

The identification of multiple amino-acid coordination sites which can bind to the radiopharmaceutical synthon fac-[Re(CO)3]+ is reported.
Keywords: rhenium, technetium, radiopharmaceutical agents, fragment-based design, two X-ray wavelengths
Abstract
Multiple possibilities for the coordination of fac-[Re(CO)3(H2O)3]+ to a protein have been determined and include binding to Asp, Glu, Arg and His amino-acid residues as well as to the C-terminal carboxylate in the vicinity of Leu and Pro. The large number of rhenium metal complex binding sites that have been identified on specific residues thereby allow increased target identification for the design of future radiopharmaceuticals. The core experimental concept involved the use of state-of-art tuneable synchrotron radiation at the Diamond Light Source to optimize the rhenium anomalous dispersion signal to a large value (f′′ of 12.1 electrons) at its L I absorption edge with a selected X-ray wavelength of 0.9763 Å. At the Cu Kα X-ray wavelength (1.5418 Å) the f′′ for rhenium is 5.9 electrons. The expected peak-height increase owing to the optimization of the Re f′′ was therefore 2.1. This X-ray wavelength tuning methodology thereby showed the lower occupancy rhenium binding sites as well as the occupancies of the higher occupancy rhenium binding sites.
1. Introduction
Rhenium-188/186 and technetium-99m tricarbonyl complexes have shown much potential as therapeutic and diagnostic radiopharmaceuticals. The synthetic kit of the fac-[M(CO)3]+ [where M is rhenium(I) or technetium(I)] core, based in water, makes this starting synthon a highly attractive pharmaceutical model which coordinates to multiple ligands (Alberto et al., 1999 ▸; Schibli et al., 2000 ▸). We report here the coordination of multiple fac-[Re(CO)3]+ complex fragments to a protein that was selected to provide a full range of exposed amino acids. Both monodentate and bidentate complex coordination were observed to the aspartic acid, glutamic acid, arginine, leucine, proline and histidine residues, indicating preferential binding to neutral and anionic N and O atom donors with pK a values varying from 3.71 to 4.15 (Cantor & Schimmel, 1980 ▸; Betts & Russell, 2003 ▸). These structural observations are critical for the development of radiopharmaceutical drug design using the fac-[M(CO)3]+ (where M is technetium-99m, rhenium-186 or rhenium-188) radionuclide core (Alberto et al., 1995 ▸, 1998 ▸, 2001 ▸), in particular considering the fragment-based drug-design (FBDD) method (Joseph-McCarthy et al., 2014 ▸; Erlanson, 2012 ▸; Murray et al., 2012 ▸), as specific protein–ligand binding of low-molecular-weight fragments can now be exploited to derive a model for possible drug-like lead compounds.
The recent crystal structure studies of cisplatin and carboplatin, chemically transformed into iodoplatin, gave new leads for radiation therapy and emphasized the importance of three-dimensional structural knowledge in new compound discovery, as an alternative treatment in tumour radiation therapy was suggested based on the X-ray crystal structure analyses (Tanley & Helliwell, 2014 ▸). In this new study, we provide the three-dimensional information required for new drug-lead development for rhenium-compound interactions with a wide range of amino acids, which was derived from X-ray crystallography supplemented with extensive analysis of the Cambridge Structural Database (CSD; Allen, 2002 ▸) and was supported by the precise distance measurements which can be obtained by using the diffraction precision index (DPI; Gurusaran et al., 2014 ▸; Kumar et al., 2015 ▸). The combination of using small-molecule rhenium crystallographic data from the CSD with protein crystallography allows us to pinpoint the coordination mode (i.e. monodentate, bidentate or tridentate coordination). Both the iodoplatin and rhenium studies utilize X-ray absorption at an X-ray-absorbing centre. The iodoplatin studies allowed two X-ray absorption edges to be harnessed, the I K and Pt K edges, each offering different penetrating powers for X-rays into a tumour. The rhenium study here again relies on X-ray absorption, now with a higher total occupancy owing to interaction with more amino-acid types than previously observed. One parameter used extensively during this study was the ‘quasi bite angle’ (QBA), defined here as the angle formed between the rhenium metal and cognate amino-acid residue atoms, which gives increased insight to the binding mode compared with small-molecule rhenium bite angles and related bond distances.
Current protein crystallography studies reporting rhenium coordination are rare and report a binding preference for the histidine imidazole side chain (Binkley et al., 2011 ▸; Zobi & Spingler, 2012 ▸; Santoro et al., 2012 ▸; Takematsu et al., 2013 ▸). For radiopharmaceutical applications, it was noted that coordination could be achieved by first reacting the fac-[Re(CO)3]+ core, followed by ligand coordination to the metal after protein binding (Zobi & Spingler, 2012 ▸; Santoro et al., 2012 ▸). This inverted [2+1] approach (Mundwiler et al., 2004 ▸) is troubling from the aspect of site-specific drug interaction, as the open coordination sites of fac-[M(CO)3]+ may first lead to high retention in the kidneys, liver and blood pool (Schibli et al., 2000 ▸). The results obtained here in this study, whereby rhenium coordination is also observed to aspartic acid, glutamic acid, arginine and leucine residues, arising from the technical innovation of using tuned X-ray wavelengths to improve the detectability of Re-atom binding, opens new alternatives for preferential binding other than to an imidazole moiety of histidine alone.
2. Experimental
2.1. Crystallization
Standard crystallization conditions for hen egg-white lysozyme (20 mg) consisting of pH 4.7, 10% NaCl, 0.04 M sodium acetate with fac-[Et4N]2[Re(CO)3(Br)3] at 0.03 M in 1.4 ml water in sitting-drop conditions, initially without DMSO but with silicone oil as cryoprotectant, led to crystals with no diffraction. With the inclusion of DMSO at 7.5%(v/v), cryoprotection of the crystals with silicone oil yielded consistent and good diffraction. The use of Paratone oil caused decomposition of the crystals upon contact with the Paratone oil in conditions either without or with DMSO; this was plainly visible under a visible-light microscope. Pure silicone oil was thus utilized. The crystal was transferred into the oil on a microscope slide and moved for ∼3 s to allow complete coating. The crystals grew over a period of approximately three weeks. DMSO is typically used as a ‘drug vehicle’ to increase the in vitro and in vivo solubility of pharmaceuticals within the bloodstream, to increase the accessibility of drugs to cell membranes and muscle tissue, and to allow them to cross the blood–brain barrier (typically at dosages below 6 ml kg−1; Kelava et al., 2011 ▸; Swanson, 1985 ▸; Santos et al., 2003 ▸; Colucci et al., 2008 ▸). Therefore, our in vitro crystallization condition has a clinical context, although the use of DMSO in the clinical context still remains controversial (Hall et al., 2014 ▸). Carbonyl ligands in the coordinated fac-[Re(CO)3]+ to HEWL were directly confirmed as measured by their signature stretching frequencies in solid-state infrared spectra (IR, cm−1): v(CO) = 2011, 1997, 1863.
2.2. X-ray data collection, structure solution and refinement
The rhenium–HEWL data collection was first conducted on an in-house Bruker PLATINUM135 detector with the crystal sample at a distance of 50 mm from the detector. An X-ray exposure time of 10 s per 0.5° crystal rotation angle was used with an X-ray wavelength of 1.5418 Å. The data were processed using the Bruker PROTEUM2 internal software package. X-ray data were also collected on beamline I04 at Diamond Light Source (DLS) using an X-ray wavelength of 0.9763 Å so as to optimize the rhenium f′′ anomalous signal at the Re L I absorption edge. Both of the X-ray diffraction data collections were carried out at a fixed temperature of 100 K for the samples. Data and space-group validation were further confirmed with Zanuda and MOSFLM (Lebedev & Isupov, 2014 ▸; Leslie & Powell, 2007 ▸; Battye et al., 2011 ▸) in the CCP4 software suite. The structures were solved via molecular replacement, using the reported lysozyme structure with PDB code 2w1y (Cianci et al., 2008 ▸) as a molecular-replacement search model within Phaser (McCoy et al., 2007 ▸), and were then refined in REFMAC5 (Vagin & Teplyakov, 2010 ▸) in CCP4i (Winn et al., 2011 ▸). Protein model refinement was initially conducted in the tetragonal space group P43212. Owing to apparent unresolvable symmetry indications near the Leu129 residue, space-group validation was considered in triclinic (P1) and then orthorhombic (P212121) symmetry utilizing Zanuda (Lebedev & Isupov, 2014 ▸) and MOSFLM (Leslie & Powell, 2007 ▸) in the CCP4 software suite (Winn et al., 2011 ▸). The orthorhombic P212121 space group was selected based on reasonable rhenium–amino-acid bond distances as found in the CSD and better clarity of the electron density for the coordinated ligands. The R int values were basically indistinguishable [0.077 (1.453) in P212121 versus 0.077 (2.066) in P43212], which we attribute to the protein being in the higher symmetry, while the lower symmetry space group is a better choice for the rhenium-compound binding with respect to feasible bond distances. The successful placement of two individual protein subunits using the same search model (PDB entry 2w1y) was a direct confirmation of the crystal-packing layout. A similar orthorhombic space-group allocation has indeed been seen before, for example in an HEWL structure containing cisplatin (Tanley et al., 2012 ▸). With respect to the archived data, the authors make the following statement of rationale for the finalized data files that we have firstly deposited in the PDB and secondly deposited as Supporting Information to this article, guided by a referee and the editor as well as our own views.
The PDB is focused on the protein. The protein refines best, with the most sensible ADPs, in the tetragonal space group. In the orthorhombic space group, the protein ADPs are flagged as poorer by PARVATI (http://skuld.bmsc.washington.edu/parvati/; Merritt, 1999 ▸), but the rhenium–ligand distances and the best defined electron density refined in the orthorhombic space group more sensibly fit the prior knowledge from the CSD. To resolve this dichotomy, and to respect everyone’s wish to deposit an acceptable set of coordinates in the PDB, whilst also not compromising the prior knowledge in the CSD, we deposited in the PDB the tetragonal refined coordinates for the synchrotron data with the Re atoms but with the ligand atoms removed. The ligand model of the rheniums bound to the protein is however best served in the orthorhombic space group, and we deposit as Supporting Information to this article the best model, both chemically and with respect to fit to the X-ray diffraction data, along with the synchrotron diffraction data. Since the home-laboratory Cu Kα data provided an important second X-ray wavelength, and were therefore important in decisions about Re-atom placement, but were not concerned with protein details, we provide the orthorhombic refined home-laboratory data model and structure factors with this article as Supporting Information. Therefore, in summary, the best protein model refinement has been deposited in the PDB (synchrotron) with the highest symmetry for the protein (tetragonal). The best model for the rhenium ligand–protein interactions is attached to this article (synchrotron, orthorhombic).
Model building and adjustment were conducted within the Coot molecular-graphics program (Emsley & Cowtan, 2004 ▸) alternating with cycles of REFMAC5 in CCP4i. Alternatively, refinement for software comparison was conducted in PHENIX (Afonine et al., 2012 ▸). Ligand-binding occupancies were calculated using SHELXTL (Sheldrick, 2008 ▸), with further manual adjustment guided by residual F o − F c electron-density peak evidence.
The ‘quasi bite angle’ (QBA), in addition to the specific bond distances measured (and supported by the diffraction precision index; Gurusaran et al., 2014 ▸; Kumar et al., 2015 ▸), provides increased insight into the binding mode when compared with the small-molecule rhenium bite angles and related bond distances. We have also made extensive use of the Cambridge Structural Database using the rigorous search tools that the CSD provides.
The PDB deposition code for the DLS tetragonal refinement is 5nbj. In each refinement the finalized protein model coordination and the respective diffraction data sets (structure factors) and the PDB validation reports were provided to the editor for their use and for the referees. The raw diffraction images for the synchrotron and home laboratory data sets are available at Zenodo (Brink & Helliwell, 2017). Table 1 ▸ provides a summary of the data and the model refinements: column 1, tetragonal, synchrotron data, model of the protein with rheniums (deposited in the PDB); column 2, orthorhombic, synchrotron data, model of the protein refined with rheniums and ligands; column 3, home-laboratory Cu Kα data, model of the protein refined with rheniums and ligands.
Table 1. X-ray crystallographic data and final protein model refinement statistics for Diamond Light Source (DLS) data (refined in tetragonal and orthorhombic space groups) and Cu Kα data (orthorhombic).
Overall diffraction resolution values are given, with values for the outer diffraction resolution shell in parentheses.
| DLS (λ = 0.9763 Å) (tetragonal; PDB code 5nbj) | DLS (λ = 0.9763 Å) (orthorhombic)† | Cu Kα (λ = 1.5418 Å) (orthorhombic)† | |
|---|---|---|---|
| Data reduction | |||
| Space group | P43212 | P212121 | P212121 |
| Unit-cell parameters‡ (Å, °) | a = 79.89 (1), b = 79.89 (1), c = 37.00 (2), α = β = γ = 90 | a = 36.98 (3), b = 79.80 (1), c = 79.92 (1), α = β = γ = 90 | a = 79.70 (1), b = 79.71 (1), c = 36.83 (3), α = β = γ = 90 |
| Molecular mass (Da) | 14700 | 14700 | 14700 |
| Molecules per asymmetric unit | 1 | 2 | 2 |
| Detector | Dectris PILATUS 6M-F | Dectris PILATUS 6M-F | Bruker APEX II |
| Crystal-to-detector distance (mm) | 135 | 135 | 40 |
| X-ray wavelength (Å) | 0.97625 | 0.97625 | 1.5418 |
| Observed reflections | 735148 (31186) | 735464 (99591) | 647723 (22460) |
| Unique reflections | 32463 (1660) | 63838 (9126) | 22610 (3107) |
| Resolution (Å) | 39.95–1.27 | 56.47–1.26 | 39.86–1.79 |
| Completeness (%) | 99.9 (98.3) | 99.9 (99.5) | 99.4 (96.4) |
| R merge | 0.077 (2.066) | 0.077 (1.453) | 0.142 (0.750) |
| 〈I/σ(I)〉 | 20.9 (1.7) | 14.7 (1.6) | 17.52 (1.92) |
| Multiplicity | 22.6 (18.8) | 11.5 (10.9) | 28.48 (10.9) |
| Mn(I) half-set correlation CC1/2 | 0.999 (0.556) | 0.998 (0.536) | § |
| Cruickshank DPI (Å) | 0.049 | 0.050 | ¶ |
| Average B factor (Å2) | 21.0 | 22.8 | 20.45 |
| Refinement | |||
| R factor/R free (%) | 17.22/19.6 | 17.9/22.6 | 19.4/26.6 |
| R factor, all (%) | 17.22 | 18.2 | 16.6 |
| R.m.s.d., angles (°) | 1.145 | 2.793 | 1.122 |
| Ramachandran values (%) | |||
| Most favoured | 98.4 | 96.6 | 98.8 |
| Additional allowed | 1.56 | 3.44 | 1.16 |
| Disallowed | 0 | 0 | 0 |
The raw diffraction images are available at Zenodo (Brink & Helliwell, 2017 ▸).
Note that the order of the a, b, c unit-cell parameter values in Table 1 ▸ follows the respective conventions of the two different diffraction data-processing programs that we have used.
The CC1/2 metric is more recently introduced than the Bruker software used with the APEX II instrument, which therefore does not include it. The other, much used, metric of 〈I/σ(I)〉 crossing 2 is provided.
In the case of anisotropic protein model refinement undertaken at a diffraction resolution worse than ∼1.6 Å the calculated DPI formula denominator value of [number of observations (21313) − number of refined parameters (20690)] is approaching zero and the DPI estimate thus becomes unstable. Therefore, the distance values from our Cu Kα data cannot have reliably reported e.s.d. values. Details regarding the ‘DPI webserver’ can be found in Kumar et al. (2015 ▸). We prefer to use an anisotropic refinement for the Cu Kα case as it improved the F o − F o residual density, in particular around the Re atoms and their coordinated ligands.
Additional refinement protocols and tables discussing various aspects are included in the Supporting Information. Supplementary Table S1(a): comparison of the effects on F o − F c residual electron-density map peaks by the various refinement programs of SHELXL, PHENIX and REFMAC including the proper utilization of rhenium f′ and f′′ values. Supplementary Table S1(b): comparison of the effects on F o − F c residual electron-density map peaks by the various refinement programs of PHENIX and REFMAC involving orthorhombic and tetragonal refinements. Supplementary Table S2: comparison of anomalous difference electron-density peak heights in the Cu Kα and DLS (λ = 0.9763 Å) diffraction data. Supplementary Table S3: table of Re-atom distances from their cognate specific residues as well as their metal occupancies and B factors for Cu Kα diffraction data. Supplementary Table S4: table of selected bond distances and angles derived from the Diamond Light Source (λ = 0.9763 Å) diffraction data set for the cases not discussed in the main text.
3. Results and discussion
The coordination complex fac-[Re(CO)3(H2O)3]+ readily coordinates to monodentate, bidentate and tridentate ligand systems via the three labile aqua sites (Alberto et al., 1999 ▸; Salignac et al., 2003 ▸; Grundler et al., 2004 ▸, 2006 ▸; Helm, 2008 ▸; Brink et al., 2014 ▸). The facially coordinated carbonyl ligands have chemically known coordinative stability and it is conventionally considered to be chemically unlikely that any substitution will occur at these three sites unless specific carbonyl substitution is targeted (Tisato et al., 2006 ▸; Braband et al., 2012 ▸; Rattat et al., 2001 ▸). Owing to chemical kinetic understanding of the stability of the rhenium tricarbonyl complexes, we have assumed that no exchange or substitution is currently occurring under our experimental conditions and therefore the occupancies of the ligands are the same as those of the Re atoms. Henceforth, the ligands where 2F o − F c electron density is present (i.e. His15) were refined with the same occupancy value as the Re atom in the orthorhombic refinement. The occupancy of any ligands in a cif monomer which show partial but not complete 2F o − F c electron density have been refined with an occupancy of zero. This was performed in order to abide with the current chemical and kinetic understanding of fac-[Re(CO)3(H2O)3]+ complexes whilst respecting the weak electron density (presumed to be due to ligand mobility). In sites where the position of the rhenium metal centre is clearly defined by the anomalous difference density map, i.e. Leu129, but where no/little 2F o − F c electron density occurs above the 1.3σ level, only the position of the rhenium metal has been placed.
Rhenium binding to aspartic acid occurs at residues Asp119A, Asp18A, Asp18B, Asp52A and Asp52B. Binding to glutamic acid occurs at residues Glu7B, Glu35A and Glu35B. Binding to arginine occurs at residues Arg125B and Arg61B (however, the position of Arg61B is relatively uncertain owing to poor 2F o − F c density). Binding to leucine occurs at the Leu125A and Leu129B residues. Rhenium complexes also occur in the vicinity of residues Leu129B, between Pro70A and Arg61A, and in the region of Arg14B as nonbinding entities. We note therefore the large number of repeated binding observations at identical amino-acid types in the protein A and B subunits. We confirm the octahedral environment of the rhenium at the His15 residues in both the A and B subunits of the protein (Fig. 1 ▸), which has the highest refined metal occupancy. Where the metal occupancy is lower, not all of the ligand positions of fac-[Re(CO)3(H2O)n]+ (n ≤ 3) are defined by electron density. We have therefore refined with a monomer cif file (labelled as RRE) in sites which show incomplete electron density for the coordinative ligands, or have simply placed the Re atom in position according to the anomalous density map, as its core evidence.
Figure 1.

Composite OMIT maps of the binding site at His15 in chains A (a) and B (b) coordinating to fac-[Re(CO)3(H2O)2N] (the N atom is from His15). The 2F o − F c electron-density map contoured at 1.2 r.m.s. is shown in blue and the F o − F c electron-density map contoured at 5.0σ (the Coot default; Emsley & Cowtan, 2004 ▸) is shown in green; the anomalous electron-density map contoured at 3.0σ is shown in orange. This figure was prepared using CCP4mg (McNicholas et al., 2011 ▸).
The rhenium tricarbonyl complex coordinates to the His15A and His15B side chains, with bond distances of 2.25 (8) and 2.36 (8) Å. The electron-density map is well defined around His15A, whereas a break in the 2F o − F c density along the trans axial carbonyl ligand is observed for His15B. Related small-molecule fac-[Re(CO)3Nimidazole] complexes show bond distances ranging from 2.174 (4) to 2.197 (5) Å (Schibli et al., 2000 ▸; Garcia et al., 2000 ▸; Fernández-Moreira et al., 2014 ▸; Brink et al., 2013 ▸). The protein study here and the CSD values therefore agree within statistical precision, which is naturally limited by the 1.26 Å resolution X-ray model refinement of the protein. The occupancies of the Re atoms are 83% for both chains A and B (Fig. 1 ▸). Reports by Binkley et al. (2011 ▸) and Zobi & Spingler (2012 ▸) similarly mention preferential binding to the histidine imidazole moiety and describe well the structural environment around this rhenium tricarbonyl fragment as well as the ligand coordination.
Rhenium binding to aspartic acid residues occurs repeatedly, namely Asp119A (Fig. 2 ▸), Asp18A and Asp18B, and Asp52A and Asp52B. Close proximity of Arg125A NH2 to the O atom of the carbonyl ligand [2.13 (8) Å] is observed for the Re3H complex (also visible in Fig. 2 ▸). The interspatial distance [3.96 (9) Å] between the respective NH2 and Re atom is, within the bond-distance error of 0.09 Å, a van der Waals interaction (i.e. 3.7 Å). Similarly, for chain B a close interaction is observed between Arg125B NH2 and the Re atom [NH2⋯Re4D = 1.98 (9) Å], which likewise in turn shows coordination to Asp119B.
Figure 2.

Composite OMIT maps of the Asp119A binding site coordinating to fac-[Re(CO)3(H2O)2]. This figure clearly shows the close proximity of the Arg125 residue to the rhenium core. The electron-density maps are contoured as in Fig. 1 ▸. F o − F c density is indicated for the axial CO and H2O ligands, which are refined according to the monomer cif library. However, no electron density is apparent for the trans coordinating CO ligand. The CO has therefore been refined with zero occupancy but must be chemically present, in accordance with the current chemical understanding of fac-[M(CO)3]+ complexes as previously stated. The quasi bite angle (QBA) at Asp119A is 41 (2)° for OD2—Re3H⋯OD1. The distances to the Re atom are 2.5 (1) and 3.26 (9) Å, respectively. The coordination environment at Asp119B is similar, with bond distances as listed in Supplementary Table S4. This figure was prepared using CCP4mg (McNicholas et al., 2011 ▸).
An unusual cyclic dimer is found in the CSD (CSD refcode UDENAU) in which one Re atom binds bidentately to the N (α-amino) and O (α-carboxylate) atoms of an aspartic acid subunit (Supplementary Fig. S1). The third available position is occupied by the O atom (β-carboxylate) from a second Asp unit (Nayak et al., 2013 ▸). The Re—O1(β-carboxylate) and Re-O(α-carboxylate) bond distances are both 2.149 (4) Å. The distance between the noncoordinated O2(β-carboxylate) and the Re atom is 3.35 Å. The QBA for O1(β-carboxylate)—Re⋯O2(β-carboxylatenoncoordinated) is measured as 41.5°, a very close approximation to the QBA between Re and Asp119. A short contact of 3.306 (8) Å is observed for the dimer between the (α-amino) NH2 and the CO ligand of the neighbouring rhenium complex. Another small-molecule crystal structure in the CSD is an aspartic N-monoacetic acid coordinated tridentately to fac-[Re(CO)3(H2O)3]+, which has average Re—O bond distances of 2.138 (2) Å (CSD refcode CEJSOB; Klenc et al., 2012 ▸); these are within the range of that found in this protein crystallographic study.
The rhenium metal atom coordination at Asp18A has an occupancy of 33%. The bond distances to the Re atom are 2.06 (9) and 3.00 (8) Å, with a QBA of 47 (2)°. The coordination at Asp52A has an occupancy of 42%. The bond distances from the carboxylic O atom to the Re atom are 2.2 (1) and 3.5 (1) Å, with a QBA of 34 (1)°. A second rhenium complex coordinated to Glu35A (rhenium occupancy 30%) lies in close proximity, with definable F o − F c density and an Re—OE1 bond distance of 2.62 (8) Å, a distance which is shorter than the sum of the van der Waals radii (3.67 Å; Desiraju & Steiner, 2006 ▸). Interestingly, the rhenium metal centres of Re6H and Re7H are separated by 3.8 (1) Å, which is also within the range of a van der Waals interaction (4.3 Å; Figs. 3 ▸ and 4 ▸).
Figure 3.
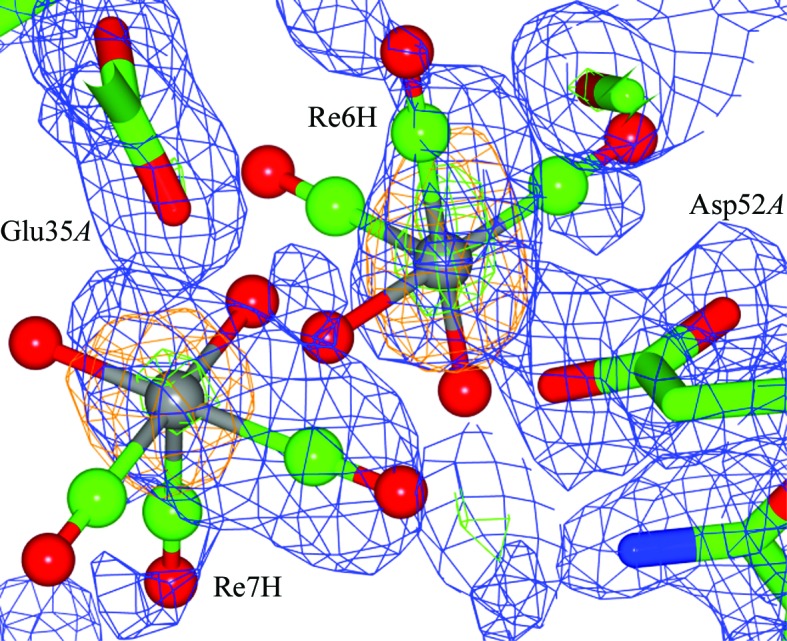
Composite OMIT maps of the Asp52A and Glu35A binding sites coordinating to fac-[Re(CO)3(H2O)2]+, indicating the close proximity of the two Re atoms to each other. The relative positions of the Re atoms are clearly defined by the anomalous map to σ levels of 14.3 and 14.8. Clashes occur between the carbonyl and aqua ligands of the two Re atoms. The respective orientation of the fac-[Re(CO)3]+ moieties has been placed as well as possible, taking into account the limited 2F o − F c density. An F o − F c electron-density peak (6σ) is found in the vicinity, but its assignment is chemically uncertain and was therefore not made. The likely explanation for this layout, since their summed occupancies is less than 100%, is that a fraction of the unit cells in the crystals have a rhenium in one location and another fraction favours the nearby location. An alternative is that it is a dirhenium compound, for which there are several possibilities such as oxo-bridged, carbonyl bridged or metal–metal complexes. Electron-density maps are contoured as in Fig. 1 ▸. This figure was prepared using CCP4mg (McNicholas et al., 2011 ▸).
Figure 4.

Anomalous density accuracy in an overlay fit between the Cu Kα and DLS models. (a) The composite OMIT electron-density map for the Cu Kα laboratory data at Re6 and Re5, illustrating the relative positions of the Re atoms (as determined by the anomalous difference density map) and water solvent molecules (H2O34 and H2O17, as determined by the F o − F c and 2F o − F c electron-density maps). (b) Identical view as in (a) for the DLS data. An overlay fit between the Cu Kα and DLS models gave an r.m.s.d. of 0.14 Å for this subunit, i.e. closely identical in all respects. The fac-[Re(CO)3(H2O)2(X)] cif file (RRE) indicating the positions of the Re atoms in the DLS data as determined by the DLS X-ray wavelength optimized anomalous difference electron-density map. Notice the near-perfect overlay position of the respective Re atoms, i.e. Re6 and Re5 versus RRE6 and RRE7, as well as for H2O34 and H2O17 versus O2 for RRE6 and RRE7.
Related small-molecule structures of fac-[Re(CO)3]+ bound to aspartic acid fragments have Re—O bond distances varying from 2.138 (2) to 2.149 (4) Å (Klenc et al., 2012 ▸; Nayak et al., 2013 ▸), whereas the Re—OH2 bond distances for small-molecule fac-[Re(CO)3(H2O)3]+ complexes typically vary from 2.171 (5) to 2.21 (1) Å (Alberto et al., 1999 ▸). Bite angles that are formed from four coordinated κ2O,O′ complexes range from 59.4 (1) to 59.8 (2)° (Gibson et al., 1994 ▸, 1999 ▸).
An Re atom is found in the vicinity of Glu7B with a distance of 3.30 (8) Å between the OE2 and Re atoms. Direct coordination is considered to be unlikely owing to the reported Re—O bond distance of the small-molecule Re(κ-N,O) glutamic acid complex (Mundwiler et al., 2004 ▸) being 2.163 (4) Å (CSD refcode EXIWUD). However, a possible interaction may occur between the atoms as the distance is less than the sum of the van der Waals radii (3.67 Å). Only the placement of the Re atom is defined, as no 2F o − F c density is observed for the aqua or carbonyl ligands (the anomalous difference peak height from the DLS data is 8.3σ versus 4.7σ for the Cu Kα data), i.e. as expected owing to the X-ray wavelength optimization of the rhenium f′′, thus further confirming its identification.
Additionally, binding to arginine is observed at Arg61B, with respective Re—N distances of 2.4 (2) Å and (a not chemically possible) 0.8 (2) Å (NH1); however, the positions of Arg61B and Arg61A are relatively uncertain owing to poorly defined 2F o − F c density, in particular for NH1. The occupancy of the Re atom is 40% [anomalous difference peak-height evidence from DLS data = 9.4σ (Arg61A) and 9.7σ (Arg61B)]. There are, however, to date no small-molecule crystal structures of fac-[M(CO)3]+ (M = Re, Tc) bonded to arginine listed in the CSD to allow a direct comparison (Allen, 2002 ▸).
Binding to leucine occurs at the Leu129A and Leu129B residues, with Re—O bond distances of 2.5 (1) Å (occupancy of 31%) and 2.8 (1) Å (occupancy of 21%), respectively. A van der Waals interaction occurs between the Leu129B OXT atom and Re [spatial distance of 3.23 (8) Å, which is less than the sum of the van der Waals radii (3.67 Å)]. No rhenium(I)–leucine complexes are found in the CSD database; however, a rhenium(V)–leucine complex (Melián et al., 2000 ▸) (CSD refcode LOPTOZ) has been reported with typical Re—Oleucine and Re—Nleucine bond distances of 2.12 (1) and 2.22 (3) Å, respectively.
Rhenium anomalous difference density also occurs in the vicinity of residues Leu129B [3.47 (7) Å], where a 40% occupancy rhenium complex occurs. The electron density is partially explained by the allocation of two fractionally disordered molecules, the positions of which have an Re⋯Re distance of 1.5 (1) Å as indicated by the anomalous map. The complex is unlikely to be a Re—Re chemical dimer owing to the too short distance value and the stable oxidation state of fac-[ReI(CO)3]+ as well as the following results from the CSD. Utilizing Mogul (Bruno et al., 2004 ▸) to search for all possible Re⋯Re interactions involving the fac-[Re(CO)3]+ fragment yields 15 hits in the CSD with minimum/maximum bond distances ranging from 2.837 to 3.233 Å (Fig. 5 ▸ a). If all Re⋯Re interactions are considered, irrespective of their oxidation states and coordination mode, a search yield of 1546 hits is obtained with minimum/maximum bond distances ranging from 2.245 to 3.497 Å (Fig. 5 ▸ b).
Figure 5.

Comparison of all possible small-molecule Re⋯Re interactions. (a) Mogul search plots of the number of small-molecule hits utilizing Re⋯Re interactions containing the fac-[Re(CO)3]+ fragment as a search criterion found in the CSD database. Colour bars indicate the number of structure hit entries with respect to bond length (Å) in the CSD version update 5.37 data library. The minimum/maximum bond distances had a standard deviation of 0.129 Å and a mean value of 3.065 Å. (b) Data analysis of all small-molecule hits {not necessarily containing the fac-[Re(CO)3]+ fragment as illustrated in (a)} for Re⋯Re interactions as a search criterion in the CSD database. The longest Re⋯Re distance [3.4934 (6) Å] is found in a cyclic pentakis(μ-hydrido)-icosacarbonyl-pentarhenium complex (CSD refcode PORYIE; Bergamo et al., 1998 ▸), whereas the shortest is for a carboxylate-dirhenium complex (CSD refcode GUVTUO; Golichenko & Shtemenko, 2015 ▸).
In addition to the diversity of binding to these various amino acids, as described in detail above, we also note that nonbonding rhenium entities are found between residues in the vicinity of Pro70A [Re⋯O = 3.6 (1) Å] and Arg61A with an occupancy of 60% and in the region of Arg14B [Re⋯N = 6.2 (1) Å] with an occupancy of 40%.
4. Conclusions
In summary, we have found multiple binding sites for a rhenium metal complex on different amino acids, findings that have never been reported previously. Specifically, we see that there are numerous chemical coordination possibilities for the fundamental synthon fac-[ReI(CO)3(H2O)3]+ to Asp, Glu, Arg and His amino-acid residues as well as to the C-terminal carboxylate in the vicinity of Leu and Pro. Naturally, we confirm that the highest occupancy binding is to the histidine imidazole group. Rhenium and technetium have similar chemical properties but distinctly different radioactive signatures, with 188Re and 186Re being used for therapy and 99mTc being used for imaging across all diseases. This diversity of amino-acid interactions that we have uncovered, in addition to the ability to precisely indicate bond-distance and angle ranges for specific three-dimensional structure development, will create more new Re/Tc lead compounds for site-specific binding to protein tissue in radiopharmaceutical applications. Furthermore, the total sum of the binding of rhenium to the various amino acids that has been discerned would immediately lead to the possibility of a reduced, or better yet targeted, radiation dose with respect to medical radiation treatment.
Supplementary Material
PDB reference: hen egg-white lysozyme with bound rhenium, 5nbj
Supplementary Information. DOI: 10.1107/S2052252517003475/lt5001sup1.pdf
Diamond Light Source orthorhombic data. Zip file contains DLS orthorhombic protein crystallographic structure factors, coordinates, ligand cif file and anomalous density map.. DOI: 10.1107/S2052252517003475/lt5001sup2.zip
Cu K alpha orthorhombic data. Zip file contains structure factors, coordinates, ligand cif file and anomalous map.. DOI: 10.1107/S2052252517003475/lt5001sup3.zip
DLS tetragonal refinement : data submitted to PDB (PDB entry 5nbj).. DOI: 10.1107/S2052252517003475/lt5001sup4.zip
Raw diffraction images URL: https://doi.org/10.5281/zenodo.345364
Acknowledgments
AB wishes to acknowledge the South African National Research Foundation, the SASOL University Collaboration Initiative, The University of the Free State (UFS) Prestige Scholar Program and Professor Andreas Roodt at UFS for financial assistance. We are grateful to the University of Manchester for general support and its X-ray diffraction facilities, to the Diamond Light Source for access to the I04 beamline (within the University of Manchester Block Allocation of beamtime; BAG MX8997-29 i04-1), which was vital to the results presented here, and to Dr Colin Levy and the Manchester Protein Structure Facility for assistance during data collection at Diamond Light Source. AB thanks Dr Simon W. M. Tanley for early discussions on protein model refinement, as well as Professor David Collison for helpful support in fulfilling the research visit to the University of Manchester. We thank, in particular, Dr Louise Natrajan and her research group for the provision of laboratory facilities and chemicals at the University of Manchester. We wish to thank the referees for their time and the sharing of their expertise. We also thank the Co-editor Professor Phil Lightfoot.
References
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Alberto, R., Ortner, K., Wheatley, N., Schibli, R. & Schubiger, P. A. (2001). J. Am. Chem. Soc. 123, 3135–3136. [DOI] [PubMed]
- Alberto, R., Schibli, R., Egli, A., Schubiger, P. A., Herrmann, W. A., Artus, G., Abram, U. & Kaden, T. A. (1995). J. Organomet. Chem. 493, 119–127.
- Alberto, R., Schibli, R., Egli, A., Schubiger, P. A., Abram, U. & Kaden, T. A. (1998). J. Am. Chem. Soc. 120, 7987–7988. [DOI] [PubMed]
- Alberto, R., Schibli, R., Waibel, R., Abram, U. & Schubiger, A. P. (1999). Coord. Chem. Rev. 190–192, 901–919.
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Bergamo, M., Beringhelli, T., D’Alfonso, G., Mercandelli, P., Moret, M. & Sironi, A. (1998). Angew. Chem. Int. Ed. 37, 2128–2131. [DOI] [PubMed]
- Betts, M. J. & Russell, R. B. (2003). Bioinformatics for Geneticists, edited by M. R. Barnes & I. C. Gray, pp. 289–316. Chichester: John Wiley & Sons. https://doi.org/10.1002/0470867302.ch14.
- Binkley, S. L., Leeper, T. C., Rowlett, R. S., Herrick, R. S. & Ziegler, C. J. (2011). Metallomics, 3, 909–916. [DOI] [PubMed]
- Braband, H., Imstepf, S., Benz, M., Spingler, B. & Alberto, R. (2012). Inorg. Chem. 51, 4051–4057. [DOI] [PubMed]
- Brink, A. & Helliwell, J. R. (2017). New leads for fragment based design of Re/Tc radiopharmaceutical agents: Raw Diffraction Images. Zenodo, https://doi.org/10.5281/zenodo.345364.
- Brink, A., Visser, H. G. & Roodt, A. (2013). Polyhedron, 52, 416–423.
- Brink, A., Visser, H. G. & Roodt, A. (2014). Inorg. Chem. 53, 12480–12488. [DOI] [PubMed]
- Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D. S., Purkis, L. H., Smith, B. R., Taylor, R., Cooper, R. I., Harris, S. E. & Orpen, A. G. (2004). J. Chem. Inf. Comput. Sci. 44, 2133–2144. [DOI] [PubMed]
- Cantor, C. R. & Schimmel, P. R. (1980). Biophysical Chemistry. Part I: The Conformation of Biological Macromolecules. San Francisco: W. H. Freeman & Co.
- Cianci, M., Helliwell, J. R. & Suzuki, A. (2008). Acta Cryst. D64, 1196–1209. [DOI] [PubMed]
- Colucci, M., Maione, F., Bonito, M. C., Piscopo, A., Di Giannuario, A. & Pieretti, S. (2008). Pharmacol. Res. 57, 419–425. [DOI] [PubMed]
- Desiraju, G. R. & Steiner, T. (2006). The Weak Hydrogen Bond. Oxford University Press.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Erlanson, D. A. (2012). Top. Curr. Chem. 317, 1–32. [DOI] [PubMed]
- Fernández-Moreira, V., Marzo, I. & Gimeno, M. C. (2014). Chem. Sci. 5, 4434–4446.
- Garcia, R., Paulo, A., Domingos, Â., Santos, I., Ortner, K. & Alberto, R. (2000). J. Am. Chem. Soc. 122, 11240–11241.
- Gibson, D. H., Ding, Y., Miller, R. L., Sleadd, B. A., Mashuta, M. S. & Richardson, J. F. (1999). Polyhedron, 18, 1189–1200.
- Gibson, D. H., Mehta, J. M., Ye, M., Richardson, J. F. & Mashuta, M. S. (1994). Organometallics, 13, 1070–1072.
- Golichenko, A. A. & Shtemenko, A. V. (2015). Acta Cryst. E71, 1219–1221. [DOI] [PMC free article] [PubMed]
- Grundler, P. V., Helm, L., Alberto, R. & Merbach, A. E. (2006). Inorg. Chem. 45, 10378–10390. [DOI] [PubMed]
- Grundler, P. V., Salignac, B., Cayemittes, S., Alberto, R. & Merbach, A. E. (2004). Inorg. Chem. 43, 865–873. [DOI] [PubMed]
- Gurusaran, M., Shankar, M., Nagarajan, R., Helliwell, J. R. & Sekar, K. (2014). IUCrJ, 1, 74–81. [DOI] [PMC free article] [PubMed]
- Hall, M. D., Telma, K. A., Chang, K.-E., Lee, T. D., Madigan, J. P., Lloyd, J. R., Goldlust, I. S., Hoeschele, J. D. & Gottesman, M. M. (2014). Cancer Res. 74, 3913–3922. [DOI] [PMC free article] [PubMed]
- Helm, L. (2008). Coord. Chem. Rev. 252, 2346–2361.
- Joseph-McCarthy, D., Campbell, A. J., Kern, G. & Moustakas, D. (2014). J. Chem. Inf. Model. 54, 693–704. [DOI] [PubMed]
- Kelava, T., Ćavar, I. & Čulo, F. (2011). Period. Biol. 113, 311–320.
- Klenc, J., Lipowska, M., Taylor, A. T. & Marzilli, L. G. (2012). Eur. J. Inorg. Chem. 2012, 4334–4341. [DOI] [PMC free article] [PubMed]
- Kumar, K. S. D., Gurusaran, M., Satheesh, S. N., Radha, P., Pavithra, S., Thulaa Tharshan, K. P. S., Helliwell, J. R. & Sekar, K. (2015). J. Appl. Cryst. 48, 939–942.
- Lebedev, A. A. & Isupov, M. N. (2014). Acta Cryst. D70, 2430–2443. [DOI] [PubMed]
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Melián, C., Kremer, C., Suescun, L., Mombrú, A. W., Mariezcurrena, R. A. & Kremer, E. (2000). Inorg. Chim. Acta, 306, 70–77.
- Merritt, E. A. (1999). Acta Cryst. D55, 1109–1117. [DOI] [PubMed]
- Mundwiler, S., Kündig, M., Ortner, K. & Alberto, R. (2004). Dalton Trans., pp. 1320–1328. [DOI] [PubMed]
- Murray, C. W., Verdonk, M. L. & Rees, D. C. (2012). Trends Pharmacol. Sci. 33, 224–232. [DOI] [PubMed]
- Nayak, D. K., Halder, K. K., Baishya, R., Sen, T., Mitra, P. & Debnath, M. C. (2013). Dalton Trans. 42, 13565–13575. [DOI] [PubMed]
- Rattat, D., Schubiger, P. A., Berke, H. G., Schmalle, H. & Alberto, R. (2001). Cancer Biother. Radiopharm. 16, 339–343. [DOI] [PubMed]
- Salignac, B., Grundler, P. V., Cayemittes, S., Frey, U., Scopelliti, R., Merbach, A. E., Hedinger, R., Hegetschweiler, K., Alberto, R., Prinz, U., Raabe, G., Kölle, U. & Hall, S. (2003). Inorg. Chem. 42, 3516–3526. [DOI] [PubMed]
- Santoro, G., Blacque, O. & Zobi, F. (2012). Metallomics, 4, 253–259. [DOI] [PubMed]
- Santos, N. C., Figueira-Coelho, J., Martins-Silva, J. & Saldanha, C. (2003). Biochem. Pharmacol. 65, 1035–1041. [DOI] [PubMed]
- Schibli, R., La Bella, R., Alberto, R., Garcia-Garayoa, E., Ortner, K., Abram, U. & Schubiger, P. A. (2000). Bioconjug. Chem. 11, 345–351. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Swanson, B. N. (1985). Rev. Clin. Basic Pharm. 5, 1–33. [PubMed]
- Takematsu, K., Williamson, H., Blanco-Rodríguez, A. M., Sokolová, L., Nikolovski, P., Kaiser, J. T., Towrie, M., Clark, I. P., Vlček, A., Winkler, J. R. & Gray, H. B. (2013). J. Am. Chem. Soc. 135, 15515–15525. [DOI] [PMC free article] [PubMed]
- Tanley, S. W. M. & Helliwell, J. R. (2014). Acta Cryst. F70, 1127–1131. [DOI] [PMC free article] [PubMed]
- Tanley, S. W. M., Schreurs, A. M. M., Kroon-Batenburg, L. M. J., Meredith, J., Prendergast, R., Walsh, D., Bryant, P., Levy, C. & Helliwell, J. R. (2012). Acta Cryst. D68, 601–612. [DOI] [PubMed]
- Tisato, F., Porchia, M., Bolzati, C., Refosco, F. & Vittadini, A. (2006). Coord. Chem. Rev. 250, 2034–2045.
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zobi, F. & Spingler, B. (2012). Inorg. Chem. 51, 1210–1212. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: hen egg-white lysozyme with bound rhenium, 5nbj
Supplementary Information. DOI: 10.1107/S2052252517003475/lt5001sup1.pdf
Diamond Light Source orthorhombic data. Zip file contains DLS orthorhombic protein crystallographic structure factors, coordinates, ligand cif file and anomalous density map.. DOI: 10.1107/S2052252517003475/lt5001sup2.zip
Cu K alpha orthorhombic data. Zip file contains structure factors, coordinates, ligand cif file and anomalous map.. DOI: 10.1107/S2052252517003475/lt5001sup3.zip
DLS tetragonal refinement : data submitted to PDB (PDB entry 5nbj).. DOI: 10.1107/S2052252517003475/lt5001sup4.zip
Raw diffraction images URL: https://doi.org/10.5281/zenodo.345364