

Abstract
Painful nerve injury disrupts Ca2+ signaling in primary sensory neurons by elevating plasma membrane Ca2+-ATPase (PMCA) function and depressing sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) function, which decreases endoplasmic reticulum (ER) Ca2+ stores and stimulates store-operated Ca2+ entry (SOCE). The extracellular matrix glycoprotein thrombospondin-4 (TSP4), which is increased after painful nerve injury, decreases Ca2+ current (ICa) through high-voltage–activated Ca2+ channels and increases ICa through low-voltage–activated Ca2+ channels in dorsal root ganglion neurons, which are events similar to the effect of nerve injury. We therefore examined whether TSP4 plays a critical role in injury-induced disruption of intracellular Ca2+ signaling. We found that TSP4 increases PMCA activity, inhibits SERCA, depletes ER Ca2+ stores, and enhances store-operated Ca2+ influx. Injury-induced changes of SERCA and PMCA function are attenuated in TSP4 knock-out mice. Effects of TSP4 on intracellular Ca2+ signaling are attenuated in voltage-gated Ca2+ channel α2δ1 subunit (Cavα2δ1) conditional knock-out mice and are also Protein Kinase C (PKC) signaling dependent. These findings suggest that TSP4 elevation may contribute to the pathogenesis of chronic pain following nerve injury by disrupting intracellular Ca2+ signaling via interacting with the Cavα2δ1 and the subsequent PKC signaling pathway. Controlling TSP4 mediated intracellular Ca2+ signaling in peripheral sensory neurons may be a target for analgesic drug development for neuropathic pain.
Keywords: Neuropathic pain, Intracellular calcium signaling, Thrombospondin-4, Plasma membrane Ca2+-ATPase, Ca2+ stores and stimulates store-operated Ca2+ entry, Sarco-endoplasmic reticulum Ca2+-ATPase
1. Introduction
Neuronal cytoplasmic Ca2+ is the essential regulator of numerous physiological functions, such as excitation, synaptic transmission, synaptic plasticity, and neuronal differentiation and survival (Catterall et al., 2013; Gemes et al., 2011; Paschen, 2001). When neurons are activated, free cytoplasmic Ca2+ concentration ([Ca2+]i) increases due to Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) or ligand-gated Ca2+ channels on the plasma membrane. This may be supplemented by release of Ca2+ from intracellular stores upon activation of ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate receptors (IP3R) on the membrane of endoplasmic reticulum (ER). In addition, Ca2+ can enter neurons through channels that are indirectly activated by the depletion of ER Ca2+ stores in the process known as store-operated Ca2+ entry (SOCE) (Gemes et al., 2011). After neuronal activation, elevated [Ca2+]i recovers back to resting levels by Ca2+ extrusion through the plasma membrane Ca2+-ATPase (PMCA) (Duncan et al., 2013) and Na+-Ca2+ exchanger (NCX), and by Ca2+ sequestration into the ER by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) (Gemes et al., 2012). We have previously observed that, after nerve injury, sensory neurons of the dorsal root ganglia (DRG) develop decreased resting [Ca2+]i (Fuchs et al., 2005), depressed Ca2+ influx through VGCCs (Hogan et al., 2000), and decreased SERCA function (Duncan et al., 2013), as well as elevated Ca2+ clearance from the cytoplasm by PMCA function (Gemes et al., 2012) and enhanced Ca2+ influx through SOCE due to depletion of ER Ca2+ stores (Gemes et al., 2011). However, the pathological effector driving these events is undefined.
Thrombospondins (TSP) are a family of large oligomeric, extracellular matrix glycoproteins that mediate interactions between cells and interactions of cells with underlying matrix components (Adams, 2001; Risher and Eroglu, 2012). The TSP family consists of five members (TSP 1–5) divided into two subgroups (TSP1/2 vs. TSP3/4/5) according to their functional domains (Risher and Eroglu, 2012). TSP4 is expressed by astrocytes and neurons, in which it can promote neurite outgrowth and synaptogenesis (Arber and Caroni, 1995; Eroglu et al., 2009; Pan et al., 2015). Recently, TSP4 has been found to be a factor contributing to neuropathic pain (Crosby et al., 2014; Kim et al., 2012). Specifically, TSP4 gene and protein expression is elevated in DRGs after peripheral nerve injury, and intrathecal administration of TSP4 protein amplifies excitatory pre-synaptic transmission in the dorsal horn via promoting excitatory synaptogenesis by binding to its receptor, voltage-gated Ca2+ α2δ1 subunit (Cavα2δ1) (Kim et al., 2012; Pan et al., 2015; Park et al., 2016). However, specific cellular and molecular mechanisms by which TSP4 acts are still unclear.
Various observations support the view that actions of TSP4 may contribute to the pathological effects of axotomy in sensory neurons. Specifically, application of TSP4 in vitro reproduces the in vivo effects of nerve injury on VGCCs (Hogan et al., 2000; Pan et al., 2016). Additionally, expression of TSP4 protein is elevated in axotomized sensory neurons following peripheral nerve injury (Pan et al., 2015), which suggests that TSP4 may have an important role in regulating intracellular signaling. Here, we test our hypothesis that elevated TSP4 after nerve injury leads to depletion of ER Ca2+ stores and disordered function of PMCA, SERCA, and SOCE in primary sensory neurons.
2. Materials and methods
2.1. Animals
Male adult mice (129S1/SvImJ) and TSP4 gene knock-out (KO) mice (B6.129P2-Thbs4tm1Dgen/J) were obtained from The Jackson Laboratory. The Cavα2δ1 advillin conditional knock-out (CKO) mice were generated by crossing mice with Cre-recombinase expression in advillin positive cells to 129/sv background mice with exon 6 of the Cavα2δ1 gene (MGI ID: 88295) floxed with loxP sites (Park et al., 2016). CKOAdv-Cre+/− mice have conditionally knocked out Cavα2δ1 in sensory neurons. CKOAdv-Cre−/− littermates mice were used as wildtype (WT) control. Male Sprague Dawley rats weighing 170–200g used in some experiments were obtained from The Taconic Biosciences. All animals were housed in a room with a 12/12 h day/night cycle and free access to food and water. Male mice were used for initial studies and female mice were used to confirm major findings. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (AUA00001809 for experiments on mice and AUA00002752 for experiments on rats). Those animals were maintained and used according to the NIH Guide for the Care and Use of Laboratory Animals, and in compliance with federal, state, and local laws. DRGs from mice and rats were rapidly harvested following deep isoflurane anesthesia and decapitation.
2.2. Neuron isolation and plating
DRGs from mice or rats were rapidly harvested following deep isoflurane anesthesia and decapitation. Ganglia were placed in a 35 mm dish containing Ca2+/Mg2+-free, cold HBBS (Life Technologies) and cut into four to six pieces that were incubated in in 0.01% blendzyme 2 (Roche Diagnostics, Indianapolis, IN) for 26 min followed by incubation in 0.25% trypsin (Sigma Aldrich, St. Louis, MO) and 0.125% DNAse (Sigma) for 30 min, both dissolved in Dulbecco’s modified Eagle’s medium (DMEM)/F12 with glutaMAX (Invitrogen, Carlsbad, CA). After exposure to 0.1% trypsin inhibitor and centrifugation, the pellet was gently triturated in culture medium containing Neural Basal Media A with B27 supplement (Invitrogen), 0.5 mM glutamine, 10 ng/ml nerve growth factor 7S (Alomone Labs, Jerusalem, Israel) and 0.02 mg/ml gentamicin (Invitrogen). Dissociated neurons were plated onto poly-L-lysine coated glass cover slips (DeutschesSpiegelglas, Carolina Biological Supply, Burlington, NC) and maintained at 37°C in humidified 95% air and 5% CO2 for 2 hours, and were studied no later than 8 hours after harvest.
2.3. Measurement of cytoplasmic Ca2+ concentration
Measurement of [Ca2+]i was performed following our previously published protocols (Duncan et al., 2013; Gemes et al., 2012). In brief, unless otherwise specified, regular Tyrode’s solution (in mM: NaCl 140, KCl 4, CaCl2 2, MgCl2 2, glucose 10, HEPES 10 with an osmolarity of 297–300 mOsm and pH 7.40) was used to bathe the neurons. Stock solution of Fura-2-AM (Invitrogen) was dissolved in DMSO and subsequently diluted in the relevant bath solution such that final bath concentration of DMSO was 0.2% or less, which does not affect [Ca2+]i (Gemes et al., 2011). The 500 μl recording chamber was superfused by gravity-driven flow at a rate of 3 ml/min. Agents were delivered by directed microperfusion controlled by a computerized valve system through a 500 μm diameter hollow quartz fiber 300 μm upstream from the neurons. This flow completely displaced the bath solution, and constant flow was maintained through this microperfusion pathway by delivery of bath solution when specific agents were not being administered. Dye imaging shows that solution changes were achieved within 200 ms.
Neurons plated on cover slips were exposed to Fura-2-AM (5 μM) at room temperature in a solution that contained 2% bovine albumin to aid dispersion of the fluorophore. After 30 min, they were washed 3 times with regular Tyrode’s solution, given 30 minutes for de-esterification, and then mounted in the recording chamber. Neurons were first examined under bright field illumination, and those showing signs of lysis, crenulation or superimposed glial cells were excluded. To determine [Ca2+]i, the fluorophore was excited alternately with 340 nm and 380 nm wavelength illumination (150 W Xenon, Lambda DG-4, Sutter, Novato, CA), and images were acquired at 510 nm using a cooled 12-bit digital camera (Coolsnapfx, Photometrics, Tucson, AZ) and inverted microscope (Diaphot 200, Nikon Instruments, Melville, NY) through a 20X objective. Recordings from each neuron were obtained as separate regions (MetaFluor, Molecular Devices, Downingtown, PA) at a rate of 3 Hz. After background subtraction, the fluorescence ratio R for individual neurons was determined as the intensity of emission during 340 nm excitation (I340) divided by I380, on a pixel-by-pixel basis, and [Ca2+]i was estimated by the formula Kd•β•(R-Rmin)/(Rmax-R) where β = (I380max)/(I380min). Values of Rmin, Rmax and β were determined by in-situ calibration and were 0.38, 8.49 and 9.54, and Kd was 224 nm. Only neurons with stable baseline R traces were further evaluated. Traces were analyzed using Axograph X 1.1 (Axograph Scientific, Sydney, Australia).
Depolarization-induced Ca2+ transients were generated by microperfusion application of K+ (50 mM) for 0.3 s. To characterize the rate of recovery of the activity-induced [Ca2+]i transient, the trace was fit by a mono-exponential curve (y = y0 + k × exp (−x/τ)) from which the time constant (τ) was derived as a measure of the pace of recovery. To block PMCA function, HEPES was replaced by Trizma (10 mM) in regular Tyrode’s with pH at 8.8. To block SERCA function, thapsigargin (TG, 1μM, Sigma-Aldrich, St. Louis, MO) was added into solutions. For quantifying SERCA and PMCA activity, we limited the participation of mitochondria by excluding traces that either had a transient peak above 400nM or showed a shoulder of sustained [Ca2+]i elevation during the descending limb of the activity-induced transient (Gemes et al., 2012). To induce the release of stored Ca2+ from the ER, caffeine (20mM) was added to regular Tyrode’s solution. SOCE function was quantified as the amplitude of the transient induced by switching from Ca2+-free bath solution to solution with 2 mM Ca2+, either with or without TG (1 μM). In all TSP4 treatment groups, neurons were incubated with TSP4 (60nM) for 4 hours, which is the dose and duration of application necessary to produce effects based on our previous electrophysiological experiments (Pan et al., 2016).
2.4. IB4 staining
After the measurement of cytoplasmic Ca2+ concentration, neurons were identified by incubating with isolectin B4 (IB4) conjugated with fluorescein isothiocyanate (10 μg/ml, Sigma-Aldrich, St. Louis, MO) for 10 min and then washed with regular Tyrode’s solution. IB4-positive (IB4pos) neurons were identified as those that retained a complete ring of FITC stain around the perimeter of the soma (Barabas et al., 2012).
2.5. Injury model
Fifteen mice were subjected to spinal nerve ligation (SNL), based on the original technique (Kim and Chung, 1992) with modifications (Gemes et al., 2012; Pan et al., 2015; Rigaud et al., 2008). Briefly, during anesthesia by inhalation of isoflurane (1.5–2.5% in oxygen), the right lumbar paravertebral region was exposed through a midline incision, and the fifth lumbar (L5) transverse process was removed to expose the L4 spinal nerve, which was ligated with 6-0 silk suture and severed approximately 1 mm distal to their respective DRGs. The wound was closed in layers and the skin stapled. Control mice received skin incision and closure only. DRGs were collected for experiments 15–21 days after the surgery.
2.6. Reagents
Fura-2 AM, thapsigargin and all other common chemicals were obtained from Sigma-Aldrich. YM 244769, CGP 37157, N-[2-[[3-(4-Bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89), Calphostin C, Phorbol 12-myristate 13-acetate (PMA), (9R,10S,12S)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester (KT5720) were obtained from Tocris Bioscience. Ru360 was obtained from EMD Millipore. TSP4 proteins were expressed and purified according to our previous report (Kim et al., 2012).
2.7. Data Analysis
DRG sensory neurons are a diverse population with various functional and molecular attributes. Sensory neuron somatic diameter is broadly associated with specific sensory modalities. Whereas small unmyelinated neurons generally carry nociceptive traffic, large myelinated neurons are activated by low threshold stimuli (Ma et al., 2003). Small-diameter sensory neurons are further divided neurochemically into IB4pos and IB4-negative (IB4neg) populations, which have distinct electrophysiological properties and sensitivities to noxious stimuli (Stucky and Lewin, 1999). By defining the small-diameter population of our recorded neurons from mice as having a diameter ≤ 27μm (Barabas et al., 2012), we found that 98% of the IB4pos population were small (Fig. 1A) and 65% of small neurons were IB4pos, which is consistent with previous observations (Barabas et al., 2012; Dirajlal et al., 2003). Defining large neurons as diameter > 27μm, 4% of which were IB4pos. Those few IB4pos large neurons were ignored in further analysis. For experiments using neurons from rats, we defined the population of our recorded neurons with a diameter ≤ 34μm as small, and neurons with a diameter > 34 μm as large (Gemes et al., 2011).
Fig. 1.
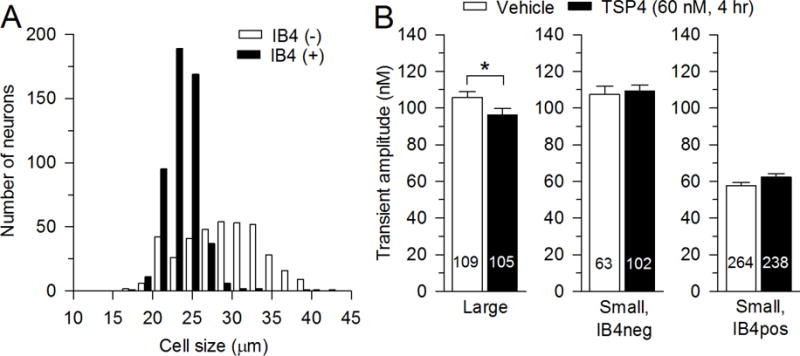
Effects of TSP4 on resting [Ca2+]i of DRG neurons. A, Classification of neurons according to size and IB4 staining. B, Summary data show effects of TSP4 incubation on resting [Ca2+] i in neurons of different groups categorized by size and IB4 binding. The numbers in the bars represent the n for neurons in groups, and bars show mean ± SEM, here and in subsequent figures. * p < 0.05.
Statistical analyses were performed with Excel (Microsoft, Redmond, WA), GraphPad Prism 6 (GraphPad Software, Inc, CA) and Sigmaplot11.0 (Systat Software Inc, Chicago, IL). Student’s t-test, and one-way or two-way analysis of variance (ANOVA) with Bonferroni post hoc analysis were used to test significance of differences between groups. When our experiment question only focuses on TSP4 effects, analyses were performed by Student’s t-test to test significance of the effect of TSP4 unless stated specifically. A p value less than 0.05 was considered significant. Data are reported as mean ± SEM.
3. Results
3.1. TSP4 reduces resting [Ca2+]i of DRG neurons
We have previously found that resting [Ca2+]i in both large- and small-sized DRG neurons is decreased after spinal nerve ligation and section (Fuchs et al., 2005). We therefore tested whether TSP4 has similar effects as nerve injury on resting [Ca2+]I, which showed that TSP4 incubation (4 hours) minimally reduced the resting [Ca2+]i in large-sized neurons but not in small neurons (Fig. 1B).
3.2. TSP4 slows recovery of depolarization-induced Ca2+ transients
Following neuronal activation by brief application of K+, the depolarization-induced Ca2+ transient returns to resting level in an exponential fashion (Fig. 2A) with a recovery τ that is controlled by PMCA and SERCA (Duncan et al., 2013; Gemes et al., 2012). TSP4 significantly increased the recovery τ of the depolarization-induced Ca2+ transient in IB4pos and IB4neg small-sized neurons, and also showed a trend towards increased recovery τ in large-sized neurons (p = 0.08) (Fig. 2A, B).
Fig. 2.
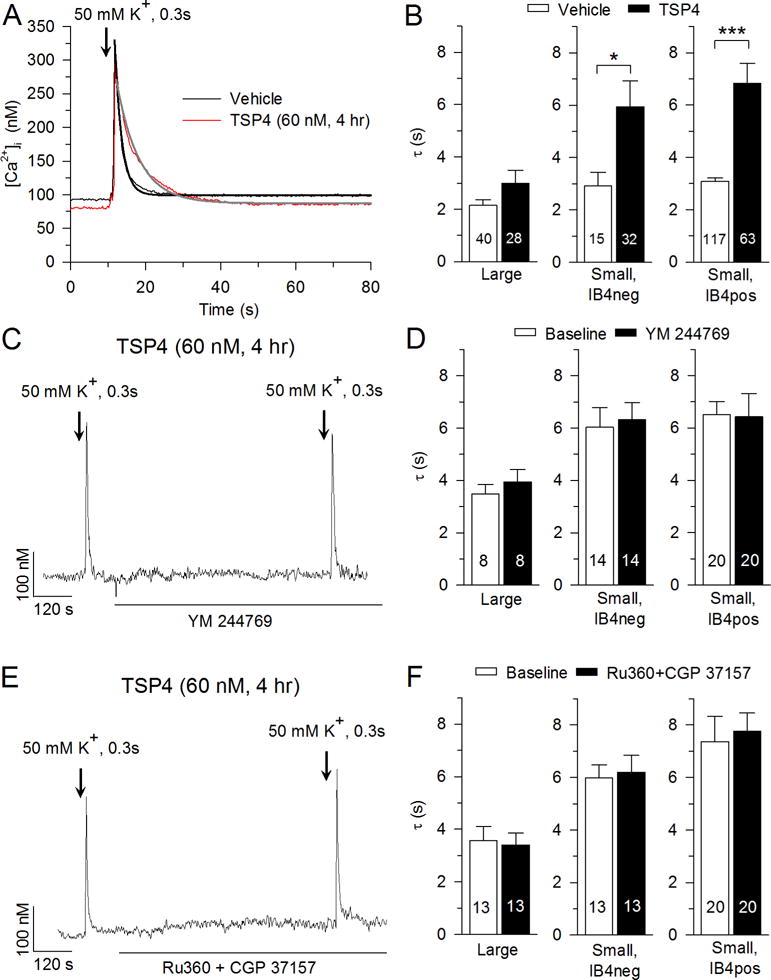
Effects of TSP4 on the depolarization-induced Ca2+ transient. A, Sample traces show the influence of TSP4 on recovery of the Ca2+ transient induced by neuronal depolarization with KCl (50 mM K+ for 0.3 s). Black and gray lines show single exponential fit of the recovery of [Ca2+] i. B, Summary data show TSP4 decreased [Ca2+] i recovery following depolarization in small-sized neurons. C, Sample trace shows the effect of NCX inhibitor, YM 244769 (5 μM), on recovery of the Ca2+ transient induced by neuronal depolarization with KCl solution. D, Summary data show that YM 244769 did not change [Ca2+] i recovery following depolarization (paired t-test). E, Sample trace shows the effect of mitochondrial Ca2+ uniporter inhibitor, Ru360 (10 μM), and mitochondrial NCX inhibitor, CGP 37157 (10 μM), on recovery of the Ca2+ transient induced by neuronal depolarization with KCl solution. F, Summary data show that Ru360 and CGP 37157 did not change [Ca2+] i recovery following depolarization (paired t-test). * p < 0.05, *** p < 0.001.
We and others (Gemes et al., 2012; Usachev et al., 2002) have previously shown that NCX contributes minimally to Ca2+ extrusion after neuronal activation in DRG sensory neurons. Here, we further confirmed this with blockade of NCX by applying NCX inhibitor YM 244769 (5 μM), which did not change recovery τ with or without TSP4 treatment (Fig. 2C, D). Additionally, we eliminated NCX function by replacing Na+ with N-methyl-d-glucamine (NMDG) in the Tyrode’s solution, which also failed to change recovery τ with or without TSP4 treatment (data not shown). In contrast, a study in which Li+ was used replace Na+ showed decreased Ca2+ clearance following depolarization in a subpopulation of DRG neurons (Lu et al., 2006). We have confirmed similar results using Li+ Tyrode’s solution in our preparations (data not shown).
While this may appear to be discordant with our findings with YM 244769 and NMDG substitution, Li+ is known to participate in Li+/Ca2+ exchange (Palty et al., 2004), and can activate Na+-activated K+ currents (Hess et al., 2007). Thus, we believe the most reliable findings indicate that NCX contributes minimally to Ca2+ transient recovery in sensory neurons, and is not a target of TSP4.
In order to clearly identify effects of TSP4 on SERCA and PMCA, we limited the participation of mitochondria by excluding traces that either had a transient peak above 400nM or showed a shoulder of sustained [Ca2+]i elevation during the descending limb of the activity-induced transient (Gemes et al., 2012). It has been reported that mitochondria might also contribute to Ca2+ buffering in sensory neurons even during small increases in [Ca2+]i. This was concluded in a study using the protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) to block uptake and release of Ca2+ in mitochondria (Lu et al., 2006). However, the use of protonophores, including CCCP and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone, also leads to robust hydrolysis of cytoplasmic ATP (Budd and Nicholls, 1996), which in turn limits PMCA function, making interpretation of these data difficult. Therefore, to further test whether mitochondria contribute to Ca2+ buffering under the conditions of our present study (transient peak less than 400 nM and without a shoulder), we used Ru360, a cell permeable and selective inhibitor of the mitochondrial Ca2+ uniporter, to block mitochondrial Ca2+ influx and CGP 37157 (a specific antagonist of mitochondrial Na+-Ca2+ exchanger) to block mitochondrial Ca2+ efflux. We found that application of Ru360 (10μM) and CGP 37157 (10μM) did not change recovery τ with TSP4 treatment (Fig. 2E, F) or without TSP4 treatment (data not shown).
On the basis of these findings, our further examination of the delayed recovery of the depolarization-induced Ca2+ transient during TSP4 treatment focused on specific regulation of PMCA and SERCA to resolve the specific effects of these interacting Ca2+ buffering systems.
3.3 TSP4 accelerates PMCA function
PMCA is the principal means by which DRG neurons extrude Ca2+ from the cytoplasm to reduce [Ca2+]i (Gemes et al., 2012). PMCA function is elevated following nerve injury by SNL, which contributes to neuronal hyperexcitability (Gemes et al., 2012). To test the regulation of PMCA by TSP4, we blocked SERCA function with TG (1μM) and measured recovery of [Ca2+]i following brief neuronal depolarization (Gemes et al., 2012). Blocking SERCA function with TG increased τ in all neuronal types (Fig. 3). Two-way ANOVA analysis showed an interaction between effects of TSP4 and TG treatments in large-sized neurons (F1,92 = 21.35, p < 0.001; Fig. 3B left), small IB4neg neurons (F1,63 = 9.00, p < 0.01; Fig. 3C left) and small IB4pos neurons (F1,225 = 8.93, p < 0.01; Fig. 3D left). To determine whether there was a difference in τ between groups treated with TG with respect to the TSP4-induced increase, data from TG-treated neurons were analyzed as a percent change from control. These data confirmed a diminished TG effect after TSP4 treatment in all neuronal groups (Fig. 3B right, C right, D right), which suggests elevated PMCA function by TSP4.
Fig. 3.
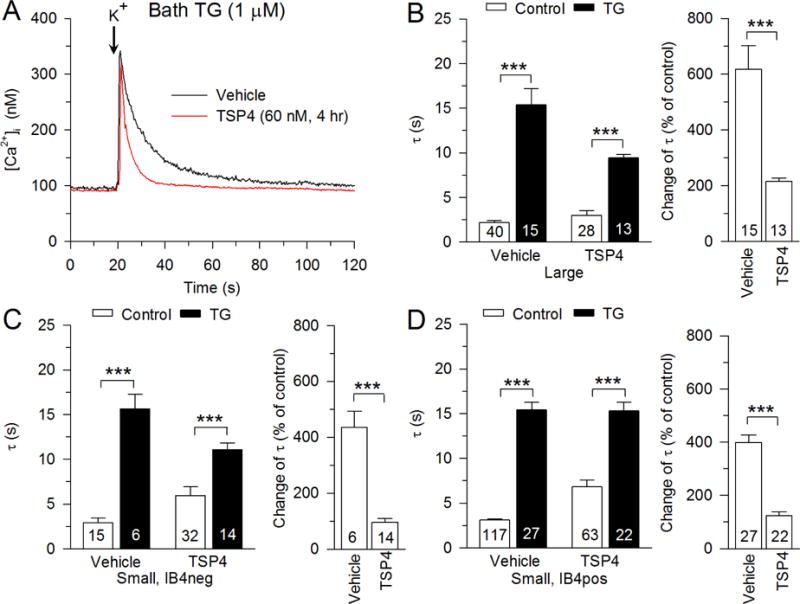
Regulation of PMCA function by TSP4. A, Sample traces show the effect of TSP4 on PMCA function, revealed by measuring recovery of the depolarization-induced transient during inhibition of SERCA function with thapsigargin (TG, 1 μM). Summary data show that effects of TSP4 on PMCA functions in large-sized neurons (B), small-sized IB4neg neurons (C), and small-sized IB4pos neurons (D). Control data are from Fig. 2. *** p < 0.001.
3.4 TSP4 inhibits SERCA function
SERCA, which resides on neuronal ER membrane, is a Ca2+ ATPase that reduces [Ca2+]i by pumping Ca2+ from the cytosol of the cell into the ER lumen. To functionally isolate SERCA, a bath Tyrode’s solution with pH 8.8 was used to eliminate PMCA Ca2+ extrusion, which requires reciprocal H+ exchange (Duncan et al., 2013). Blocking PMCA function with pH 8.8 Tyrode’s solution causes resting [Ca2+]i to increase due to unopposed SOCE function (Fig. 4A). This rise in [Ca2+]i was greater for TSP4-treated neurons in each neuronal group (Fig. 4B), which indicates that SOCE function is elevated by TSP4. To measure the effect of TSP4 on isolated SERCA function, the depolarization-induced Ca2+ transient was tested in pH 8.8 Tyrode’s solution, which blocks PMCA function (Fig. 4C). Two-way ANOVA analysis showed an interaction between effects of TSP4 and pH 8.8 solution treatments in large-sized neurons (F1,103 = 10.96, p < 0.001; Fig. 4D left), small IB4neg neurons (F1,59 = 8.29, p < 0.01; Fig. 4E left) and small IB4pos neurons (F1,300 = 34.49, p < 0.001; Fig. 4F left). To determine whether there was a difference between groups treated with pH 8.8 solution with respect to the TSP4-induced increase in τ, data from pH 8.8 solution-treated neurons were analyzed as a percent change from control. These data confirmed an increased effect of pH 8.8 solution after TSP4 treatment in all neuronal groups (Fig. 4D–F right), which suggests inhibited SERCA function by TSP4. This effect of TSP4 on SERCA, which acts to prolong the activity-induced [Ca2+]i transient, competes with the effect of TSP4 on PMCA, which acts to shorten the duration of the transient. Since the overall effect of TSP4 is to prolong the transient (Fig. 2), it appears that the TSP4 effect on SERCA is dominant. To determine if this effect of TSP4 on SERCA is influenced by sex of the animal subject, female mice were also tested. In them, TSP4 had similar effects on the recovery τ of depolarization-induced Ca2+ transient during SERCA block with pH 8.8 Tyrode’s solution in large-sized neurons (vehicle: 3.73 ± 0.56 s, n = 10; TSP4 treatment: 7.65 ± 0.77 s, n = 9; p < 0.001, t-test) and small-sized neurons (vehicle: 6.67 ± 0.92 s, n = 27; TSP4 treatment: 16.67 ± 1.9 s, n = 23; p < 0.001, t-test).
Fig. 4.
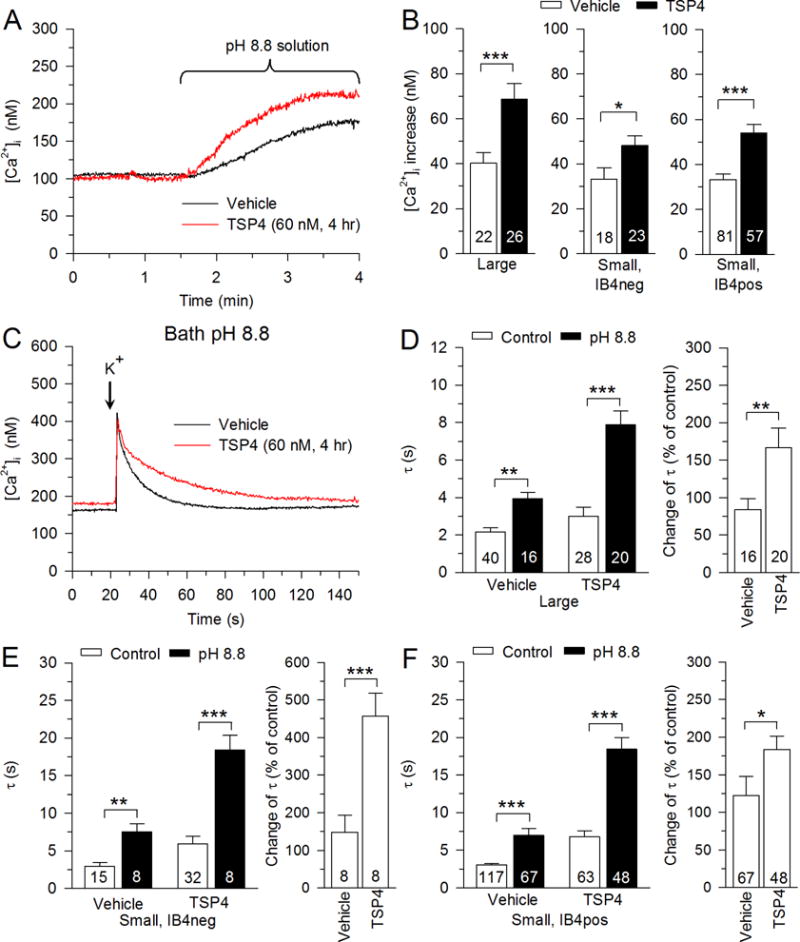
Regulation of SERCA function by TSP4. A, Sample traces show the effect of inhibition of PMCA function with pH 8.8 Tyrode’s solution on neurons with and without TSP4 treatment. B, Summary data show effects of TSP4 on resting [Ca2+] i in pH 8.8 Tyrode’s solution. C, Sample traces show the effect of TSP4 on SERCA function, revealed by measuring recovery of the depolarization-induced transient recovery during inhibition of PMCA function with pH 8.8 Tyrode’s solution. Summary data show that TSP4 decreased SERCA functions in large-sized neurons (D), small-sized IB4neg neurons (E), and small-sized IB4pos neurons (F). Control data are from Fig. 2. * p < 0.05, ** p < 0.01, *** p < 0.001.
3.5 TSP4 reduces ER Ca2+ stores
Since nerve injury depletes ER Ca2+ stores and SERCA’s function is necessary to replenish ER Ca2+ stores (Rigaud et al., 2009), we next examined the effect of TSP4 on this parameter. The ryanodine receptor Ca2+ release channels (RyRs) are the major pathway of both constitutive and Ca2+-induced Ca2+ release from ER in DRG neurons (Shmigol et al., 1995; Usachev et al., 1993). By activating the RyRs with caffeine (20mM), the resulting transient elevation of [Ca2+]i can be measured as a representation of Ca2+ transfer from the ER pool into the cytoplasm. The caffeine induced transient amplitude reflects the initial release from the ER while the duration of the transient is influenced by other Ca2+ regulatory processes including PMCA, SOCE and mitochondrial Ca2+ buffering. We therefore quantified transient amplitude as the best reflection of cytoplasmic Ca2+ load released from the ER. The caffeine-induced transient of large-sized neurons and IB4neg small-sized neurons was suppressed by TSP4, indicating reduced Ca2+ stores in ER (Fig. 5A, B). Interestingly, IB4pos small-sized neurons showed minimal response to caffeine. It has been previously reported that small-sized DRG neurons from mice have little or no response to caffeine application (Shmigol et al., 1994), whereas we have previously seen caffeine induced ER Ca2+ release in small-sized DRG neurons from rats that on average was even greater than in large-sized neurons (Rigaud et al., 2009). To confirm that these differences are species dependent, we again tested the response to caffeine in DRG neurons from rats. Consistent with our previous findings, caffeine induced larger transient in small-sized IB4pos (Fig. 5C). In contrast to mouse neurons, TSP4 had no effect on large-sized and IB4pos small-sized rat neurons, whereas TSP4 suppressed caffeine-releasable Ca2+ stores in IB4neg small neurons (Fig. 5C). These findings show that TSP4 treatment reduces ER Ca2+ stores in sensory neuron in a subtype-specific fashion, but the sensitivity of specific neuronal subtypes is species-dependent.
Fig. 5.
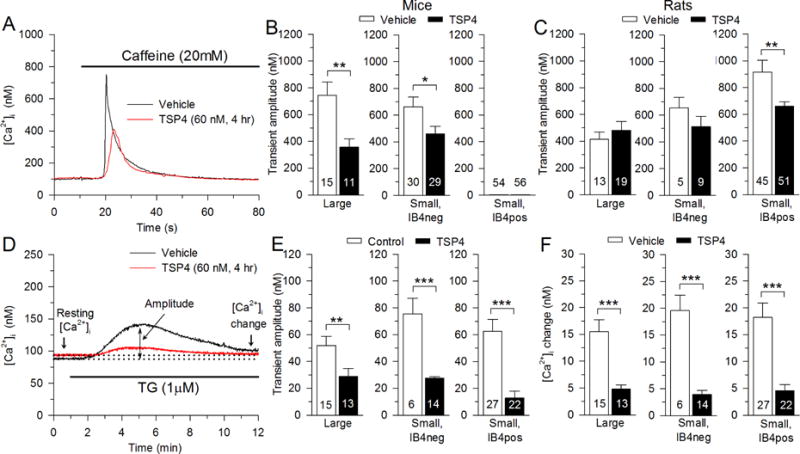
Regulation of ER Ca2+ store by TSP4. A, Sample traces show the effect of TSP4 on the Ca2+ transient induced by release of ER stores by caffeine (20 mM). B, Summary data show reduced releasable Ca2+ stores, measured as Ca2+ transient amplitude, in mouse neurons incubated in TSP4. C, Summary data show TSP4 effects in rat neurons. D, Sample traces show the effect of TSP4 on the Ca2+ transient induced by the release of stores by the SERCA blocker TG, 1 μM. E, Summary data show TSP4 suppresses the TG-induced Ca2+ transient amplitude. F, Summary data show TSP4 suppresses the TG-induced elevation of resting [Ca2+] i. * p <0.05, ** p < 0.01, *** p < 0.001.
An alternative approach to measure intracellular Ca2+ stores is by TG application, which triggers a Ca2+ release transient by blocking SERCA and leaving constitutive Ca2+ release through RyRs unopposed. Under this condition, the Ca2+ release is a relatively slow process. We quantified Ca2+ release from ER stores by measuring transient amplitude (Rigaud et al., 2009). This revealed TSP4 suppression of Ca2+ release from ER stores in all neuronal groups (Fig. 5D, E), including those IB4pos small neurons.
3.6 TSP4 increases SOCE by depletion of ER Ca2+ stores
We have previously reported that nerve injury elevates SOCE function, which increases neuronal excitability (Gemes et al., 2011). This raises the question of whether TSP4 is involved in the regulation or SOCE. Here, we first examined the effect of TSP4 on constitutive SOCE in resting neurons. Resting [Ca2+]i is set by the opposing actions of PMCA and SOCE. Our findings above show that TSP4 activates PMCA, but the resting [Ca2+]i is affected only minimally and only in the large neuron group. This predicts that TSP4 administration may induce an enhanced level of SOCE. We tested this by incubating neurons with TG (1μM) and allowing the Ca2+ release transient to recover (10 min). This showed that resting [Ca2+]i was elevated after TG, reflecting increased activity of SOCE in all neuronal subtypes and in the presence or absence of TSP4 (Fig. 5D, E). However, this effect of TG on resting [Ca2+]i was diminished in neurons incubated with TSP4 compared to controls (Fig. 5D, F). This suggests that in the presence of added TSP4, either SOCE is already relatively activated, causing TG application to produce proportionately less effect, or that maximum inducible SOCE is diminished by TSP4.
To resolve this, we next examined SOCE directly by eliminating SOCE with Ca2+-free solution and measuring the increase of [Ca2+]i upon readdition of Ca2+ to the bath, which represents Ca2+ influx through SOCE. Bath Ca2+ withdrawal resulted in a fall of [Ca2+]i that was greater in TSP4 treated neurons in all neuronal groups (Fig. 6A, B). Ca2+ transients induced by readdition of Ca2+ were larger in neurons treated with TSP4 in all neuronal groups (Fig. 6A, C). These findings indicate that SOCE is activated in the presence of TSP4.
Fig. 6.
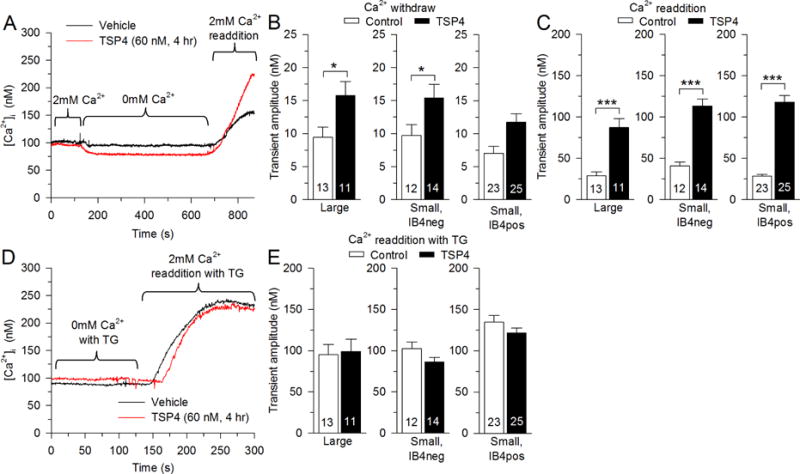
Regulation of SOCE by TSP4. A, Sample traces show the effect of TSP4 on SOCE. B, TSP4 amplifies the [Ca2+]i decrease during Ca2+-free solution, which blocks SOCE. C, TSP4 incubation amplifies the [Ca2+]i rise during bath Ca2+ readdition. D, E, TSP4 has no effect on the [Ca2+] i rise during bath Ca2+ readdition when SOCE is fully activated by SERCA blockade with TG (1 μM). * p < 0.05, *** p < 0.001.
The influence of TSP4 upon SOCE may be indirect through its effect of depleting ER Ca2+ store level (Fig. 5B, C), which is the definitive factor that regulates SOCE. To identify whether TSP4 additionally interacts directly with the SOCE channel elements, we first depleted ER Ca2+ stores by exposing neurons to TG (1μM, 8min) in Ca2+-free bath solution. Subsequent readdition of bath Ca2+ resulted in SOCE transients that were comparable in control and TSP4 groups (Fig. 6D, E), which indicates that TSP4 has no effect on fully activated SOCE channels. Taken together, these results indicate that TSP4 increases SOCE function indirectly through its depletion of ER Ca2+ stores, which is at least in part the result of TSP4 blocking SERCA function.
To summarize (Table 1), our findings indicate that TSP4 activates PMCA and suppresses SERCA, which depletes ER Ca2+ stores and thereby indirectly drives greater SOCE activity.
Table 1.
Summary of TSP4’s effects (Direction of arrow represents upregulation or downregulation).
| Large-sized Neurons | Small-sized Neurons | ||
|---|---|---|---|
| IB4neg | IB4pos | ||
| Resting [Ca2+]i | ↓ | – | – |
| Recovery | – | ↓ | ↓ |
| PMCA | ↑ | ↑ | – |
| SERCA | ↓ | ↓ | ↓ |
| ER Ca2+ stores | ↓ | ↓ | – |
| Resting SOCE | ↑ | ↑ | ↑ |
| Maximal SOCE | – | – | – |
3.7 SERCA and PMCA dysfunction after nerve injury is dependent on TSP4
We next tested the role of TSP4 in generating injury-induced loss of SERCA function and gain of PMCA and SOCE function that we have previously observed (Duncan et al., 2013; Gemes et al., 2011; Gemes et al., 2012) by examining the effects of SNL in TSP4 KO mice, compared to that in littermate WT animals. For these experiments, only small-sized DRG neurons were studied, without further differentiation by IB4 binding. Two-way ANOVA analysis showed an interaction between nerve injury and effects of TSP4 (F1,395 = 4.15, p < 0.05; Fig. 7A, B, C left). To determine whether there was a difference between different TSP4 genotype groups with respect to the nerve injury induced decrease in Ca2+ transient amplitude, data from SNL rats were analyzed as a percent change from control. These data confirmed that decreased [Ca2+]i transient amplitude after SNL was absent in DRG neurons from TSP4 KO mice (Fig. 7C right), which is consistent with our previous findings of TSP4-dependent loss of ICa after neuronal injury (McCallum et al., 2006; Pan et al., 2015). SNL increased the recovery τ of depolarization-induced Ca2+ transients in neurons from WT mice, but this effect was absent in DRG neurons from TSP4 KO mice (two-way ANOVA, F1,395 = 10.79, p < 0.01; Fig. 7D left). SNL injury also specifically decreased SERCA function (two-way ANOVA, F1,205 = 8.59, p < 0.01; Fig. 8A, B left) and elevated PMCA function (two-way ANOVA, F1,205 = 3.96, p < 0.05; Fig. 8C left, D left), both of which were eliminated in neurons from TSP4 KO mice (Fig. 8C right, D right) when data from SNL mice were analyzed as a percent change from control. We infer from these results that TSP4 is a critical mediator of the effects of injury upon intracellular Ca2+ signaling. In the absence of injury, [Ca2+]i transient amplitude (Fig. 7C left) and SERCA function (Fig. 8B left) is also elevated in neurons from TSP4 KO mice compared with neurons from TSP4 WT mice, which indicates that constitutive TSP4 has a physiological role in maintaining SERCA function and intracellular Ca2+ homeostasis.
Fig. 7.
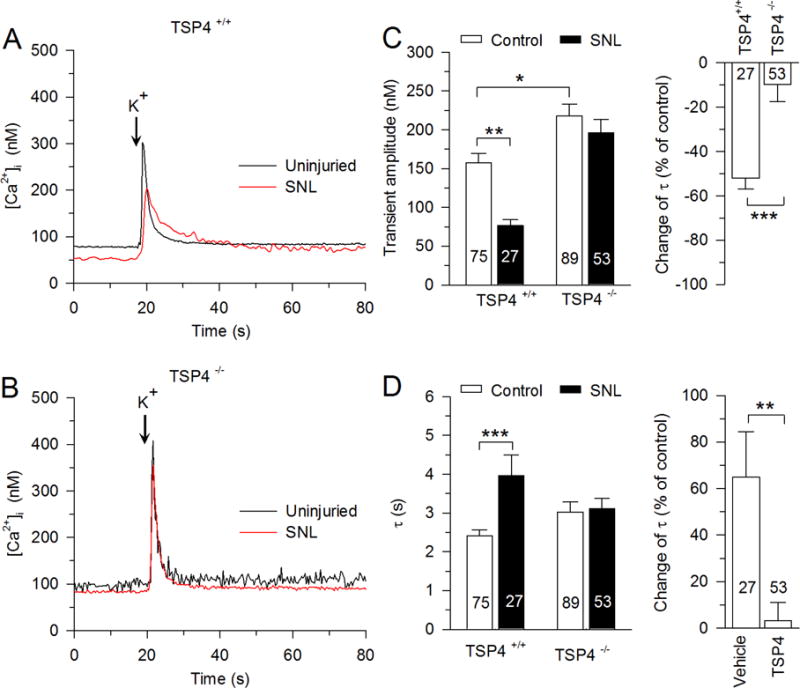
The influence of nerve injury (spinal nerve ligation, SNL) on the depolarization-induced Ca2+ transient depends on TSP4. A, SNL slows the recovery of the Ca2+ transient in small-sized neurons from WT mice. B, SNL has no effect on Ca2+ transient recovery in small-sized neurons from TSP4 KO mice. C, Summary data show that the transient amplitude is reduced after SNL in WT mice (left), but this effect of injury is absent in TSP4 KO mice (right). D, Summary data show that the transient recovery τ is reduced after SNL in WT mice (left), but this effect of injury is absent in TSP4 KO mice (right). * p < 0.05, ** p < 0.01, *** p < 0.001.
Fig. 8.
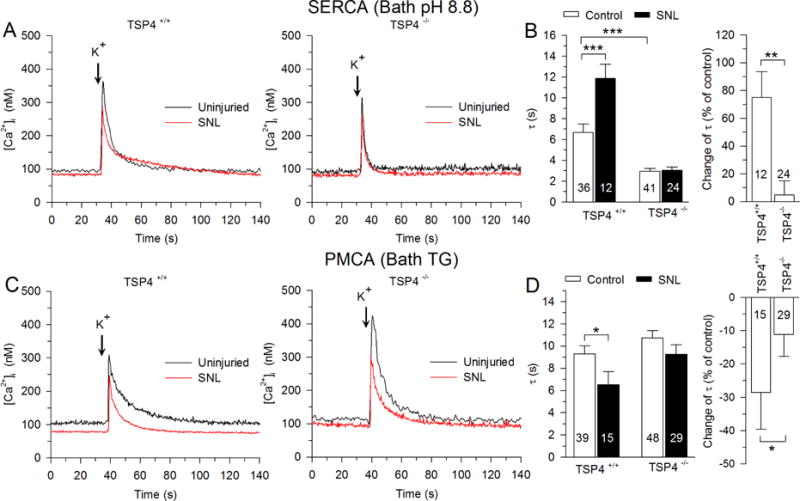
Injury-induced inhibition of SERCA function and elevation of PMCA function depends on TSP4. Sample traces (A,) and summary data (B, left) show that SERCA function was reduced by injury (SNL) in small-sized neurons from WT mice. Similarly, sample traces (C,) and summary data (D, left) show that PMCA function is enhanced by injury. In TSP4 KO animals, these effects of injury are absent for both SERCA (B, right) and PMCA (D, left). * p < 0.05, ** p < 0.01, *** p < 0.001.
3.8 TSP4 regulates [Ca2+]i by binding with the Cavα2δ1 subunit
The VGCC α2δ1 subunit is the binding site of both TSP4 (Eroglu et al., 2009; Park et al., 2016) and the analgesic gabapentin (GBP) (Hendrich et al., 2008; Pan et al., 2016) although they might work on different domains of Cavα2δ1 subunit (Lana et al., 2016). This suggests that TSP4 may have its intracellular effects by binding with the Cavα2δ1. We therefore examined whether genetic deletion of the Cavα2δ1 can block TSP4’s effects on cytoplasmic Ca2+. In sensory neurons dissociated from CKOAdv-Cre−/− control mice, TSP4 increased the recovery τ of the depolarization-induced Ca2+ transient (2.16 ± 0.20 s, n = 62; Fig. 9A). This is somewhat faster than the recovery in wildtype animals in Fig. 2 (3.00 ± 0.32 s, n = 132, p < 0.05), which may be due to somewhat different backgrounds as they were obtained from different breeding colonies at different vendors and have different ability to clear intracellular Ca2+. Two-way ANOVA analysis showed an interaction between Cavα2δ1 knockout and effects of TSP4 on the recovery τ of the depolarization-induced Ca2+ transient (F2,272 = 4.25, p < 0.05; Fig. 9B left). Further analysis of data from TSP4-treated neurons, using percent change from vehicle control, revealed that prolongation of the Ca2+ transient by TSP4 was eliminated (Fig. 9B right) in sensory neurons dissociated from the CKOAdv-Cre+/− mice that lack Cavα2δ1, suggesting that TSP4 regulates SERCA by initially binding with the Cavα2δ1 subunit.
Fig. 9.
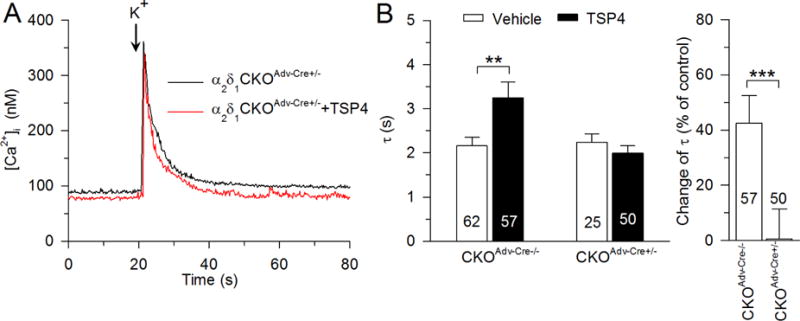
The Cavα2δ1 is required for TSP4’s regulation of recovery of the depolarization-induced Ca2+ transient. Sample traces (A,) and summary data (B) show that TSP4-induced increase of [Ca2+] i the recovery τ was eliminated in small-sized DRG neurons from Cavα2δ1 conditional KO mice. ** p < 0.01, *** p < 0.001.
3.9 Regulation of SERCA function by TSP4 is PKC signaling dependent
Both TSP4 and GBP can bind with the VGCC α2δ1 subunit. GBP can potently block TSP-induced formation of excitatory synapses (Dolphin, 2013; Eroglu et al., 2009; Hendrich et al., 2008). Additionally, GBP can modulate PKC signaling in the spinal cord dorsal horn (Yeh et al., 2011; Zhang et al., 2015) and regulate NMDA receptor function via PKC (Gu and Huang, 2001). Furthermore, activation of PKC can accelerate SERCA Ca2+ uptake into ER (Usachev et al., 2006). We therefore examined whether PKC signaling is involved in the TSP4’s regulation on SERCA function. Two-way ANOVA analysis showed an interaction between PKC signaling regulation and effects of TSP4 in regular solution (F3,204 = 9.37, p < 0.001; Fig. 10A left) and in pH8.8 solution (F3,204 = 3.60, p < 0.05, Fig. 10B left), and post hoc testing revealed that incubation (4hr) with PKC inhibitors GF109203X (5 μM) or calphostin C (500 nM) increased the recovery τ of brief depolarization-induced Ca2+ transients both in regular solution (Fig. 10A) and in pH8.8 solution (PMCA blocked, Fig. 10B), and both inhibitors blocked the effect of TSP4 on the recovery τ of brief depolarization-induced Ca2+ transients (one-way ANOVA, F3,100 = 25.42, p < 0.001; Fig. 10A right) or TSP4 suppression of SERCA function (one-way ANOVA, F3,100 = 16.6, p < 0.001; Fig. 10B right). These observations suggest that TSP4 regulates SERCA via PKC signaling. To test this further, we examined the effect of PKC activation on TSP4’s effects on SERCA function. We found that the PKC agonist PMA (1 μM) accelerated SERCA function and occluded the further action of TSP4 (Fig. 10 A, B). These findings together support a role of PKC in mediating the action of TSP4 on SERCA.
Fig. 10.
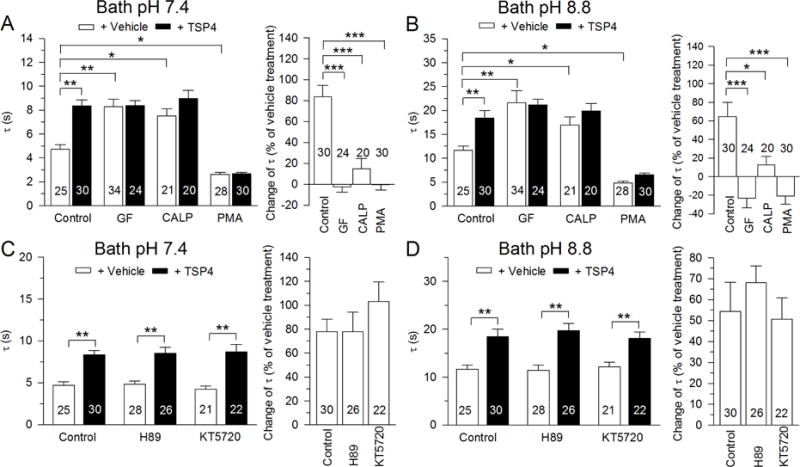
TSP4’s regulation of depolarization-induced Ca2+ transient is PKC-dependent. The recovery τ is prolonged and the effect of TSP4 is eliminated by PKC inhibitors GF 109203X (GF, 5 μM) and Calphostin C (CALP, 500nM) in both normal bath solution (A, left) and after isolating SERCA function by bath pH 8.8 (B, left) in small-sized DRG neurons. The PKC activator (PMA, 1 μM) enhances transient recovery and eliminates the effect of TSP4 in both normal bath solution (A, right) and after isolating SERCA function by bath pH 8.8 (B, right). PKA blockers H89 (10 μM) and KT5720 (1 μM) had no effect on TSP4 in both normal bath solution (C) or after isolating SERCA function by bath pH 8.8 (D) in small-sized DRG neurons. * p < 0.05, ** p < 0.01, *** p < 0.001.
GBP has been reported to activate cAMP-dependent protein kinase A (PKA) signaling in various tissues (Lee et al., 2008; Takasu et al., 2008). In cardiomyocytes, SERCA function is regulated by PKA via phospholamban (Dutta et al., 2002; Kranias and Hajjar, 2012; Veglia and Cembran, 2013), which is not expressed in neurons (Plessers et al., 1991), although similar proteins are (Dou and Joseph, 1996). We therefore tested whether PKA signaling took part in TSP4’s effects on SERCA function. Two-way ANOVA analysis showed an interaction between PKA signaling regulation and effects of TSP4 in regular solution (F2,146 = 3.96, p < 0.05; Fig. 10C left) and in pH8.8 solution (F2,146 = 3.37, p < 0.05; Fig. 10D left), while further testing revealed that PKA blockers H89 (10 μM) and KT5720 (1 μM) had no effect on TSP4 on the recovery τ of brief depolarization-induced Ca2+ transients (one-way ANOVA, F2,75 = 0.95, p = 0.39; Fig. 10C right) or TSP4 suppression of SERCA function (one-way ANOVA, F2,75 = 0.62, p = 0.54; Fig. 10D right). From these findings, we conclude that the action of TSP4 on SERCA is not dependent on PKA signaling.
4 Discussion
The experiments reported here have investigated the effects of TSP4 on intracellular Ca2+ homeostasis regulation in primary sensory neurons. Our findings show that TSP4 accelerates the function of PMCA and inhibits the SERCA function, which results in reduced ER Ca2+ stores that in turn triggers increased SOCE function (Fig. 11). We further confirmed that increased TSP4 after nerve injury contributes to the altered PMCA and SERCA function through interacting with the Cavα2δ1.
Fig. 11.
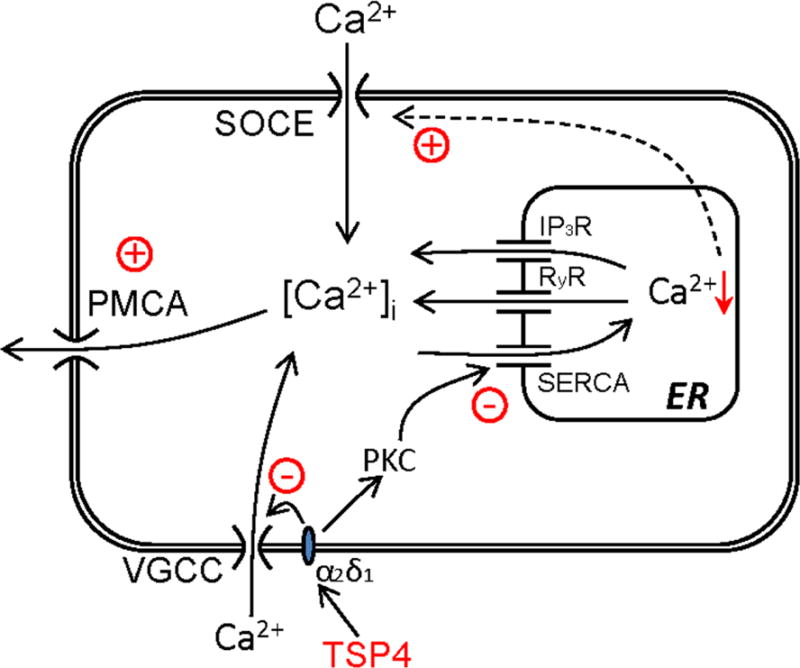
Model of TSP4 regulation of [Ca2+] i signaling. TSP4 first binds with the Cavα2δ1 and possibly with activated PKC. Activated PKC in turn decreases SERCA function, which indirectly increases SOCE function by depleting calcium stores in ER. Additionally, TSP4 amplifies PMCA activity, but to a lesser extent than the effect on SERCA, such that [Ca2+]i is minimally altered.
DRG sensory neurons are a diverse population with different functions that can be characterized by neuronal size and neurochemical staining, such as IB4pos or IB4neg (Dirajlal et al., 2003; Ma et al., 2003; Stucky and Lewin, 1999). We found that IB4pos and IB4neg populations of small neurons differ in resting [Ca2+]i levels and response to caffeine application (Fig. 1, 5). Furthermore, the subpopulations of neurons show different responses to caffeine application mice and rats (Fig. 5). A possible explanation for this discrepancy is that neurons with different IB4 staining from different species have different expression levels of RyRs isoforms, and thus different sensitivities to caffeine. All three isoforms (RyR1, RyR2 and RyR3) (Lanner et al., 2010) can be co-expressed in most of the tissues, but RyR1 is primarily expressed in skeletal muscle and RyR2 is in cardiac muscles, while RyR3 is mainly expressed in brain and DRG neurons (Lanner et al., 2010; Ooashi et al., 2005). These RyRs isoforms have different sensitivities to caffeine (Fessenden et al., 2000; Lanner et al., 2010), so further study may determine whether differential expression accounts for the distinct Ca2+ signaling features we have identified in DRG neuronal subgroups. Despite the differences in caffeine release of stores, our thapsigargin experiments nonetheless show that Ca2+ stores in IB4pos small neurons are present, and are depressed by TSP4, suggesting that TSP4 suppresses SERCA function in this neuronal subpopulation as well.
Nerve injury disrupts Ca2+ signaling in DRG neurons. Both inward Ca2+ current through VGCCs and resting [Ca2+]i are reduced after nerve trauma (Fuchs et al., 2005; Hogan et al., 2000), PMCA function is increased, and SERCA is impaired, culminating in depleted ER Ca2+ stores and reduced [Ca2+] i in the injured neurons (Duncan et al., 2013; Gemes et al., 2011). Resting [Ca2+]i level is regulated by Ca2+ influx through SOCE, uptake into ER by SERCA, expulsion by PMCA, and sequestration by mitochondria. Our previous studies of DRG neurons from SNL injury rats indicate that elevated PMCA function contributes to depressed resting [Ca2+]i (Duncan et al., 2013; Fuchs et al., 2005). In the present study, we found only minimally effects of TSP4 on resting [Ca2+] i in large sized neurons (Fig. 1), despite substantial influences on SERCA, PMCA and SOCE. One possible explanation is that TSP4 has offsetting effects, such that TSP4-induced SERCA depression and consequent SOCE enhancement increases resting [Ca2+]i, while TSP4-induced PMCA activation drives resting [Ca2+]i down, resulting in minimal or no change overall. Thus, factors other than TSP4 must account for the strong depression of resting [Ca2+]i after injury (Fuchs et al., 2005).
SERCA plays an important role in maintaining cellular homeostasis, including in sensory neurons (Gemes et al., 2011). For instance, blocking SERCA with thapsigargin is a trigger for ER stress through depletion of ER Ca2+ stores (Luciani et al., 2009). Our present study shows that TSP4 reduces SERCA function, depletes ER Ca2+ stores, and elevates SOCE, which are key features of ER stress. A consequence of ER stress is activation of extracellular signal-regulated kinase (ERK), JNK, and p38 MAPK, all of which are activated in sensory neurons after peripheral nerve injury (Jin et al., 2003; Zhuang et al., 2006), supporting the view that ER stress is a component of neuropathic pain. Furthermore, depleting ER Ca2+ stores increases sensory neuron excitability (Gemes et al., 2009). These observations together suggest that the TSP4 signaling pathway may account for ER stress in neuropathic pain. This is supported by our observation of enhanced TSP4 expression in painful nerve injury models (Pan et al., 2015).
Finally, ER stress in cardiac myocytes can itself trigger the generation of TSP4, perhaps as an ER-resident adaptive (Lynch et al., 2012). If this also occurs in sensory neurons, then a positive feedback loop could result in which increased expression of TSP4 produces ER stress, which then drives further production of TSP4.
The Cavα2δ1 has previously been identified as the major binding partner for TSP4 synaptogenesis (Eroglu et al., 2009; Park et al., 2016). TSP4 has weak binding with Cavα2δ1 intracellularly and robust binding with membrane low density lipoprotein receptor-related protein 1 receptors in a tsA201 cell culture system (Lana et al., 2016). In HEK 293 cell culture system, TSP4 can also bind with intracellular stromal interaction molecule 1, which can bind with cartilage oligomeric matrix protein to regulate intracellular Ca2+ homeostasis (Duquette et al., 2014). Our data also suggest that binding of TSP4 to Cavα2δ1 is required for the observed modulation of SERCA and PMCA. Like TSP4, expression of Cavα2δ1 is also upregulated after nerve injury in neurons of the involved DRGs (Luo et al., 2001; Valder et al., 2003; Wang et al., 2002), and Cavα2δ1 knock-out delays the onset of mechanical hypersensitivity after nerve injury (Patel et al., 2013). VGCCs, which are the main pathway for Ca2+ entry during neuronal activation, are composed of a pore-forming α1 subunit as well as auxiliary β, α2δ and γ subunits (Catterall, 2000). Phosphorylation of the Cavα2δ1 can modulate N-type VGCCs via an ERK-dependent mechanism (Martin et al., 2006). We previously found that TSP4 regulates ICa through VGCCs by binding with Cavα2δ1 (Pan et al., 2016). Thus it appears that the Cavα2δ1 is a key element for TSP4 signaling in general.
We have found that activation of sensory neuron PKC accelerates Ca2+ uptake into ER by accelerating SERCA function, consistent with a previous report (Usachev et al., 2006), and we additionally showed blocking PKC inhibits SERCA function. By binding with α2δ1 calcium subunit, TSP4 modulates SERCA and PMCA through a PKC dependent pathway that links activation of α2δ1 Ca2+ subunit and inhibition of SERCA function. Additional evidence of this role of PKC comes from a report that shows GBP regulation of PKC signaling in the spinal cord of rats underlying neuropathic pain (Yeh et al., 2011).
Taken together, our findings show that TSP4 regulates Ca2+ homeostasis in primary sensory neurons by affecting Ca2+ influx and buffering, including SERCA and PMCA, via PKC signaling after binding to Cavα2δ1. Elevated TSP4 expression may be a key factor that disrupts Ca2+ homeostasis after neuronal injury, and may serve as a valuable target for treatment of neuropathic pain.
Highlights.
Thrombospondin-4 (TSP4) inhibits sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) function.
Effects of TSP4 on SERCA function are attenuated in voltage-gated Ca2+ channel Cavα2δ1 subunit knockout mice.
Nerve injury-induced depression of SERCA function is attenuated in TSP4 knockout mice.
Effects of TSP4 on SERCA function are dependent on protein kinase C signaling.
Acknowledgments
The study was supported by National Institutes of Health Grant R01DE021847 (to Z.D.L. & Q.H. H.). We would like to thank Dr. Fan Wang for the advillin-Cre mice, Dr. Frank Zaucke for the TSP4 cDNA construct, and Van Nancy Trinh for her assistance in recombinant TSP4 expression and purification.
Abbreviations
- TSP4
thrombospondin-4
- Cavα2δ1
voltage-gated calcium channel α2δ1 subunit
- [Ca2+]i
cytoplasmic Ca2+ concentration
- DRG
dorsal root ganglion
- VGCCs
Voltage-gated calcium channels
- SERCA
sarco-endoplasmic reticulum Ca2+-ATPase
- PMCA
plasma membrane Ca2+-ATPase
- NCX
Na+-Ca2+ exchanger
- ER
endoplasmic reticulum
- SOCE
Ca2+ stores and stimulates store-operated Ca2+ entry
- RyRs
ryanodine receptors
- IP3R
inositol 1,4,5-trisphosphate receptors
- KO
knock-out
- CKO
conditional knock-out
- Wildtype
WT
- SNL
spinal nerve ligation
- PKC
protein kinase C
- PKA
protein kinase A
- IB4
isolectin B4
- ERK
Extracellular signal-regulated kinase
- GBP
gabapentin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J Cell Biol. 1995;131:1083–1094. doi: 10.1083/jcb.131.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabas ME, Kossyreva EA, Stucky CL. TRPA1 is functionally expressed primarily by IB4-binding, non-peptidergic mouse and rat sensory neurons. PLoS One. 2012;7:e47988. doi: 10.1371/journal.pone.0047988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Leal K, Nanou E. Calcium channels and short-term synaptic plasticity. J Biol Chem. 2013;288:10742–10749. doi: 10.1074/jbc.R112.411645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby ND, Zaucke F, Kras JV, Dong L, Luo ZD, Winkelstein BA. Thrombospondin-4 and excitatory synaptogenesis promote spinal sensitization after painful mechanical joint injury. Exp Neurol. 2014;264C:111–120. doi: 10.1016/j.expneurol.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirajlal S, Pauers LE, Stucky CL. Differential response properties of IB(4)-positive and -negative unmyelinated sensory neurons to protons and capsaicin. J Neurophysiol. 2003;89:513–524. doi: 10.1152/jn.00371.2002. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. The alpha2delta subunits of voltage-gated calcium channels. Biochim Biophys Acta. 2013;1828:1541–1549. doi: 10.1016/j.bbamem.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Dou D, Joseph R. Cloning of human neuronatin gene and its localization to chromosome-20q 11.2–12: the deduced protein is a novel “proteolipid’. Brain Res. 1996;723:8–22. doi: 10.1016/0006-8993(96)00167-9. [DOI] [PubMed] [Google Scholar]
- Duncan C, Mueller S, Simon E, Renger JJ, Uebele VN, Hogan QH, Wu HE. Painful nerve injury decreases sarco-endoplasmic reticulum Ca(2)(+)-ATPase activity in axotomized sensory neurons. Neuroscience. 2013;231:247–257. doi: 10.1016/j.neuroscience.2012.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette M, Nadler M, Okuhara D, Thompson J, Shuttleworth T, Lawler J. Members of the thrombospondin gene family bind stromal interaction molecule 1 and regulate calcium channel activity. Matrix Biol. 2014;37:15–24. doi: 10.1016/j.matbio.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta K, Carmody MW, Cala SE, Davidoff AJ. Depressed PKA activity contributes to impaired SERCA function and is linked to the pathogenesis of glucose-induced cardiomyopathy. J Mol Cell Cardiol. 2002;34:985–996. doi: 10.1006/jmcc.2002.2035. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessenden JD, Wang Y, Moore RA, Chen SR, Allen PD, Pessah IN. Divergent functional properties of ryanodine receptor types 1 and 3 expressed in a myogenic cell line. Biophys J. 2000;79:2509–2525. doi: 10.1016/S0006-3495(00)76492-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Lirk P, Stucky C, Abram SE, Hogan QH. Painful nerve injury decreases resting cytosolic calcium concentrations in sensory neurons of rats. Anesthesiology. 2005;102:1217–1225. doi: 10.1097/00000542-200506000-00023. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Rigaud M, Hogan QH. Painful nerve injury shortens the intracellular Ca2+ signal in axotomized sensory neurons of rats. Anesthesiology. 2007;107:106–116. doi: 10.1097/01.anes.0000267538.72900.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Oyster KD, Pan B, Wu HE, Bangaru ML, Tang Q, Hogan QH. Painful nerve injury increases plasma membrane Ca2+-ATPase activity in axotomized sensory neurons. Mol Pain. 2012;8:46. doi: 10.1186/1744-8069-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Rigaud M, Weyker PD, Abram SE, Weihrauch D, Poroli M, Zoga V, Hogan QH. Depletion of calcium stores in injured sensory neurons: anatomic and functional correlates. Anesthesiology. 2009;111:393–405. doi: 10.1097/ALN.0b013e3181ae63b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Huang LY. Gabapentin actions on N-methyl-D-aspartate receptor channels are protein kinase C-dependent. Pain. 2001;93:85–92. doi: 10.1016/S0304-3959(01)00297-4. [DOI] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A. 2008;105:3628–3633. doi: 10.1073/pnas.0708930105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess D, Nanou E, El Manira A. Characterization of Na+-activated K+ currents in larval lamprey spinal cord neurons. J Neurophysiol. 2007;97:3484–3493. doi: 10.1152/jn.00742.2006. [DOI] [PubMed] [Google Scholar]
- Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, Bosnjak ZJ. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Li KW, Boroujerdi A, Peter Yu Y, Zhou CY, Deng P, Park J, Zhang X, Lee J, Corpe M, Sharp K, Steward O, Eroglu C, Barres B, Zaucke F, Xu ZC, Luo ZD. Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J Neurosci. 2012;32:8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana B, Page KM, Kadurin I, Ho S, Nieto-Rostro M, Dolphin AC. Thrombospondin-4 reduces binding affinity of [(3)H]-gabapentin to calcium-channel alpha2delta-1-subunit but does not interact with alpha2delta-1 on the cell-surface when co-expressed. Sci Rep. 2016;6:24531. doi: 10.1038/srep24531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Tsai TS, Liou HH. Gabapentin activates ROMK1 channels by a protein kinase A (PKA)-dependent mechanism. Br J Pharmacol. 2008;154:216–225. doi: 10.1038/bjp.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, Jeffrey KD, Sampaio AV, Underhill TM, Johnson JD. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes. 2009;58:422–432. doi: 10.2337/db07-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, Lynch KA, Okada T, Aronow BJ, Osinska H, Prywes R, Lorenz JN, Mori K, Lawler J, Robbins J, Molkentin JD. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149:1257–1268. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Shu Y, Zheng Z, Chen Y, Yao H, Greenquist KW, White FA, LaMotte RH. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J Neurophysiol. 2003;89:1588–1602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- Martin SW, Butcher AJ, Berrow NS, Richards MW, Paddon RE, Turner DJ, Dolphin AC, Sihra TS, Fitzgerald EM. Phosphorylation sites on calcium channel alpha1 and beta subunits regulate ERK-dependent modulation of neuronal N-type calcium channels. Cell Calcium. 2006;39:275–292. doi: 10.1016/j.ceca.2005.11.002. [DOI] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology. 2006;105:160–168. doi: 10.1097/00000542-200607000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooashi N, Futatsugi A, Yoshihara F, Mikoshiba K, Kamiguchi H. Cell adhesion molecules regulate Ca2+-mediated steering of growth cones via cyclic AMP and ryanodine receptor type 3. J Cell Biol. 2005;170:1159–1167. doi: 10.1083/jcb.200503157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004;279:25234–25240. doi: 10.1074/jbc.M401229200. [DOI] [PubMed] [Google Scholar]
- Pan B, Guo Y, Wu HE, Park J, Trinh VN, Luo ZD, Hogan QH. Thrombospondin-4 divergently regulates voltage gated Ca2+ channel subtypes in sensory neurons after nerve injury. Pain. 2016 doi: 10.1097/j.pain.0000000000000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Yu H, Park J, Yu YP, Luo ZD, Hogan QH. Painful nerve injury upregulates thrombospondin-4 expression in dorsal root ganglia. J Neurosci Res. 2015;93:443–453. doi: 10.1002/jnr.23498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Yu YP, Zhou CY, Li KW, Wang D, Chang E, Kim DS, Vo B, Zhang X, Gong N, Sharp K, Steward O, Vitko I, Perez-Reyes E, Eroglu C, Barres B, Zaucke F, Feng G, Luo ZD. Central mechanisms mediating thrombospondin-4 induced pain states. J Biol Chem. 2016 doi: 10.1074/jbc.M116.723478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W. Dependence of vital cell function on endoplasmic reticulum calcium levels: implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium. 2001;29:1–11. doi: 10.1054/ceca.2000.0162. [DOI] [PubMed] [Google Scholar]
- Patel R, Bauer CS, Nieto-Rostro M, Margas W, Ferron L, Chaggar K, Crews K, Ramirez JD, Bennett DL, Schwartz A, Dickenson AH, Dolphin AC. alpha2delta-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci. 2013;33:16412–16426. doi: 10.1523/JNEUROSCI.1026-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessers L, Eggermont JA, Wuytack F, Casteels R. A study of the organellar Ca2(+)-transport ATPase isozymes in pig cerebellar Purkinje neurons. J Neurosci. 1991;11:650–656. doi: 10.1523/JNEUROSCI.11-03-00650.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Barabas ME, Chernoff DI, Abram SE, Stucky CL, Hogan QH. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201. doi: 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Weyker PD, Cruikshank JM, Kawano T, Wu HE, Hogan QH. Axotomy depletes intracellular calcium stores in primary sensory neurons. Anesthesiology. 2009;111:381–392. doi: 10.1097/ALN.0b013e3181ae6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Eroglu C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol. 2012;31:170–177. doi: 10.1016/j.matbio.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Kostyuk P, Verkhratsky A. Role of caffeine-sensitive Ca2+ stores in Ca2+ signal termination in adult mouse DRG neurones. Neuroreport. 1994;5:2073–2076. doi: 10.1097/00001756-199410270-00021. [DOI] [PubMed] [Google Scholar]
- Shmigol A, Verkhratsky A, Isenberg G. Calcium-induced calcium release in rat sensory neurons. J Physiol. 1995;489(Pt 3):627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Lewin GR. Isolectin B(4)-positive and -negative nociceptors are functionally distinct. J Neurosci. 1999;19:6497–6505. doi: 10.1523/JNEUROSCI.19-15-06497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu K, Ono H, Tanabe M. Gabapentin produces PKA-dependent pre-synaptic inhibition of GABAergic synaptic transmission in LC neurons following partial nerve injury in mice. J Neurochem. 2008;105:933–942. doi: 10.1111/j.1471-4159.2008.05212.x. [DOI] [PubMed] [Google Scholar]
- Usachev Y, Shmigol A, Pronchuk N, Kostyuk P, Verkhratsky A. Caffeine-induced calcium release from internal stores in cultured rat sensory neurons. Neuroscience. 1993;57:845–859. doi: 10.1016/0306-4522(93)90029-f. [DOI] [PubMed] [Google Scholar]
- Usachev YM, DeMarco SJ, Campbell C, Strehler EE, Thayer SA. Bradykinin and ATP accelerate Ca(2+) efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca(2+) pump isoform 4. Neuron. 2002;33:113–122. doi: 10.1016/s0896-6273(01)00557-8. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Marsh AJ, Johanns TM, Lemke MM, Thayer SA. Activation of protein kinase C in sensory neurons accelerates Ca2+ uptake into the endoplasmic reticulum. J Neurosci. 2006;26:311–318. doi: 10.1523/JNEUROSCI.2920-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valder CR, Liu JJ, Song YH, Luo ZD. Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J Neurochem. 2003;87:560–573. doi: 10.1046/j.1471-4159.2003.02016.x. [DOI] [PubMed] [Google Scholar]
- Veglia G, Cembran A. Role of conformational entropy in the activity and regulation of the catalytic subunit of protein kinase A. FEBS J. 2013;280:5608–5615. doi: 10.1111/febs.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun H, Della Penna K, Benz RJ, Xu J, Gerhold DL, Holder DJ, Koblan KS. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–546. doi: 10.1016/s0306-4522(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Yeh CY, Chung SC, Tseng FL, Tsai YC, Liu YC. Biphasic effects of chronic intrathecal gabapentin administration on the expression of protein kinase C gamma in the spinal cord of neuropathic pain rats. Acta Anaesthesiol Taiwan. 2011;49:144–148. doi: 10.1016/j.aat.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang YB, Guo ZD, Li MY, Fong P, Zhang JG, Zhang CW, Gong KR, Yang MF, Niu JZ, Ji XM, Lv GW. Gabapentin Effects on PKC-ERK1/2 Signaling in the Spinal Cord of Rats with Formalin-Induced Visceral Inflammatory Pain. PLoS One. 2015;10:e0141142. doi: 10.1371/journal.pone.0141142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]