APP-WT+NT, n=8 for APP-WT+PTX, n=7 for APP-2A+NT, and n=10 for APP-2A+PTX). (h) Quantification of f (n=12 neurons for APP-WT, n=11 for APP-2A, and n=7 for APP-2E). (i) Neurons infected with Sindbis-hAPP-WT or −2A were treated with PTX, followed by surface biotinylation. Total and surface hAPP-WT/−2A were immunoblotted with 6E10. (j) Quantification of i (n=3 dishes). Molecular weights in kDa. (k) APP fluorescence internalization assay. Neurons were transfected with hAPP and empty vector or HA-epitope tagged Plk2 as indicated at top and stained for intracellular APP (hAPPint) and cytoplasmic Plk2 as indicated at left. Note the right-most column was left unpermeabilized (no perm.) and showed no staining, demonstrating that all signals are intracellular. (l) Quantification of k (n=14 for hAPP+vector and n=17 for hAPP+Plk2). ***P<0.001, **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test (except j and l, unpaired Student’s t-test). A.U., arbitrary units. Experiments were performed in at least duplicate unless otherwise noted. Data are means± SEM. Cropped blots are displayed for concise presentation. Full blots are shown in Supplementary Fig. 10.
4. Conclusions and Discussion
We elucidated a critical role for Plk2 in synaptic activity-dependent APP metabolism. Plk2 was necessary and sufficient to induce BACE-1-mediated APP amyloidogenic processing following overexcitation, associated intimately with APP, and directly phosphorylated the APP C-terminus. These results establish Plk2 as a previously unknown molecular linkage between neuronal activity and Aβ production.
Our study also provides evidence that endogenous APP resides at excitatory synapses, consistent with previous immunocytochemical studies (Hoe et al., 2009; Hoey et al., 2009). This finding has been controversial as another group found that several APP antibodies showed lack of specificity and generated high background in APP KO hippocampal neurons (Guo et al., 2012). However, this analysis did not specifically examine synapses, images were taken at low magnification, and fixation conditions were not optimized for synaptic staining, so no conclusions could be drawn from their study regarding APP synaptic localization. Under our experimental conditions, APP staining in dendrites and synapses with 7 different APP antibodies was absent in APP KO cultures, validating antibody specificity and demonstrating that these antibodies do not give high background when immunostaining is conducted appropriately for synapses.
Surprisingly, endogenous APP was detected by rodent antibodies against distinct epitopes in different loci. APP N-terminal antibodies (A8967 and RB9023) recognized postsynaptic APP, but antibodies recognizing Aβ sites (M3.2 and 4G8) and C-terminus (A8717, Y188, and C1/6.1) did not. Our interpretation is that various APP epitopes are unmasked at different subcellular locations. N-terminal epitopes seem available only at postsynapses, while C-terminal/Aβ ones are exposed only within dendrites. C-terminal and Aβ site antibodies may detect different pools of APP as well, such as trafficking full-length APP or intracellular APP processing fragments that lack N-terminal sequences (Fig. 1e). The precise subsynaptic localization of different APP puncta will be interesting for future analysis using higher resolution methods. We conclude that endogenous APP at synapses is detectable only with N-terminal antibodies, possibly explaining inconsistencies in the field regarding synaptic distribution of APP as results would depend on the particular antibody employed. These data support the general idea that different epitope “fingerprints” of APP are associated with various subcellular locations (presynaptic, postsynaptic, dendritic), suggestive of context-specific conformation or assemblies of protein-protein interactions at these sites. Our results also provide a useful resource for exploiting various epitopes to examine different clusters of APP selectively at specific subcellular loci.
An important mechanistic insight presented here was the identification of both T668 and S675 as essential residues for Plk2- and activity-mediated APP surface loss and processing during heightened synaptic activity. T668 of APP has previously garnered attention as a phosphosite that may stimulate amyloidogenic processing (Lee et al., 2003; Pastorino et al., 2006). However, these results are controversial as a T668A knock-in mouse mutation failed to affect constitutive rodent Aβ levels although activity-inducible formation was not measured (Barbagallo et al., 2010; Sano et al., 2006). The lack of effect of the T668A knock-in mutation is indeed consistent with and predicted from our findings that showed no effect of the single T668A mutation on activity-dependent APP processing. Thus, a single T668A mutation is not sufficient, but requires the additional S675A mutation to block activity-inducible amyloidogenic APP processing. Like T668, S675 is phosphorylated in AD brain (Lee et al., 2003), but this site has not been well characterized and does not lie within recognized functional motifs. Further analysis of the molecular function of this phosphorylation site is therefore warranted. In general, multiple APP phosphosites may be differentially recruited in a combinatorial fashion, imparting greater versatility to accommodate APP functions in varied physiological contexts (Hoe et al., 2012).
We further observed that Plk2 robustly promotes APP internalization. As the APP C-terminus interacts with multiple cytoplasmic adaptor proteins in a phosphorylation-dependent manner (Schettini et al., 2010), T668/S675 phosphorylation may alter the conformation of this domain, potentially controlling accessibility of internalization or scaffolding motifs. This interpretation is supported by the increased basal surface expression of APP-2A and its inability to undergo surface removal with sustained overactivity. Since endocytosis of APP is considered a prerequisite for amyloidogenic cleavage (Cirrito et al., 2008) and internalized APP is known to sort into BACE-1-containing endosomes upon activity induction (Das et al., 2013), we surmise that Plk2-dependent internalization of APP could increase its availability for BACE-1 during activity-dependent APP β-processing.
Several additional kinases have been implicated in APP processing (Schettini et al., 2010) such as glycogen synthase kinase-3α (GSK-3α) (Phiel et al., 2003), although these findings are also controversial (Jaworski et al., 2011). Contradictory findings may arise because appropriate physiological contexts have not been adequately considered. For instance, Plk2 was required specifically for hyperactivity-driven amyloidogenic processing, but not constitutive Aβ generation. Other kinases and their cognate phosphorylation site(s) may mediate APP processing only under specific conditions: basally, in different cell types, subcellular compartments, or developmental states, or in response to diverse stimuli such as proliferation or stress. Further work will be required to unravel the complex interplay among different kinases in normal APP processing and AD pathogenesis.
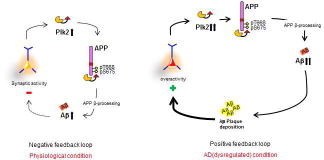
Based on the known role of Plk2 as a homeostatic regulator, we propose a model in which Plk2 and APP comprise a homeostatic negative feedback circuit that may normally function to prevent neuronal overexcitation (see graphic abstract; negative feedback loop). Chronic synaptic overactivity induces somatodendritic expression of Plk2, which binds directly to the intracellular tail of APP and phosphorylates T668/S675, stimulating endocytosis of surface APP and subsequent Aβ production. This mechanism occurs over a slow time course, concordant with the observation that many forms of homeostatic plasticity occur on relatively slow timescales (reviewed in Queenan et al., 2012). On a molecular level, this delay may reflect the time required to induce Plk2 expression, ~6–12 hrs (Seeburg and Sheng, 2008). We speculate this response may contribute to negative feedback homeostasis to dampen overactivity, although this possibility requires further investigation. The observation that activity-dependent loss of PSD-95 staining requires beta-secretase and gamma-secretase inhibitors lends support to the idea that APP processing is involved in synaptic homeostasis, a topic of interest for future study.
However, Aβ aggregation may subvert the physiological homeostatic system during pathogenesis, yielding plaque-associated hyperexcitability instead (Busche et al., 2008; Minkeviciene et al., 2009; Palop et al., 2007; Rudinskiy et al., 2012). This situation could create a positive feedback loop (see graphic abstract; positive feedback loop) that produces further aggregates and synaptic overactivity. It will be important to confirm the proposed model in vivo, by testing whether inhibition of Plk2 kinase function prevents Aβ production, plaque formation, and memory deficits in AD mouse models.
Notably, Aβ and Plk2 are both synaptotoxic and lead to synapse loss, a strong correlate of cognitive decline in AD (Hardy and Selkoe, 2002). The mechanism described here may present therapeutic opportunities for addressing excess synapse loss and possibly memory impairments in AD by engaging physiological regulatory networks of controlling APP metabolism.
Supplementary Material
Supplementary Figure 1. Localization of APP in cultured hippocampal neurons and antibody verification. (a) Immunocytochemistry of full-length endogenous APP in primary rat hippocampal neurons (21 days in vitro (DIV)) using N-terminal-directed polyclonal antibody (APP-N A8967, recognizing a conserved peptide sequence in 695- and 770-amino acid splice variants of human and rodent APP) (magenta), co-stained with post-synaptic marker PSD-95 or pre-synaptic markers Synaptotagmin 1 or VGlut1 (green), with merge at top. (b) Quantification of APP colocalization with PSD-95 or VGlut1, indicating APP detected by APP N-terminal antibodies are substantially located at postsynapses, and not at presynapses (n=37 neurons for PSD-95 and n=29 for VGlut1). (c) Endogenous APP in rat hippocampal neurons (21–24 DIV) using APP-N (A8967) (magenta), co-stained with excitatory synaptic markers SPAR or GluA2 (green), with merge at right. (d) (Top) Endogenous APP in rat hippocampal culture neurons (14 DIV) was co-immunostained using APP C-terminal antibody (APP C C1/6.1) (magenta) and APP N-terminal antibody (APP N A8967) (green). (Bottom) APP N or APP C (magenta) was co-stained with dendritic marker MAP2 (green) as indicated, with merge at right. Scale bars, 10 μm. ***P<0.001, Student’s t-test. Data are means±SEM.
Supplementary Figure 2. Verification of APP antibodies.
(a, b) Forebrain lysates from APP-SwDI transgenic and APP knockout (KO) mice were immunoblotted using C-terminal-directed antibody (APP C) and N-terminal-directed polyclonal antibody (APP N); β-actin used as loading control. (c) Neurons (21–24 DIV) were transfected with PLL3.7 (empty vector expressing only GFP), APP shRNA in PLL3.7, or left untransfected for 3 days, then immunolabeled with APP N A8967 antibodies to detect endogenous APP and with GFP antibodies to identify transfected neurons and visualize neuronal morphology. Note dramatic loss of dendritic spines due to APP knockdown. (d) Quantification of APP shRNA knockdown efficiency (***P<0.001, Student’s t-test; n=8 neurons for PLL3.7 and n=6 for shRNA). A.U., arbitrary units. Data are means±SEM. Experiments were performed in at least duplicate. Scale bars, 10 μm.
Supplementary Figure 3. Synaptic loss of APP by chronic overactivity is rescued by inhibition of β-secretase or Plk2. (a) Representative dendrites of rat hippocampal neurons (DIV 20–24) treated with picrotoxin (PTX, 25 μM, 18–20 h) or vehicle (NT) and co-treated with Plk inhibitor BI2536 (50 nM) as indicated, then co-immunolabeled with APP-N antibody and PSD-95. APP intensity was measured only at synapses (i.e. colocalizing with PSD-95, as indicated by circles). (b) Quantification of a (n=29 neurons for NT, 30 for PTX, 38 for BI2536, 30 for PTX+BI2536). (c) Quantification of synaptic APP levels from neurons treated with PTX in the absence or presence of β-secretase inhibitor (BSI II, 1 μM) (n=29 neurons for NT, 30 for PTX, 23 for BSI, 19 for PTX+BSI). Data are means±SEM. ***P<0.001, ANOVA and Tukey’s post hoc test.
Supplementary Figure 4. Time- and dose-dependent loss of dendritic and somatic APP with PTX. (a,b) Hippocampal neurons (21 DIV) were treated with picrotoxin (PTX, 25 μM) for different time periods as indicated, and then immunolabeled for endogenous rat APP (using APP-N antibodies) within (a) dendrites and (b) somata (acquired with shorter exposure time). Scale bars, 20 μm for wide views, 10 μm for dendrites. (c,d) Quantification of APP dendritic and somatic intensity from a,b as indicated (n=15 neurons for 0 hr, n=15 for 4 hrs, n=16 for 8 hrs, n=23 for 12 hrs, n=23 for 16 hrs, and n=15 for 20 hrs). (e) Quantification of dendritic APP intensity treated with different PTX concentrations as indicated for 20 hrs (n=14 neurons for 0 μM, n=8 for 10 μM, n=10 for 20 μM, n=9 for 50 μM, n=7 for 100 μM). ***P<0.001, **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test. Data are mean±SEM. Experiments were performed in at least duplicate.
Supplementary Figure 5. Activity-dependent reduction in APP requires ionotropic glutamate receptors but not α- or γ-secretases. (a–l) Neurons (18–24 DIV) were treated with PTX (25 μM, 20 h) or vehicle, and co-treated with vehicle (DMSO), APV (50 μM) plus CNQX (40 μM) (a,b), α-secretase inhibitors GM6001 (5 μM) (d,e) or TAPI-1 (1 μM) (g,h), or γ-secretase inhibitor (GSI) DAPT (1 μM) (j,k) and then immunolabeled with APP-N antibodies. (c) Quantification of a,b (n=32 neurons for NT, n=33 for PTX, n=41 for APV+CNQX, and n=35 for PTX+APV+CNQX). (f) Quantification of d,e (n=13 neurons for NT, n=13 for PTX, n=11 for NT+GM6001, and n=10 for PTX+GM6001). (i) Quantification of g,h (n=12 neurons for control, n=10 for PTX, n=8 for TAPI-1, and n=10 for PTX+ TAPI-1). (l) Quantification of j,k (n=10 neurons for control, n=12 for PTX, n=11 for GSI, and n=13 for PTX+GSI). ***P<0.001, **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate. Scale bars, 20 μm for wide views, 10 μm for dendrites.
Supplementary Figure 6. Activity-dependent APP depletion is not due to toxicity. (a,b) Cultured rat hippocampal neurons (21–24 DIV) were treated with PTX (25 μM, 20 h) or vehicle (no treatment, NT) and co-immunolabeled for endogenous rat APP (using APP N antibodies) and for MAP2, with representative dendrites shown below wide field images. (c) Quantification of a,b (n=9 neurons for APP+NT, n=6 for APP+PTX, n=10 for MAP2+NT, and n=6 for MAP2+PTX). (d,e) Time- (d) and dose-dependent (e) effect of PTX on viability of hippocampal neurons (20–25 DIV), expressed as percentages of untreated control values (NT or 0 h) (n=3–5 cultures). ***P<0.001, *P<0.05; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate.
Supplementary Figure 7. Activity-dependent APP depletion is not caused by loss of synapses. (a–f) Cultured hippocampal neurons (DIV21–24) were stimulated with PTX (25 μM, 20 h) or vehicle control, and co-treated with vehicle (DMSO), γ-secretase inhibitor (GSI) DAPT (1 μM) or β-secretase inhibitor (BSI) as indicated. Stimulated neurons were immunolabeled for endogenous PSD-95, with representative dendrites shown below wide-field images. (c) Quantification of a,b (n=8 neurons for control, n=9 for PTX, n=11 for GSI, and n=8 for PTX+GSI). (f) Quantification of d,e (n=21 neurons for control, n=25 for PTX, n=22 for BSI, and n=24 for PTX+BSI). ***P<0.001, **P<0.01; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate.
Supplementary Figure 8. β-secretase is required for Plk2-mediated hAPP depletion in COS-7 cells. (a) GFP-tagged human APP770 was co-expressed in COS-7 cells with empty vector, myc-Plk2-KD or myc-Plk2-WT, in the presence or absence of BSI II (1 μM, 24 h) as indicated, and cells were immunostained with antibodies against GFP (green) and myc (magenta). Arrowhead, example of colocalized GFP-APP and Plk2-KD. Arrow, Plk2-WT-transfected cell with nearly absent APP. (b) Quantification of APP intensity from a (n=10 cells). (c) Human myc-tagged APP770 or APP-MV was co-transfected in COS-7 cells with vector, myc-Plk2-KD or myc-Plk2-CA in the presence of vehicle (DMSO), BSI (1 μM, 24 h) or GM6001 (5 μM, 24 h) as indicated. Expression of APP and Plk2 were measured by immunoblotting with APP C-terminus and myc antibodies, respectively. (d) Quantification of c (n=10 independent cultures for APP WT, n=4 for APP+BSI, and n=2 for APP M671V). (e) Myc tagged-human α- or β-CTF was co-expressed with vector, myc-Plk2-KD, or myc-Plk2-CA in COS-7 cells. CTFs and Plk2 expression were detected with myc antibody. (f) Quantification of e (n=5 cultures for vector and Plk2-CA and n=6 for Plk2-KD). (g) HA epitope-tagged ApoE receptor 2 (ApoER2), GFP-tagged amyloid beta precursor-like protein 1 (APLP1), or GFP-tagged low-density lipoprotein receptor-related protein (LRP) were co-expressed with vector, myc-Plk2-KD or myc-Plk2-CA in COS-7 cells and each expression product detected with respective tag antibodies by immunoblotting. (h) Quantification of g (n=4 cultures for ApoER2, LRP, and BACE-1 and n=8 for APLP1). ***P<0.001, **P<0.01, *P<0.05 compared to empty vector; ANOVA with Tukey’s post hoc test. Data are mean±SEM. Experiments were performed in at least duplicate. Molecular weights in kDa.
Supplementary Figure 9. Site-directed mutagenesis of APP C-terminus and immunopurification of hAPP. (a) APP WT or APP 5A constructs were co-transfected with vector, myc-Plk2-KD or myc-Plk2-CA in COS-7 cells and lysates analyzed by immunoblotting for full-length APP (APP C-terminus antibody) and Plk2 (myc). (b) Quantification of a (n=10 dishes for APP-WT and n=4 for APP-5A). (c) Hippocampal neurons (21–24 DIV) were transfected with human APP-WT or APP-5A, treated with vehicle (NT) or PTX (25 μM, 20 h), and immunolabeled for GFP and human/rodent APP using N-terminal APP antibodies. (d) Quantification of c (n=7 cells for APP-WT+NT, n=6 for APP-WT+PTX, n=6 for APP-5A+NT, and n=8 for APP-5A+PTX). (e) Myc-tagged APP WT or APP single phosphomutants (T654A, S655A, T668A, S675A and T686A; APP695 numbering used throughout for consistency) were co-transfected with vector (vec), myc-Plk2-KD or myc-Plk2-CA in COS-7 cells. APP and Plk2 expression were analyzed by myc antibody immunoblotting. (f) Quantification of APP intensity from e (n=8 dishes). (g) Myc epitope-tagged APP 4A, 3A1 or 3A2 mutants (green) were co-transfected with HA epitope-tagged empty vector, Plk2-KD or Plk2-WT (magenta) in COS-7 cells and expression levels analyzed by double immunocytochemistry with respective epitope tag antibodies. (h) Quantification of APP intensity from g (APP-4A: n=9 cells for vector, n=7 for KD, n=6 for WT; APP-3A1: n=6 for vector, n=7 for KD, n=5 for WT; APP-3A2: n=4 for vector, n=5 for KD, n=4 for WT). ***P<0.001, **P<0.01, *P<0.05 compared to empty vector or to non-treated controls; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate. Molecular weights in kDa.
Supplementary Figure 10. Uncropped western blots.
Regions of blots used for figures are shown boxed in red. Molecular weights are in kDa.
Research highlights.
Only N-terminal antibodies recognize postsynaptic pools of APP in cultured neurons.
Plk2 is required for activity-dependent β-site processing of synaptic APP.
Plk2 interacts directly with and phosphorylates APP at Ser675 and Thr688.
APP dual phosphorylation by Plk2 regulates its surface expression and β-processing.
Acknowledgments
We thank members of the Pak laboratory for comments. We acknowledge the Biacore Molecular Interaction Shared Resource (BMISR) for surface plasmon resonance.
Funding
This work was supported by Georgetown Memory Disorders Program and National Institute of Health [grant numbers NS048085, DTSP]. The Shared Resources are partially supported by NIH/NIC CCSG [grant numbers P30-CA051008].
Footnotes
Author contributions
The authors have made the following declaration about their contribution: performed experiments: YL, JSL, KJL, and DTSP. Designed experiments and wrote the paper: YL, JSL, RST and DTSP. The authors declare no competing conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbagallo APM, Weldon R, Tamayev R, Zhou D, Giliberto L, Foreman O, D’Adamio L. Tyr(682) in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS One. 2010;5:e15503. doi: 10.1371/journal.pone.0015503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin GMJ, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011;14:750–6. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–9. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Das U, Scott Da, Ganguly A, Koo EH, Tang Y, Roy S. Activity-Induced Convergence of APP and BACE-1 in Acidic Microdomains via an Endocytosis-Dependent Pathway. Neuron. 2013;79:447–60. doi: 10.1016/j.neuron.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti M Lou, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(Pt 1):1857–70. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/S0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- Evers DM, Matta JA, Hoe HS, Zarkowsky D, Lee SH, Isaac JT, Pak DTS. Plk2 attachment to NSF induces homeostatic removal of GluA2 during chronic overexcitation. Nat Neurosci. 2010;13:1199–207. doi: 10.1038/nn.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Li H, Gaddam SSK, Justice NJ, Robertson CS, Zheng H. Amyloid precursor protein revisited: neuron-specific expression and highly stable nature of soluble derivatives. J Biol Chem. 2012;287:2437–45. doi: 10.1074/jbc.M111.315051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DTS, Rebeck GW. The effects of amyloid precursor protein on postsynaptic composition and activity. J Biol Chem. 2009;284:8495–506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe HS, Lee HK, Pak DTS. The upside of APP at synapses. CNS Neurosci Ther. 2012;18:47–56. doi: 10.1111/j.1755-5949.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci. 2009;29:4442–60. doi: 10.1523/JNEUROSCI.6017-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Lechat B, Gees M, Kremer A, Demedts D, Borghgraef P, Devijver H, Kügler S, Patel S, Woodgett JR, Van Leuven F. GSK-3α/β kinases and amyloid production in vivo. Nature. 2011;480:E4–5. doi: 10.1038/nature10615. discussion E6. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP Processing and Synaptic Function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kauselmann G, Weiler M, Wulff P, Jessberger S, Konietzko U, Scafidi J, Staubli U, Bereiter-Hahn J, Strebhardt K, Kuhl D. The polo-like protein kinases Fnk and Snk associate with a Ca(2+)- and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J. 1999;18:5528–39. doi: 10.1093/emboj/18.20.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Lee Y, Rozeboom A, Lee JY, Udagawa N, Hoe HS, Pak DTS. Requirement for Plk2 in orchestrated ras and rap signaling, homeostatic structural plasticity, and memory. Neuron. 2011;69:957–73. doi: 10.1016/j.neuron.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Moussa CEH, Lee Y, Sung Y, Howell BW, Turner RS, Pak DTS, Hoe HS. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience. 2010;169:344–56. doi: 10.1016/j.neuroscience.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Südhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279:10542–50. doi: 10.1074/jbc.M310001200. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–62. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DTS, Sheng M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science. 2003;302:1368–73. doi: 10.1126/science.1082475. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–40. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–34. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VMY, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–9. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–21. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan BN, Lee KJ, Pak DTS. Wherefore art thou, homeo(stasis)? Functional diversity in homeostatic synaptic plasticity. Neural Plast. 2012;2012:1–12. doi: 10.1155/2012/718203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudinskiy N, Hawkes JM, Betensky RA, Eguchi M, Yamaguchi S, Spires-Jones TL, Hyman BT. Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer’s disease. Nat Neurosci. 2012;15:1422–9. doi: 10.1038/nn.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Nakaya T, Pedrini S, Takeda S, Iijima-Ando K, Iijima K, Mathews PM, Itohara S, Gandy S, Suzuki T. Physiological mouse brain Abeta levels are not related to the phosphorylation state of threonine-668 of Alzheimer’s APP. PLoS One. 2006;1:e51. doi: 10.1371/journal.pone.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schettini G, Govoni S, Racchi M, Rodriguez G. Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: signal transduction and/or transcriptional role--relevance for Alzheimer pathology. J Neurochem. 2010;115:1299–1308. doi: 10.1111/j.1471-4159.2010.07044.x. [DOI] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DTS, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–83. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Pak D, Sheng M. Polo-like kinases in the nervous system. Oncogene. 2005;24:292–298. doi: 10.1038/sj.onc.1208277. [DOI] [PubMed] [Google Scholar]
- Seeburg DP, Sheng M. Activity-Induced Polo-Like Kinase 2 Is Required for Homeostatic Plasticity of Hippocampal Neurons during Epileptiform Activity. J Neurosci. 2008:28. doi: 10.1523/JNEUROSCI.1853-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–22. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J Biol Chem. 1996;271:9390–7. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- Verges DK, Restivo JL, Goebel WD, Holtzman DM, Cirrito JR. Opposing Synaptic Regulation of Amyloid-{beta} Metabolism by NMDA Receptors In Vivo. J Neurosci. 2011;31:11328–37. doi: 10.1523/JNEUROSCI.0607-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Arnim CAF, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, Spoelgen R, Hshieh TT, Ranganathan S, Battey FD, Liu CX, Bacskai BJ, Sever S, Irizarry MC, Strickland DK, Hyman BT. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem. 2005;280:17777–85. doi: 10.1074/jbc.M414248200. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Localization of APP in cultured hippocampal neurons and antibody verification. (a) Immunocytochemistry of full-length endogenous APP in primary rat hippocampal neurons (21 days in vitro (DIV)) using N-terminal-directed polyclonal antibody (APP-N A8967, recognizing a conserved peptide sequence in 695- and 770-amino acid splice variants of human and rodent APP) (magenta), co-stained with post-synaptic marker PSD-95 or pre-synaptic markers Synaptotagmin 1 or VGlut1 (green), with merge at top. (b) Quantification of APP colocalization with PSD-95 or VGlut1, indicating APP detected by APP N-terminal antibodies are substantially located at postsynapses, and not at presynapses (n=37 neurons for PSD-95 and n=29 for VGlut1). (c) Endogenous APP in rat hippocampal neurons (21–24 DIV) using APP-N (A8967) (magenta), co-stained with excitatory synaptic markers SPAR or GluA2 (green), with merge at right. (d) (Top) Endogenous APP in rat hippocampal culture neurons (14 DIV) was co-immunostained using APP C-terminal antibody (APP C C1/6.1) (magenta) and APP N-terminal antibody (APP N A8967) (green). (Bottom) APP N or APP C (magenta) was co-stained with dendritic marker MAP2 (green) as indicated, with merge at right. Scale bars, 10 μm. ***P<0.001, Student’s t-test. Data are means±SEM.
Supplementary Figure 2. Verification of APP antibodies.
(a, b) Forebrain lysates from APP-SwDI transgenic and APP knockout (KO) mice were immunoblotted using C-terminal-directed antibody (APP C) and N-terminal-directed polyclonal antibody (APP N); β-actin used as loading control. (c) Neurons (21–24 DIV) were transfected with PLL3.7 (empty vector expressing only GFP), APP shRNA in PLL3.7, or left untransfected for 3 days, then immunolabeled with APP N A8967 antibodies to detect endogenous APP and with GFP antibodies to identify transfected neurons and visualize neuronal morphology. Note dramatic loss of dendritic spines due to APP knockdown. (d) Quantification of APP shRNA knockdown efficiency (***P<0.001, Student’s t-test; n=8 neurons for PLL3.7 and n=6 for shRNA). A.U., arbitrary units. Data are means±SEM. Experiments were performed in at least duplicate. Scale bars, 10 μm.
Supplementary Figure 3. Synaptic loss of APP by chronic overactivity is rescued by inhibition of β-secretase or Plk2. (a) Representative dendrites of rat hippocampal neurons (DIV 20–24) treated with picrotoxin (PTX, 25 μM, 18–20 h) or vehicle (NT) and co-treated with Plk inhibitor BI2536 (50 nM) as indicated, then co-immunolabeled with APP-N antibody and PSD-95. APP intensity was measured only at synapses (i.e. colocalizing with PSD-95, as indicated by circles). (b) Quantification of a (n=29 neurons for NT, 30 for PTX, 38 for BI2536, 30 for PTX+BI2536). (c) Quantification of synaptic APP levels from neurons treated with PTX in the absence or presence of β-secretase inhibitor (BSI II, 1 μM) (n=29 neurons for NT, 30 for PTX, 23 for BSI, 19 for PTX+BSI). Data are means±SEM. ***P<0.001, ANOVA and Tukey’s post hoc test.
Supplementary Figure 4. Time- and dose-dependent loss of dendritic and somatic APP with PTX. (a,b) Hippocampal neurons (21 DIV) were treated with picrotoxin (PTX, 25 μM) for different time periods as indicated, and then immunolabeled for endogenous rat APP (using APP-N antibodies) within (a) dendrites and (b) somata (acquired with shorter exposure time). Scale bars, 20 μm for wide views, 10 μm for dendrites. (c,d) Quantification of APP dendritic and somatic intensity from a,b as indicated (n=15 neurons for 0 hr, n=15 for 4 hrs, n=16 for 8 hrs, n=23 for 12 hrs, n=23 for 16 hrs, and n=15 for 20 hrs). (e) Quantification of dendritic APP intensity treated with different PTX concentrations as indicated for 20 hrs (n=14 neurons for 0 μM, n=8 for 10 μM, n=10 for 20 μM, n=9 for 50 μM, n=7 for 100 μM). ***P<0.001, **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test. Data are mean±SEM. Experiments were performed in at least duplicate.
Supplementary Figure 5. Activity-dependent reduction in APP requires ionotropic glutamate receptors but not α- or γ-secretases. (a–l) Neurons (18–24 DIV) were treated with PTX (25 μM, 20 h) or vehicle, and co-treated with vehicle (DMSO), APV (50 μM) plus CNQX (40 μM) (a,b), α-secretase inhibitors GM6001 (5 μM) (d,e) or TAPI-1 (1 μM) (g,h), or γ-secretase inhibitor (GSI) DAPT (1 μM) (j,k) and then immunolabeled with APP-N antibodies. (c) Quantification of a,b (n=32 neurons for NT, n=33 for PTX, n=41 for APV+CNQX, and n=35 for PTX+APV+CNQX). (f) Quantification of d,e (n=13 neurons for NT, n=13 for PTX, n=11 for NT+GM6001, and n=10 for PTX+GM6001). (i) Quantification of g,h (n=12 neurons for control, n=10 for PTX, n=8 for TAPI-1, and n=10 for PTX+ TAPI-1). (l) Quantification of j,k (n=10 neurons for control, n=12 for PTX, n=11 for GSI, and n=13 for PTX+GSI). ***P<0.001, **P<0.01, *P<0.05; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate. Scale bars, 20 μm for wide views, 10 μm for dendrites.
Supplementary Figure 6. Activity-dependent APP depletion is not due to toxicity. (a,b) Cultured rat hippocampal neurons (21–24 DIV) were treated with PTX (25 μM, 20 h) or vehicle (no treatment, NT) and co-immunolabeled for endogenous rat APP (using APP N antibodies) and for MAP2, with representative dendrites shown below wide field images. (c) Quantification of a,b (n=9 neurons for APP+NT, n=6 for APP+PTX, n=10 for MAP2+NT, and n=6 for MAP2+PTX). (d,e) Time- (d) and dose-dependent (e) effect of PTX on viability of hippocampal neurons (20–25 DIV), expressed as percentages of untreated control values (NT or 0 h) (n=3–5 cultures). ***P<0.001, *P<0.05; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate.
Supplementary Figure 7. Activity-dependent APP depletion is not caused by loss of synapses. (a–f) Cultured hippocampal neurons (DIV21–24) were stimulated with PTX (25 μM, 20 h) or vehicle control, and co-treated with vehicle (DMSO), γ-secretase inhibitor (GSI) DAPT (1 μM) or β-secretase inhibitor (BSI) as indicated. Stimulated neurons were immunolabeled for endogenous PSD-95, with representative dendrites shown below wide-field images. (c) Quantification of a,b (n=8 neurons for control, n=9 for PTX, n=11 for GSI, and n=8 for PTX+GSI). (f) Quantification of d,e (n=21 neurons for control, n=25 for PTX, n=22 for BSI, and n=24 for PTX+BSI). ***P<0.001, **P<0.01; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate.
Supplementary Figure 8. β-secretase is required for Plk2-mediated hAPP depletion in COS-7 cells. (a) GFP-tagged human APP770 was co-expressed in COS-7 cells with empty vector, myc-Plk2-KD or myc-Plk2-WT, in the presence or absence of BSI II (1 μM, 24 h) as indicated, and cells were immunostained with antibodies against GFP (green) and myc (magenta). Arrowhead, example of colocalized GFP-APP and Plk2-KD. Arrow, Plk2-WT-transfected cell with nearly absent APP. (b) Quantification of APP intensity from a (n=10 cells). (c) Human myc-tagged APP770 or APP-MV was co-transfected in COS-7 cells with vector, myc-Plk2-KD or myc-Plk2-CA in the presence of vehicle (DMSO), BSI (1 μM, 24 h) or GM6001 (5 μM, 24 h) as indicated. Expression of APP and Plk2 were measured by immunoblotting with APP C-terminus and myc antibodies, respectively. (d) Quantification of c (n=10 independent cultures for APP WT, n=4 for APP+BSI, and n=2 for APP M671V). (e) Myc tagged-human α- or β-CTF was co-expressed with vector, myc-Plk2-KD, or myc-Plk2-CA in COS-7 cells. CTFs and Plk2 expression were detected with myc antibody. (f) Quantification of e (n=5 cultures for vector and Plk2-CA and n=6 for Plk2-KD). (g) HA epitope-tagged ApoE receptor 2 (ApoER2), GFP-tagged amyloid beta precursor-like protein 1 (APLP1), or GFP-tagged low-density lipoprotein receptor-related protein (LRP) were co-expressed with vector, myc-Plk2-KD or myc-Plk2-CA in COS-7 cells and each expression product detected with respective tag antibodies by immunoblotting. (h) Quantification of g (n=4 cultures for ApoER2, LRP, and BACE-1 and n=8 for APLP1). ***P<0.001, **P<0.01, *P<0.05 compared to empty vector; ANOVA with Tukey’s post hoc test. Data are mean±SEM. Experiments were performed in at least duplicate. Molecular weights in kDa.
Supplementary Figure 9. Site-directed mutagenesis of APP C-terminus and immunopurification of hAPP. (a) APP WT or APP 5A constructs were co-transfected with vector, myc-Plk2-KD or myc-Plk2-CA in COS-7 cells and lysates analyzed by immunoblotting for full-length APP (APP C-terminus antibody) and Plk2 (myc). (b) Quantification of a (n=10 dishes for APP-WT and n=4 for APP-5A). (c) Hippocampal neurons (21–24 DIV) were transfected with human APP-WT or APP-5A, treated with vehicle (NT) or PTX (25 μM, 20 h), and immunolabeled for GFP and human/rodent APP using N-terminal APP antibodies. (d) Quantification of c (n=7 cells for APP-WT+NT, n=6 for APP-WT+PTX, n=6 for APP-5A+NT, and n=8 for APP-5A+PTX). (e) Myc-tagged APP WT or APP single phosphomutants (T654A, S655A, T668A, S675A and T686A; APP695 numbering used throughout for consistency) were co-transfected with vector (vec), myc-Plk2-KD or myc-Plk2-CA in COS-7 cells. APP and Plk2 expression were analyzed by myc antibody immunoblotting. (f) Quantification of APP intensity from e (n=8 dishes). (g) Myc epitope-tagged APP 4A, 3A1 or 3A2 mutants (green) were co-transfected with HA epitope-tagged empty vector, Plk2-KD or Plk2-WT (magenta) in COS-7 cells and expression levels analyzed by double immunocytochemistry with respective epitope tag antibodies. (h) Quantification of APP intensity from g (APP-4A: n=9 cells for vector, n=7 for KD, n=6 for WT; APP-3A1: n=6 for vector, n=7 for KD, n=5 for WT; APP-3A2: n=4 for vector, n=5 for KD, n=4 for WT). ***P<0.001, **P<0.01, *P<0.05 compared to empty vector or to non-treated controls; ANOVA with Tukey’s post hoc test. Data are means±SEM. Experiments were performed in at least duplicate. Molecular weights in kDa.
Supplementary Figure 10. Uncropped western blots.
Regions of blots used for figures are shown boxed in red. Molecular weights are in kDa.