

Abstract
INTRODUCTION
Insulin is secreted by pancreatic β cells in response to glucose stimulation. Its release is controlled by the interplay of calcium and phosphoinositide signaling pathways. A rapid release phase, in which insulin containing granules that are already docked and primed at the plasma membrane (PM) undergo exocytosis, is followed by slow release. In this second phase, granules are docked and primed and then released in a series of bursts, each triggered by a spike in cytosolic Ca2+.
RATIONALE
To better understand the molecular basis underlying insulin secretion, we characterized TMEM24, a protein enriched in neuroendocrine cells previously suggested to be required for a normal secretory response.
RESULTS
We found that TMEM24 is an endoplasmic reticulum (ER) protein that concentrates at ER-PM contact sites, where it tethers the two bilayers. TMEM24 binding “in trans” to the PM is negatively regulated by phosphorylation in response to elevation of cytosolic Ca2+, so that TMEM24 transiently dissociates from the PM as Ca2+ concentration spikes and then reassociates with this membrane upon dephosphorylation. Additionally, TMEM24 contains a lipid transport module of the synaptotagmin-like, mitochondrial and lipid-binding protein (SMP) family, which we structurally characterized and showed to bind glycerolipids with a preference for phosphatidylinositol (PI). Thus, TMEM24 helps deliver PI, which is synthesized in the ER, to the PM, where it is converted to phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] to replenish pools of this lipid hydrolyzed during glucose-stimulated signaling. Supporting a key role of TMEM24 in the coordination of Ca2+ and phosphoinositide signaling, the lipid transport function of TMEM24 is essential for sustaining the intracellular Ca2+ oscillations that trigger bursts of insulin granule release and hence insulin secretion. PI(4,5)P2 is required for Ca2+-dependent exocytosis. It also controls the activity of PM ion channels that regulate cytosolic Ca2+ levels and is the precursor of IP3, which also helps to modulate cytosolic Ca2+ by triggering Ca2+ release from the ER. Thus, in insulin-secreting cells, TMEM24 participates in coordinating Ca2+ and phosphoinositide signaling pathways to cause pulsatile insulin secretion (see the figure).
CONCLUSION
Our findings implicate ER-PM contact sites and an ER resident lipid-transfer protein in the direct regulation of PM phosphoinositide pools, offering fresh insights into the mechanisms of cellular phosphoinositide dynamics. More specifically, they elaborate the mechanisms underlying insulin secretion, which is impaired in patients with type II diabetes, and may ultimately have therapeutic ramifications.
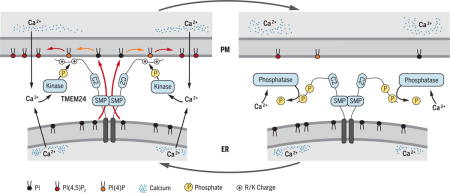
TMEM24 activity cycle at ER-PM contacts
Glucose stimulation of insulin-secreting cells triggers Ca2+ influx, phospholipase C–dependent PI(4,5)P2 cleavage, and granule exocytosis. Ca2+-stimulated phosphorylation causes TMEM24 dissociation from the PM and interruption of SMP-mediated PI transfer that allows PI(4,5)P2 resynthesis. Lowered PI(4,5)P2 levels attenuate the excitatory response. Dephosphorylation allows TMEM24 to return to the PM to replenish PI(4,5)P2, permitting a new cycle of Ca2+ elevation and secretion.
Graphical abstract
Eukaryotic cells harbor close appositions (10 to 30 nm) between the plasma membrane (PM) and the endoplasmic reticulum (ER), the organelle where the synthesis of most membrane lipids begins. These ER-PM contact sites are important in cellular calcium dynamics and also play critical roles in lipid homeostasis and signaling (1–4). Identification and functional characterization of the protein machinery at ER-PM appositions, including lipid-transfer proteins that carry lipids between membranes, have provided key insights into the molecular mechanisms underlying processes that occur at these sites (5–10).
Here, we investigated the properties of TMEM24, one of two similar proteins, TMEM24/C2CD2L and C2CD2, which bioinformatics studies predict to contain a module typically found in proteins that act at membrane contact sites (11, 12). TMEM24, which is highly expressed in professional neurosecretory cells, is a regulator of insulin secretion (13). Insulin is produced by pancreatic β cells, where it is generated in the Golgi complex via the cleavage of proinsulin, then packaged into granules and stored (14). A rise in blood glucose activates tightly coupled calcium and phosphoinositide signaling pathways in these cells and leads to granule exocytosis, which occurs in two phases (15, 16). Immediately after glucose stimulation, a rise in intracellular calcium levels triggers secretion of the readily releasable granule pool (~5%), consisting of granules already docked and primed at the PM (17). Next, a reserve granule pool undergoes docking and priming steps and is therefore released more gradually in a series of small bursts dependent on pulsatile rises in calcium levels (15, 18). Insulin secretion is impaired in patients with type II diabetes, highlighting the importance of understanding the mechanisms that underlie this process (14).
Here, we probed the molecular mechanism by which TMEM24 modulates insulin exocytosis and identified TMEM24 as a lipid transport protein resident at ER-PM contact sites implicated in the cross-talk between calcium and PM phosphoinositide dynamics that regulates insulin release. We found that TMEM24 plays a role in transporting glycerolipids with a preference for phosphatidylinositol (PI) from its site of synthesis in the ER to the PM, where it is a precursor for phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2]. At the PM, PI(4,5)P2 has multiple roles in the cell, including in signal transduction (19, 20) and in the direct control of insulin granule exocytosis (14, 21–23) and PM ion channel function (14, 22, 23). Whereas TMEM24 localization to ER-PM contact sites and its activity in transporting PI to the PM are regulated by calcium dynamics, the lipid transfer function of TMEM24 is required for calcium pulsatility.
Results and discussion
TMEM24 is an ER-anchored protein that localizes at ER-PM contacts
Bioinformatics studies predict that the transmembrane protein TMEM24 comprises a lipid-binding module of the tubular lipid-binding (TULIP) superfamily (12) (Fig. 1A), which we confirmed by crystallography (see below). All previously characterized members of the intracellular subfamily of TULIP proteins containing this module—known as synaptotagmin-like, mitochondrial, and lipid-binding protein (SMP) domain—are concentrated at membrane contact sites (10, 11, 24), suggesting that TMEM24 might also localize to sites of membrane apposition. To address this possibility, we assessed localization of versions of the human protein [TMEM24-enhanced green fluorescent protein (EGFP)] in INS-1 cells, a rat insulinoma cell line that, like pancreatic islets, exhibits high levels of endogenous TMEM24 (13) (fig. S1, A and B), or in HeLa cells (TMEM24-mCherry) that are not specialized for triggered secretion and have undetectable endogenous levels of TMEM24 (13) (fig. S1, A and B). Confocal microscopy of the transfected cells showed that moderate levels of fluorescent TMEM24 colocalized with cotransfected ER markers, mRFP-Sec61β or ER-oxGFP, indicating a localization throughout the ER (Fig. 1, B and C, left insets in C). In addition, hot spots of fluorescence were observed at the cell periphery, in a pattern consistent with a concentration at ER-PM contacts (24) (Fig. 1, B and C, right insets in C).
Fig. 1. TMEM24 is a tether at endoplasmic reticulum (ER)–plasma membrane (PM) contact sites.
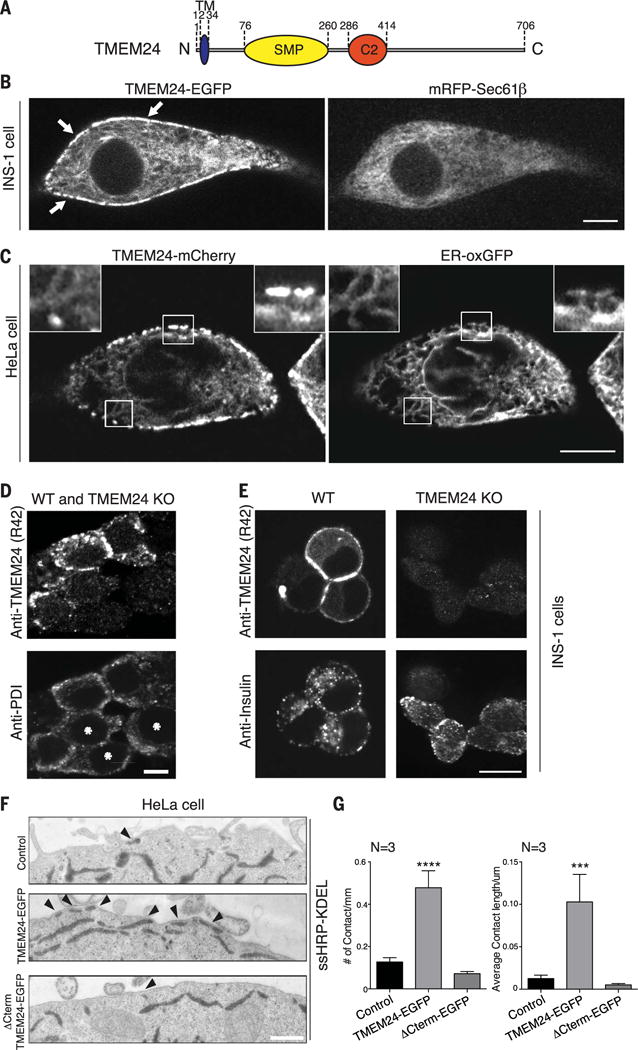
(A) Schematic representation of TMEM24. (B) Overexpression of human TMEM24-EGFP in INS-1 insulinoma cells monitored by confocal microscopy. TMEM24-EGFP partially colocalizes with ER marker mRFP-Sec61β throughout the ER and is additionally concentrated at hot spots at the cell periphery (arrows), as expected for ER-PM contact sites. (C) Overexpression of TMEM24-mCherry in HeLa cells monitored by confocal microscopy. TMEM24-mCherry colocalizes with ER marker ER-oxGFP throughout the reticular and cortical ER but is greatly enriched in the cortical ER (right insets). (D) Immunofluorescence for TMEM24 and ER marker PDI in mixed TMEM24 KO (asterisks) and WT INS-1 insulinoma cells. TMEM24 mainly localizes to the periphery of the cells. (E) Immunofluorescence for TMEM24 and insulin in WT and TMEM24 KO INS-1 cells. TMEM24 immunoreactivity is only observed in WT cells, is concentrated at the cell periphery, and does not colocalize with insulin granules. (F) Representative EM images of HeLa cells transfected with ssHRP-KDEL alone or with full-length TMEM24-EGFP or ΔCTerm TMEM24-EGFP. Arrowheads indicate ER-PM contacts. Note the increase in contact site number after overexpression of TMEM24-EGFP. (G) Quantitation of experiments described in Fig. 1F. Number (left panel; mean ± SEM) and average length (right panel; mean ± SEM) of ER-PM contacts increases relative to control only when full-length TMEM24 is overexpressed. Scale bars are 5 μm for (B) and (D), 10 μm for (C), and 200 nm for (F). P values are <0.0001 and 0.0002 for (F) left and right panels, respectively.
To rule out a possible subcellular mistargeting of exogenous TMEM24 due to the fluorescent tags, we assessed the localization of the overexpressed untagged protein and of the endogenous protein using newly generated antibodies directed against TMEM24 (Fig. 1D and fig. S1D), followed by confocal microscopy. An equatorial focal acquisition plane showed that overexpressed untagged TMEM24 immunofluorescence accumulated at the cell periphery in spots with the characteristic pattern for ER-PM contact sites (fig. S1D, upper panel) and partially colocalized with the ER marker calreticulin (Fig. 1D and fig. S1D). This peripheral localization was further confirmed by image acquisition of the ventral plane of cells, which highlighted the portions of the ER in close proximity with the plasma membrane (fig. S1D, lower panel). The same localization was observed for endogenous TMEM24 (Fig. 1D), which also partially colocalized with the ER marker protein disulfide isomerase (PDI). Absence of TMEM24 immunoreactivity in INS-1 clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9–edited cells lacking TMEM24 validated the specificity of the signal (Figs. 1D and 6B and fig. S1C).
Fig. 6. Oscillatory localization of TMEM24 at the PM affects insulin secretion in insulinoma cells.
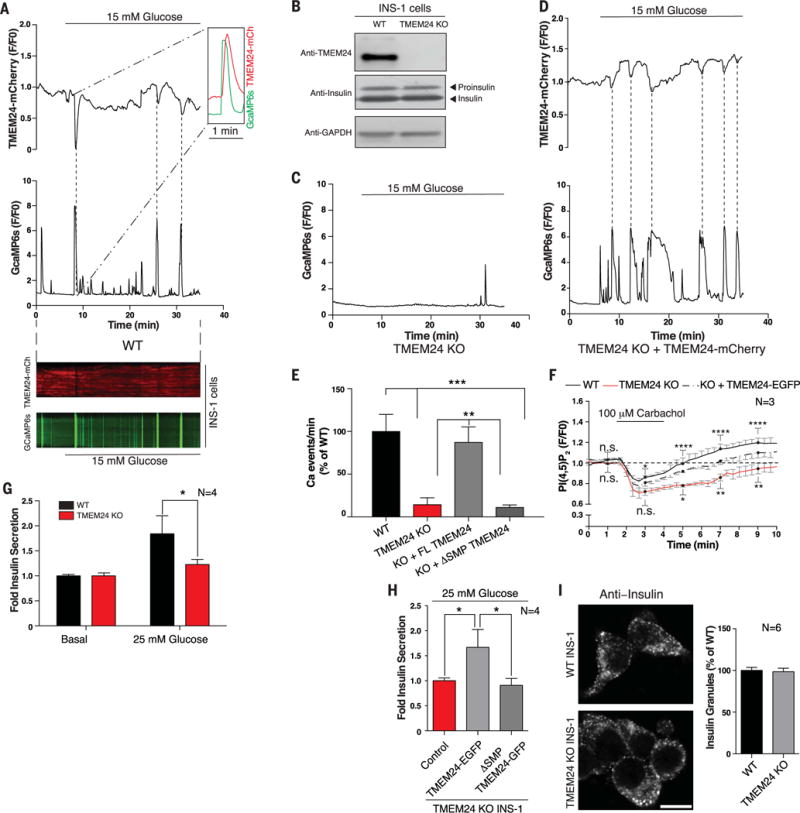
(A) (Top) High-glucose stimulation of WT INS1 cells triggers oscillations of TMEM24 (TMEM24-mCherry, top) and calcium (GcaMP6s, bottom) opposite in phase as monitored by TIRF microscopy. (Inset) Depletion of TMEM24 (peak inverted) reaches maximum after the peak of calcium influx. Dotted lines indicate corresponding peaks. (Bottom) Kymograph shows that TMEM24 and calcium oscillations are opposite in phase after glucose stimulation. (B) WT and TMEM24 KO INS1 cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against TMEM24 and insulin, showing no difference in insulin biogenesis in the KO cells. Top band in anti-insulin panel is proinsulin, whereas bottom band is insulin. GAPDH was used as a loading control. (C) Calcium oscillations (monitored by GcaMP6s) are abrogated after glucose stimulation in TMEM24 KO INS1 cells. (D) Exogenous overexpression of TMEM24-mCherry in KO cells rescues glucose-induced calcium oscillations. Dotted lines indicate corresponding peaks. (E) Quantitation of Ca2+ oscillations per minute in WT, TMEM24 KO, and TMEM24 KO INS1 cells rescued with either full-length or ΔSMP TMEM24 upon glucose stimulation. (F) Dynamics of PI(4,5)P2 recovery in WT (black) and TMEM24 KO (red) INS1 cells overexpressing PI(4,5)P2 marker iRFP-PHPLCδ after treatment with carbachol to deplete PI(4,5)P2 at the PM. TMEM24 KO cells show delayed recovery of basal PI(4,5)P2 level at the PM. This delay is rescued by exogenous overexpression of TMEM24-EGFP (dashed line). Black dots indicate time points for which significance is reported (n.s., no significant difference; *P < 0.05; **P < 0.01; ****P < 0.0001). Significances of difference for WT versus KO and KO versus KO+TMEM24 are indicated above and below traces, respectively. (G) Amount of secreted insulin from WT or TMEM24 KO INS1 cells after glucose stimulation was assayed by ELISA. Stimulated insulin secretion was normalized to secretion under basal conditions (mean ± SEM). (H) TMEM24 KO INS1 cells were transfected with full-length TMEM24-EGFP or ΔSMP-TMEM24, and insulin secretion was assayed by ELISA. Only TMEM24 containing the lipid-transfer module was able to rescue insulin secretion. (I) Anti-insulin immunofluorescence of WT (top) and TMEM24 KO (bottom) INS1 cells shows that the typical punctate distribution of insulin granules is not perturbed by loss of TMEM24 expression (as quantified at right). Scale bar is 5 μm. P values are 0.0003 for WTversus TMEM24 KO, 0.0005 for WTversus KO+ΔSMP, 0.0019 for KO versus KO+FL TMEM24, and 0.0029 for KO+FL TMEM24 versus KO+ΔSMP in (E), 0.0408 in (G), and 0.0428 for control versus FL TMEM24 and 0.0031 for FL TMEM24 versus ΔSMP in (H).
The localization of TMEM24 in the ER with a concentration in the cortical ER differs from the reported localization of endogenous TMEM24 immunoreactivity on insulin granules based on a commercially available TMEM24 antibody [Aviva #ARP47080_P050 (13)]. We confirmed this finding using the same commercial antibodies. However, we found that such immunoreactivity was still present in TMEM24 knockout (KO) INS-1 cells, suggesting that colocalization with insulin granules reflected nonspecific binding (fig. S1E). In contrast, no staining of insulin granules was produced by our new antibodies (Fig. 1E).
Several other proteins resident at ER-PM contact sites, such as the extended synaptotagmins (E-Syts) or oxysterol-binding protein-related protein (ORP) family members, function as tethers that promote the expansion of contacts when overexpressed (6, 10, 24). To assess whether TMEM24 also functions as a tether, we overexpressed TMEM24-EGFP in HeLa cells, together with horseradish peroxidase (HRP) tagged with an ER retention motif [ssHRP-KDEL (25)] and imaged the cells by electron microscopy (EM) cytochemistry. The presence of electron-dense HRP reaction product in the lumen of the ER facilitates identification of this organelle in EM images. Consistent with the tethering function of TMEM24, overexpression of TMEM24 resulted in a robust increase in PM profiles engaged in direct close apposition with the ER relative to control cells expressing ssHRP-KDEL alone (Fig. 1, E and F).
The transmembrane segment mediates TMEM24 ER association and the C-terminal region mediates PM association
TMEM24 must bind the PM to function as an ER-PM tether. The protein comprises a very short lumenal N-terminal domain, a transmembrane (TM) segment, the SMP domain, a C2 domain, and a predicted unstructured, polybasic C-terminal region (residues 415 to 706) (12, 13) (Fig. 1A). To determine which portions of TMEM24 are responsible for the tethering of the ER to the PM, we monitored the localization of EGFP fusions of TMEM24 deletion constructs and of the ER marker mRFP-Sec61β coexpressed as a control (Fig. 2). Both INS-1 (professional secretory) and COS-7 (fibroblast-like) cells were used for this analysis. The ER distribution of full-length TMEM24 was particularly striking in “en face” views of the very thin edges of COS-7 cells, where intense spots represented ER-PM contactsites (Fig. 2A). ATMEM24 construct lacking the TM segment was partially diffuse throughout the cytosol but concentrated selectively at the PM, as clearly seen in equatorial sections of INS-1 cells (Fig. 2, B and E). However, in contrast to full-length TMEM24 (Fig. 2, A and D), it decorated the PM profile continuously, rather than at hot spots, as would be the case if it were restricted to the cortical ER (Fig. 2, B and E). Thus, this construct, which lacks the TM anchor, contains the PM binding determinant(s). Conversely, a construct lacking the C-terminal region following the C2 domain localized exclusively to the reticular and not the cortical ER (Fig. 2, C and F), indicating that this region of the protein mediates association with the PM. Accordingly, overexpression of this construct does not promote the formation of contact sites, as confirmed by EM (Fig. 1, F and G). The SMP or C2 domain fragments had a diffuse cytosolic localization, and deletion of these regions from TMEM24 resulted in constructs with the same localization as full-length TMEM24 (fig. S2A). These two domains, therefore, are not required for the localization of the protein to ER-PM contact sites.
Fig. 2. TMEM24 N-terminal TM domain and C-terminal unstructured region are required for localization to ER-PM contacts.
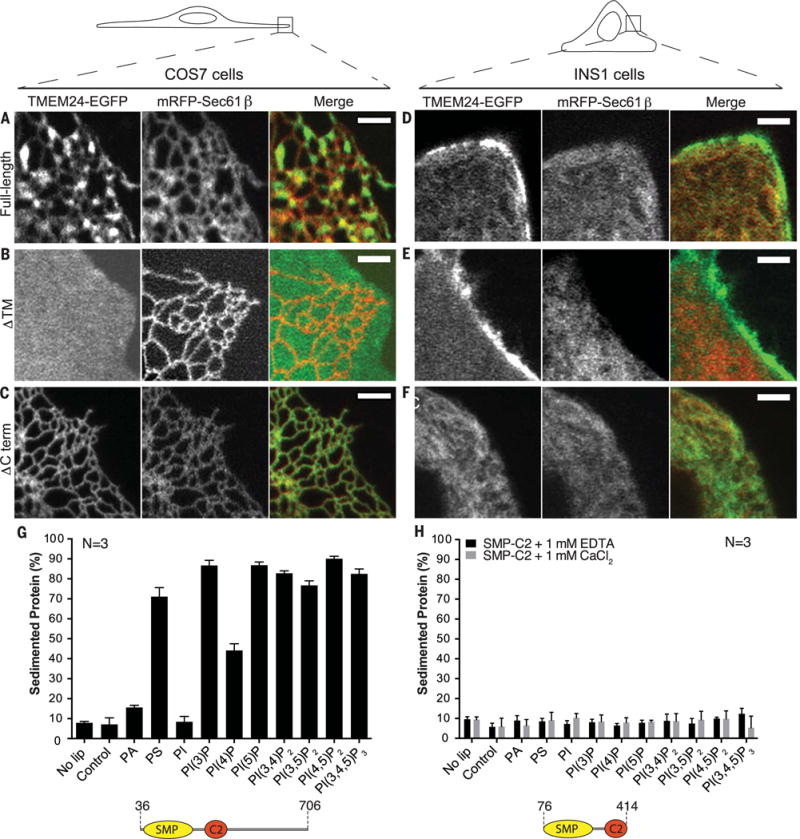
(A to F) Comparative analysis of localization of full-length, ΔTM, and ΔCTerm TMEM24-EGFP when overexpressed in COS-7 [(A) to (C)] and INS-1 [(D) to (F)] cells monitored by confocal microscopy. mRFP-Sec61β was used as ER marker. Given the different shape of the two cell types (top drawings), the plasma membrane was observed “en face” in the thin lamellipodium of the COS-7 cells but in cross section in the INS-1 cell. Full-length TMEM24 [(A) and (D)] has a diffuse localization in the ER but clusters at hot spots that represent ER-PM contact sites. The reticular shape of the ER is optimally appreciated in the “en face” view of the COS-7 cell. The construct lacking the transmembrane region (ΔTM) [(B) and (E)] localizes primarily at the PM, where it has a diffuse distribution (as clear by the different views of the COS-7 cell and the INS-1 cell). The construct lacking the C-terminal region (ΔCTerm) [(C) and (F)] precisely colocalizes with the ER marker Sec61β. Scale bar is 2 μm. (G) Purified human TMEM24 lacking the transmembrane domain (residues 36 to 706) was incubated with liposomes containing 10% PE and either 20% PS or 5% other test lipid, with the remainder PC. Liposomes were sedimented and analyzed for associated protein. ΔTM-TMEM24 directly binds acidic membranes without specific lipid preference. PA and PI are less charged than the other phosphoinositides in the panel, and their charge, closer to the hydrophobic layer, may be more shielded by the head groups of neighboring phospholipids. (H) Purified SMP-C2 fragment of TMEM24 (residues 76 to 414) does not sediment with membranes in the presence of EDTA or calcium. Liposome compositions are as in (G).
To assess whether TMEM24 might interact directly with the bilayer of the PM, rather than via a protein factor, we expressed the cytosolic portion of TMEM24 (residues 36 to 706) recombinantly and used the purified protein in binding assays with a panel of liposomes of varying compositions (Fig. 2G). TMEM24 bound acidic liposomes containing either phosphatidylserine (PS) [see also (26)] or phosphoinositides, without preference for a specific phosphoinositide, including PI(4,5)P2, a key determinant of PM identity. Supporting this hypothesis, depletion of this phosphoinositide in the PM by light-induced recruitment of the 5-phosphatase domain of Lowe oculocerebrorenal syndrome protein (OCRL) fused to cryptochrome 2 (CRY2) (27, 28) did not affect the localization of TMEM24 to contact sites in COS-7 cells (Fig. 3A). Thus, TMEM24 binds the PM selectively because the cytoplasmic leaflet of this membrane is highly acidic owing to an enrichment of PS and phosphoinositides, which function redundantly.
Fig. 3. TMEM24 localization at ER-PM contact sites is regulated by a phosphorylation-dephosphorylation mechanism.
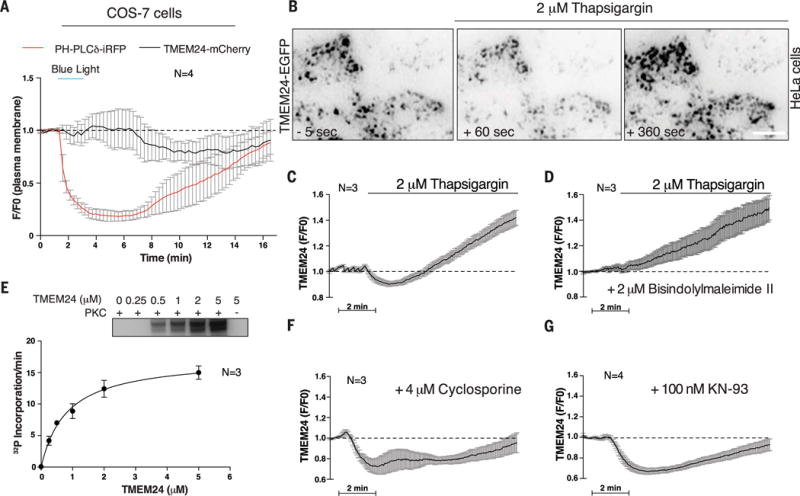
(A) TMEM24 localization at ER-PM contacts, as monitored by TIRF microscopy, is not lost upon PM PI(4,5)P2 depletion by blue light–induced CRY2-OCRL 5-phosphatase domain recruitment to the PM in COS-7 cells. (B) HeLa cells transfected with TMEM24-EGFP were analyzed by TIRF microscopy. In basal conditions, TMEM24 forms PM puncta corresponding to ER-PM contact sites (left). Treatment with thapsigargin induces partial dissociation of TMEM24 from the PM, as revealed by loss of fluorescence in the TIRF plane (middle). Subsequently, TMEM24 rapidly reassociated with the PM, overshooting basal fluorescence intensity (right). (C) Quantitation of (B). (D) HeLa cells overexpressing TMEM24-EGFP were preincubated with the PKC inhibitor bisindolylmaleimide II before stimulation with thapsigargin. PKC inhibition prevented TMEM24 dissociation from the PM but did not block excess TMEM24 PM accumulation during recovery. (E) In vitro phosphorylation of a cytosolic TMEM24 construct by purified PKC. 32P incorporation onto TMEM24 by PKC (autoradiogram in inset) (30 min, 37°C) followed Michaelis-Menten kinetics with a Km of 0.81 ± 0.085 μM. (F) Calcineurin/PP2B inhibition by pretreating HeLa cells over-expressing TMEM24-EGFP with cyclosporine before thapsigargin treatment inhibited PM reassociation of TMEM24. Thapsigargin-mediated dissociation from the PM was unaffected by cyclosporine, which did not impair PKC activity. (G) CaMK inhibition by KN-93 did not affect TMEM24 dissociation from the PM. Recovery of TMEM24 is partially attenuated under KN-93 treatment due to loss of downstream calcineurin/PP2B activation by CaMK.
Consistent with the data obtained in living cells, we found that lipid bilayer binding in vitro was dependent on the C-terminal portion of TMEM24 because a fragment comprising only the SMP and C2 domains (residues 76 to 414) did not bind acidic liposomes. Nor did the same construct bind liposomes in the presence of calcium (Fig. 2H), which induces membrane interactions for a number of previously characterized C2 domains (29). Sequences in the TMEM24 C terminus are highly conserved (fig. S2B), indicative of functional importance, and are enriched in basic residues, which are well suited to mediate interactions with acidic membranes.
Regulation of TMEM24 localization by calcium-dependent phosphorylation and dephosphorylation
The critical role of calcium dynamics in regulated secretion and the previously observed requirement of TMEM24 for efficient insulin release (13) prompted us to investigate whether calcium regulates TMEM24 localization. To this end, we treated HeLa cells transfected with TMEM24-EGFP with thapsigargin, which elevates cytosolic calcium levels by blocking the ER calcium pump (SERCA) and inducing indirectly (as a result of the depletion of calcium within the ER) the opening of store-operated calcium channels in the PM (30, 31). Upon thapsigargin treatment, TMEM24-EGFP rapidly dissociated from the PM, presumably redistributing from the cortical to the reticular ER, then reassociated with this membrane to overshoot basal levels (Fig. 3, B and C).
Inspection of polybasic sequences in the TMEM24 C-terminal region, which mediate PM association, revealed an enrichment in serine and threonine residues, including within sites that fit a consensus (32, 33) for the Ca2+-dependent protein kinase C (PKC) (fig. S2B). Phosphorylation by PKC and downstream kinases, which results in the acidification of the C-terminal segment, could account for TMEM24’s dissociation from the acidic PM. Conversely, dephosphorylation could enable TMEM24 reassociation with the PM and could result in overshooting if TMEM24 is basally phosphorylated at resting conditions. Protein serine/threonine phosphatase 2B (PP2B/calcineurin) dephosphorylates substrates, including PKC substrates, in a calcium-dependent manner and could be a player in this mechanism.
Consistent with such a scenario, TMEM24 localization to or away from ER-PM contact sites after thapsigargin treatment was altered by drugs that block either PKC or PP2B. Thus, when PKC was inhibited by the bisindolylmaleimide II (34), thapsigargin treatment no longer induced dissociation of TMEM24 from the PM (Fig. 3D). Conversely, TMEM24 reassociation with the PM after thapsigargin treatment was impaired by the PP2B inhibitor cyclosporin A (35) (Fig. 3F). Additionally, in vitro phosphorylation of purified TMEM24 (Fig. 3E) indicated that TMEM24 is a substrate for PKC.
Inhibition of another major family of calcium-dependent serine/threonine kinases, the Ca2+/calmodulin-dependent protein kinases (CaMKs), via the drug KN-93 (36), did not affect the dissociation of TMEM24 from PM upon thapsigargin treatment, arguing against the involvement of this class of Ca2+-dependent protein kinases in this effect (Fig. 3G). The loss of dephosphorylation overshoot in the presence of this inhibitor may reflect indirect effects of CaMKs on PP2B activity (37, 38).
Structural features of the SMP domain
We expressed the putative SMP domain (residues 76 to 260) of TMEM24 recombinantly in Escherichia coli, purified it by affinity and size-exclusion chromatography (SEC), crystallized it, and used the single-wavelength anomalous dispersion method to determine its structure at 3.0−Å resolution (table S1). As predicted (39), the domain adopts a TULIP fold, with two alpha helices and a highly curved antiparallel beta sheet forming a cornucopia-like structure (Fig. 4A). The fold is the same as for the SMP module present in the E-Syts (40) and predicted for components of the ER-mitochondrion encounter structure (ERMES) complex (39) (root-mean-square deviation between the TMEM24 and E-Syt2 SMP domains is 12.4 Å over 128 Cα positions).
Fig. 4. TMEM24 harbors a phosphatidylinositol (PI)–binding SMP module.
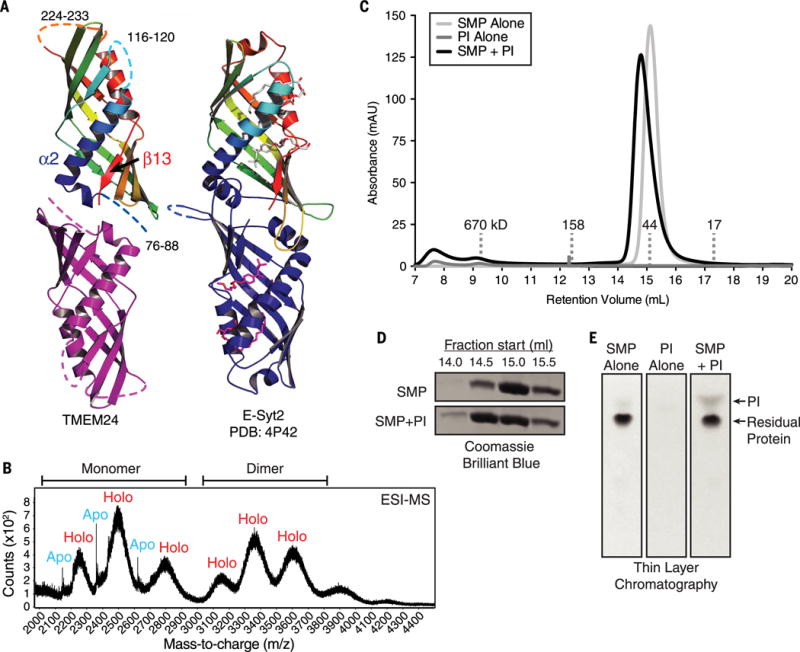
(A) TMEM24 (76 to 260) adopts an SMP domain fold. TMEM24 SMP domain is modeled as a dimer by analogy to the E-Syt2 SMP domains (at right). Disordered segments are represented as dashed lines. TMEM24 helix α2 and strand β13 move closer compared with the corresponding helix and strand in the liganded E-Syt2 structure. Four lipid molecules bound by E-Syt2 dimer are indicated. (B) Native electrospray ionization mass spectrometry (ESI-MS) analysis of TMEM24 SMP domain purified from Expi293 cells indicates dimerization, with masses consistent with one phospholipid molecule bound per monomer. Whereas the monomeric fraction exists in both apo and holo form, the dimeric fraction is mostly complexed with lipid. (C) SEC of purified TMEM24 SMP domain preincubated with liver PI indicates that PI binds and comigrates with TMEM24 SMP. Although TMEM24 SMP migrates as a dimer in solution, a small shift in apparent molecular weight upon binding to PI occurs, consistent with a change in conformation but not oligomerization state. (D) Analysis of individual fractions from 14.0 to 16.0 ml by SDS-PAGE confirms redistribution of the liganded SMP domain. (E) Fractions between 14.0 and 16.0 ml retention volume were pooled; lipid was extracted and resolved by thin-layer chromatography. The PI band did not appear in these fractions when TMEM24 SMP or liver PI was subjected separately to SEC.
In the crystal, the TMEM24 SMP domains are arranged as trimers. This oligomerization state likely is a crystal packing artifact, because both SEC and mass spectrometry analyses indicate that the domain is dimeric in solution (Fig. 4, B and C). In both E-Syt2 and the ERMES complex subunit Mmm1, the SMP domains are arranged as head-to-head dimers (40, 41), and most likely the TMEM24 TULIP modules associate similarly, with the cornucopia tips pointed away from the interface (Fig. 4A). Dimerized in this way, TMEM24 contains a cavity lined with hydrophobic residues well suited as a lipid binding pocket. The binding site is similar to that in E-Syt2, except that in E-Syt2 the cavity connects to solvent via a seam along most of its length. The structural elements bordering the seam in E-Syt2 are closer together in TMEM24 (TMEM24 helix α2 and strand β13 in Fig. 4A), eliminating the seam and narrowing the cavity as compared with E-Syt2. As a result, the particulars of lipid binding in TMEM24 may be different from those in E-Syt2. Indeed, a mass spectrometry analysis of a TMEM24-lipid complex indicates that only one glycerolipid is bound per SMP monomer (Fig. 4B) rather than two, as in E-Syt2. We have so far been unable to obtain a crystal structure of lipid-bound TMEM24, and we suspect that crystal contacts induce distortions in the TMEM24 SMP domain to make it incompatible with lipid binding. Thus, the details of how TMEM24 interacts with lipids remain unclear.
TMEM24 binds and transports glycerolipids with a preference for PI
It has been reported that other TULIP-domain proteins bind and transport lipids (5, 40, 42), suggesting that TMEM24 might as well. To identify potential lipid ligands for TMEM24, we overexpressed its SMP domain in mammalian cells (Expi293) and used mass spectrometry to identify lipids that copurify with it. A similar approach was successful in identifying the ligands of other lipid transport proteins (6, 40, 43). Similar to the E-Syts and SMP-domain proteins in the ERMES complex (40, 41), the TMEM24 SMP module bound glycerophospholipids (table S2). The proportion of the lipids bound [37.9% phosphatidylcholine (PC), 15.9% phosphatidylethanolamine (PE), 35% phosphatidylinositol (PI), 10.0% phosphatidylserine (PS), rest other glycerophospholipids] (table S2) reflects their relative abundance in cells, except for PI, which constitutes only 5 to 10% of total cellular glycerophospholipid and was enriched in the TMEM24 bound fraction (44) (table S2). These data, particularly when contrasted with the lipid analysis of the SMP domain of E-Syt2 (40), indicated PI as a potential preferred TMEM24 ligand. Accordingly, PI comigrated with a TMEM24 SMP dimer, as assessed by SEC and thin-layer chromatography (TLC), confirming binding (Fig. 4, C to E).
We carried out in vitro transfer assays to determine whether TMEM24 can transfer PI between membranes (Fig. 5A). In these experiments, a cytosolic N-terminally dodecahistidine (His12)– tagged TMEM24 construct was tethered between “heavy” (~400 nm in diameter, filled with 25% sucrose) and “light” (~100 nm in diameter, no sucrose) liposomes to permit the transfer of lipids between them. One set of liposomes also contained DOGS-NTA-Ni lipid, allowing capture of TMEM24 via the His12 tag, whereas the second set contained PS for binding the TMEM24 C terminus (Fig. 5A, top panels). After incubation, we removed the protein by proteolysis and separated the heavy and light liposomes by centrifugation, then monitored transport of selected lipids using high-performance liquid chromatography (HPLC) (to compare PI and PS transport) or scintillation counting (for PI versus PE) (Fig. 5A, lower panel). TMEM24 transferred both PE and PS less efficiently than PI, consistent with a preference for PI transport (Fig. 5B).
Fig. 5. TMEM24 functions as a lipid-transfer protein with a preference for PI.
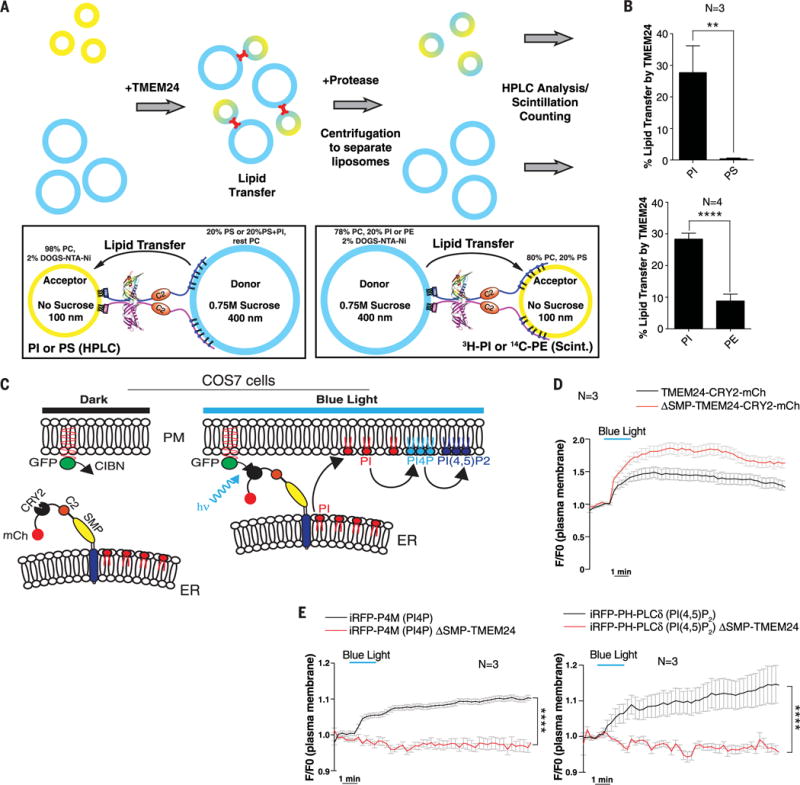
(A) In vitro lipid-transfer assays with TMEM24. (Top) Heavy donor (blue) and light acceptor (yellow) liposomes were mixed in the presence or absence of a recombinant N-terminally His12-tagged construct comprising the cytosolic portion of TMEM24 (residues 36 to 706). After lipid transfer (15 or 30 min),TMEM24 was degraded with protease and liposomes were separated by centrifugation. Donor and acceptor liposomes were separately analyzed for lipid content by HPLC or scintillation counting. (Bottom left) TMEM24 was tethered to donor liposomes containing PS via the basic C terminus and to acceptor liposomes containing 2 mole % (mol %) DOGS-NTA-Ni2+ via its N-terminal His12 tag. Transfer to acceptor liposomes of PS or PI, originally present at 20 mol % only in the donor liposomes, was measured by HPLC analysis after 30-min transfer. (Bottom right) TMEM24 was tethered to donor liposomes containing DOGS-NTA-Ni2+ via its N-terminal His12 tag and to acceptor liposomes containing PS via the basic C terminus. Transfer to acceptor liposomes of 14C-PE or 3H-PI, originally present only in the donor liposomes, was measured by scintillation counting after 15-min transfer. (B) Quantitation of the assays described in (A). TMEM24 preferentially transfers PI over PS (top panel) and PE (bottom panel). Mean ± SD indicated. (C) Schematic representation of light-induced TMEM24 PM recruitment strategy. TMEM24 was rapidly recruited to the PM by light-induced dimerization of CRY2 and CIBN modules. A version of TMEM24 with its C terminus replaced by a CRY2-mCherry module was expressed in COS-7 cells. The CIBN module is localized to the PM via a C-terminal CAAX box. (D) Dynamics of TMEM24-CRY2-mCherry (black) and ΔSMP-TMEM24-CRY2-mCherry (red) fluorescence before, during, and after blue-light stimulation (20 pulses, 200 ms each), as monitored by TIRF microscopy. (E) TIRF microscopy of PI(4)P marker iRFP-P4M (left) and the PI(4,5)P2 marker iRFP-PH-PLCδ (right) before, during, and after blue-light stimulation. Note the increase in both phosphoinositides due to transfer of PI to the PM by TMEM24 after blue-light stimulation (black). PM recruitment of ΔSMP-TMEM24-CRY2-mCherry does not affect PM levels of PI(4)P or PI(4,5)P2 (red). P values are P = 0.0049 and P < 0.0001 for (B) upper and lower panels, respectively, and P < 0.0001 for both (E) panels.
Next, we explored whether TMEM24 may transfer glycerolipids, and more specifically PI, to the PM in living cells. Enrichment of PI in the PM is lowest among the glycerolipids (5). Furthermore, this pool is rapidly metabolized by kinases and phospholipases and thus needs to be continuously replenished to maintain steady-state levels of its downstream products at rest and to compensate for acute reductions in response to physiological or pharmacological manipulations. We examined whether TMEM24 might participate in PI replenishment at the PM by determining whether its acute recruitment affected levels of its downstream metabolites phosphatidylinositol 4-phosphate (PI4P) and PI(4,5)P2, two lipids whose levels can be assessed in vivo by genetically encoded fluorescence probes. To this end, we modified the optogenetics approach described above (27): The N-terminal domain of the transcription factor CIB1 (CIBN) was fused to a C-terminal CAAX prenylation motif to induce its constitutive targeting to the PM, and the C-terminal polybasic sequence of TMEM24 was replaced by an in-frame CRY2-mCherry cassette to make its recruitment to the PM, via interaction with CIBN, inducible by blue light (Fig. 5C).
In COS-7 cells expressing these constructs, blue light induced a reversible interaction of TMEM24-CRY2-mCherry with PM-anchored CIBN. This interaction recruited ER-anchored TMEM24 to the PM contact sites (Fig. 5D). The kinetics of the recruitment were slower than previously observed for a cytosolic protein (27) because TMEM24 is tethered to the ER and could interact with the PM only by diffusion through the ER membrane. Levels of PI4P (P4M probe) (Fig. 5E, left panel) and PI(4,5)P2 (PH-PLCδ probe) (Fig. 5E, right panel) underwent rapid increases coincident with the recruitment to the PM of the TMEM-CRY2-mCherry fusion protein (Fig. 5E, black lines). In contrast, levels of these phosphoinositides did not change using a TMEM24-CRY2-mCherry construct that lacked the SMP module (Fig. 5E, red lines). The only modest, ~10% increase in PM PI4P and PI(4,5)P2 after TMEM24 PM recruitment likely reflects the occurrence of homeostatic mechanisms that prevent the excess accumulations of these lipids in the PM.
Taken together, these data support the hypothesis that TMEM24 binds and transfers PI from the ER to the PM, where it can be converted to PI4P and PI(4,5)P2 by enzymes resident there.
Pulsatile PM localization of TMEM24 after glucose stimulation in insulin-secreting cells and link to calcium oscillations
Intracellular calcium levels in insulin-secreting cells oscillate during the slow-release phase of secretion after glucose stimulation (19, 20). To elucidate whether the properties of TMEM24 as described above are relevant in a physiological context, we investigated its dynamics and effect on PI(4,5)P2 signaling upon glucose stimulation in INS-1 cells. We analyzed the localization of TMEM24 (TMEM24-mCherry) at the PM and of calcium levels (GCaMP6s) in response to glucose. Pulses of TMEM24 dissociation from the PM occurred opposite in phase with oscillations in cytosolic calcium, suggesting a clear relation between the two events (Fig. 6A and fig. S3A). Stretching the time axis, however, revealed that peaks of calcium oscillations slightly preceded peaks of TMEM24 dissociation (Fig. 6A, inset), consistent with the hypothesis that calcium-dependent phosphorylation is responsible for the dissociation of TMEM24.
It was shown that calcium oscillations are abolished upon reduction of PI(4,5)P2 below a critical level in INS-1 cells. TMEM24 could be part of the mechanism that sustains the oscillations. Indeed, calcium oscillations were abrogated in TMEM24 KO cells (Fig. 6, B to E, and fig. S3B) and were rescued in KO cells transfected with a full-length TMEM24 construct (Fig. 6, D and E, and fig. S3C). In contrast, a ΔSMP construct did not rescue calcium pulsatility (Fig. 6E), indicating a requirement for the lipid transfer function of TMEM24.
To assess more directly the contribution of TMEM24 to PI(4,5)P2 resynthesis, wild-type (WT) and TMEM24 KO cells were stimulated with carbachol, which triggers phospholipase C (PLC)– dependent PI(4,5)P2 cleavage at the PM and increases intracellular calcium concentrations (19). In WT cells, PM PI(4,5)P2 depletion, as assessed by the iRFP-PH-PLCδ signal in total internal reflection fluorescent (TIRF) microscopy, recovered and then overshot the basal levels. A similar overall dynamic was observed in TMEM24 KO cells. However, the recovery was delayed (Fig. 6F), and such delay was rescued by expression of TMEM24 (Fig. 6F), consistent with the hypothesis that TMEM24 plays a role in replenishing PM PI(4,5)P2. Because PM PI(4,5)P2 levels did eventually recover even in the absence of TMEM24, there are clearly additional TMEM24-independent mechanisms to supply phosphoinositides to the PM, as expected given the occurrence of other PI transport proteins(45).
The TMEM24 lipid transfer module is required for stimulated insulin exocytosis
A previous report implicated TMEM24 in the regulation of insulin secretion (13) but did not elucidate the molecular basis for its action. We confirmed the effect of TMEM24 on insulin release by comparing glucose-stimulated insulin secretion in WT and TMEM24 KO INS-1 cells, finding that secretion is strongly reduced in the latter (Fig. 6G). Furthermore, the lipid transfer function of TMEM24 was required for secretion because transfection of TMEM24 KO cells with a full-length TMEM24 construct rescued stimulated secretion, whereas a construct lacking the SMP lipid-transfer module did not (Fig. 6H). Similarly, overexpression of TMEM24 in WT INS-1 cells enhanced evoked insulin secretion, whereas overexpression of the SMP-deletion construct did not (fig. S3D).
We ruled out that the effect of TMEM24 on evoked secretion was because of an effect on insulin granule biogenesis, because immunoblotting of lysates of WT and TMEM24 KO INS-1 cells (Fig. 6B) for both proinsulin and insulin revealed similar levels in the two cell populations. Furthermore, immunofluorescence on WT or TMEM24 KO INS-1 cells using an antibody against insulin did not show obvious differences in insulin granule number or their overall distribution in the cell (Fig. 6I). These data, which are in agreement with observations of Pottekat et al. (13), are consistent with a secretory defect owing to impaired granule exocytosis.
We suggest that one mechanism by which ablation of this protein attenuates triggered exocytosis is through an impaired delivery of PI to the PM, a process that becomes critical for the regeneration of PI(4,5)P2 during the recovery from its PLC-dependent consumption. Lowered levels of PI(4,5)P2 produced by the absence of TMEM24 will in turn result in ablation of calcium oscillations and hence a defect of Ca2+-dependent exocytosis. Direct effects of lowered levels of PI(4,5)P2 or its metabolite diacylglycerol (DAG) on the exocytic machinery are also possible, because PI(4,5)P2 is required for calcium-triggered exocytosis (21, 46) and DAG is required for the priming steps leading to granule fusion (47, 48). Experiments on broken neuroendocrine cells have shown that efficient secretory granule exocytosis requires transport of PI to the PM and PI(4,5)P2 synthesis in this membrane, most likely to replenish PI(4,5)P2 pools degraded upon cell lysis (21, 46, 49). A cytosolic PI transport protein (PITP) plays a role in this process (46). Our results suggest that the SMP-domain protein TMEM24 may have a synergistic role with PITP in insulin-secreting cells, although TMEM24-mediated lipid transfer is additionally coordinated with calcium dynamics.
Concluding remarks
In summary, our data demonstrate that TMEM24 is an ER-PM contact site tether. After glucose stimulation in insulin-secreting cells and in response to a rise in intracellular calcium, the TMEM24 C terminus is phosphorylated and hence acidified, leading to TMEM24 disassociation from the acidic PM and ER-PM contacts. Subsequent dephosphorylation within the cell allows TMEM24 to return to these sites (Fig. 7). As TMEM24 is alternately phosphorylated and then dephosphorylated, it localizes to ER-PM contacts in a pulsatile manner opposite in phase with the variations in calcium. We show further that TMEM24 uses an SMP-family lipid transfer module to transport PI, the precursor of PI(4,5)P2, from its site of synthesis in the ER to the PM (Fig. 7) and thus helps to replenish PI(4,5)P2 pools, which are degraded during PLC signaling. Our data do not exclude that TMEM24 may also be involved in the transfer of other glycerolipids, a possibility that we have not extensively explored.
Fig. 7. Model for TMEM24 modulation of pulsatile insulin secretion through effects on phosphoinositide and calcium dynamics.
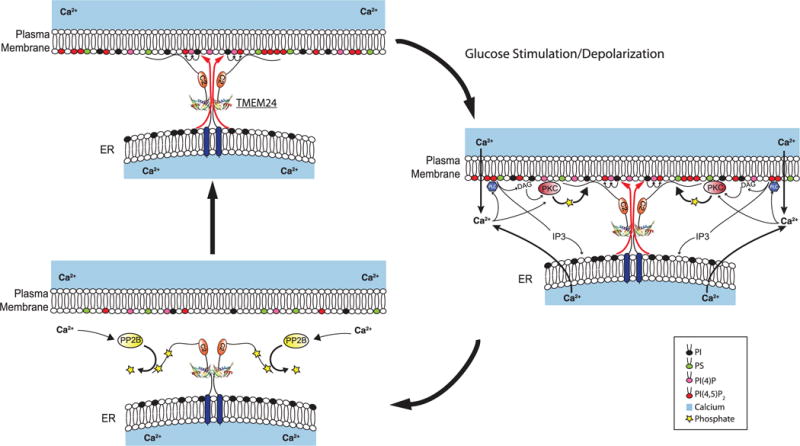
(Top left) In insulin-secreting cells at resting state, ER-anchored TMEM24 localizes to ER-PM contact sites via interaction between its basic C-terminal region and the PM. In this state, TMEM24 transfers PI from the ER to the PM, maintaining the pool of PM phosphoinositides, including PI(4)P and PI(4,5)P2. PI(4,5)P2 interacts with voltage-gated calcium channels, increasing their responsiveness to depolarization caused by glucose stimulation. PI(4,5)P2 also participates in priming of insulin secretory granules in preparation for exocytosis. (Right) Upon glucose stimulation or membrane depolarization, calcium influx through the calcium channels stimulates insulin release while activating the PKC pathway, causing phosphorylation of the TMEM24 C terminus. This C-terminal modification is responsible for the dispersion of TMEM24 from ER-PM contacts into the reticular ER. Transfer of PI to the PM by TMEM24 is abolished. Calcium also activates PLCδ, which is responsible for cleavage of PI(4,5)P2 to form DAG and IP3, partially depleting PI(4,5)P2 from the PM and further elevating levels of cytosolic calcium via activation of IP3 receptors in the ER membrane. The loss of PI(4,5)P2 eventually promotes the closed state of PM calcium channels and terminates IP3 production. (Bottom left) Calcium-induced activation of the S/T-phosphatase calcineurin/PP2B results in dephosphorylation of the TMEM24 C terminus, allowing TMEM24 to repopulate ER-PM contact sites. PI transfer to the PM resumes to replenish PI(4)P and PI(4,5)P2 pools, reenabling calcium channel opening and priming a new population of insulin granules. The oscillatory interplay between calcium and TMEM24 continues in a cyclical fashion for the duration of glucose stimulation.
Moreover, we found that the lipid transfer function of TMEM24 plays a critical role in calcium pulsatility. Because PI(4,5)P2 itself affects the activity of PM ion channels (22), including in beta cells (23), and inositol 1,4,5-trisphosphate (IP3) generated from PI(4,5)P2 modulates calcium oscillations through a cross-coupling between PLC and IP3 receptor activity (50, 51), TMEM24 most likely participates in calcium oscillations through its role in replenishing PM PI(4,5)P2 pools: Its disassociation from contact sites at high cytosolic calcium allows PM PI(4,5)P2 to decrease through PLC activity. This ensures that the IP3 receptors (regulated by IP3) and the PM ion channels that control calcium influx and are regulated by PI(4,5)P2 are inactivated, lowering cytosolic calcium levels. In response to this change, TMEM24 reassociates with the PM and helps replenish PI(4,5)P2 pools (Fig. 7 and legend). Without calcium oscillations, insulin exocytosis would be impaired, as observed in both TMEM24 knockdown (13) and KO cells (Fig. 6G).
Thus, in insulin secreting cells, TMEM24 coordinates phosphoinositide and calcium dynamics, and it likely serves a similar function in other peptide-secreting neurosecretory cells. Phosphoinositide and calcium signaling pathways are conserved in all eukaryotes, suggesting TMEM24 as a paradigm for proteins that couple these processes in other cell types. The TMEM24 paralog C2CD2 is one candidate that could have such a function, as could other lipid-transfer protein family members.
Our results clearly point to a role for ER-PM contact sites and their resident protein TMEM24 in phosphoinositide metabolism and signaling. That membrane contact sites are key participants in these processes is also supported by other recent work. As examples, Nir2 has recently been implicated in recycling PI(4,5)P2–derived phosphatidic acid (PA) from the PM to the ER for conversion to PI after signaling events (52, 53), and we recently proposed that the E-Syts may similarly be involved in the recycling of another PI(4,5)P2 metabolite, DAG (5). Additionally, ORP family members, which have been shown to counter-exchange PI4P to transport other lipids (PS and sterol) against a gradient (6, 7, 9), function in regulating PI4P levels at membrane contact sites (54). Our findings, while helping to illuminate the complex workings underlying insulin secretion and pulsatility, also implicate ER-PM contact sites and a resident lipid-transfer protein in the direct regulation of PM phosphoinositide pools, offering fresh insights into the mechanisms of cellular phosphoinositide dynamics.
Materials and methods
Antibodies and reagents
Antibodies were obtained from the following commercial sources: rabbit anti-TMEM24 (Aviva Biotechnology), mouse anti-Calreticulin (Enzo Life Science), rabbit anti-Proinsulin/Insulin, guinea pig anti-Insulin (AbCam), mouse anti-mRFP (Rockland) and mouse anti-Actin (MP Biomedical). Mouse anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was generated in our lab, while Rabbit anti-TMEM24 was generated by YenZym Antibodies LLC using a C-terminal peptide from TMEM24 with the following sequence: CLRSGTKLIFRRRPRQKE. Alexa488, Alexa594, and Alexa647 conjugated secondary antibodies and Thapsigargin were from Life Technologies; Bisindolylmaleimide II, KN-93, Cyclosporine and Carbamoylcholine chloride (Carbachol) were from Tocris. Lipids were from Avanti Polar Lipids and American Radiolabeled Chemicals.
Plasmid construction
TMEM24 coding sequences (full-length, SMP domain, C2 domain and C-terminal region) were amplified by PCR from human cDNA. The PCR product was cloned into pEGFP-N1 (BD Clontech) using XhoI or BglII and EcoRI sites. Truncated forms (ΔTM, ΔSMP, ΔC2, ΔCterm) of TMEM24 were obtained by site-directed mutagenesis (QuikChange II XL, Agilent Technology) of full-length TMEM24-EGFP. TMEM24 fragments amplified by PCR were cloned into pCDF-C6P using EcoRI or BamHI and NotI sites for bacterial expression with an N-terminal GST tag. pCMV6-AN-His (Origene) was modified to include an N-terminal His12 tag. TMEM24 ΔTM (36–706) was cloned using XhoI and NotI sites into pCMV6-AN-12His for Expi293 cell expression. TMEM24 SMP (76–260) was cloned into pcDNA3.1-3xFLAG using BamHI and NotI sites for Expi293 cell expression as an N-terminal 3xFLAG-tagged protein. The human TMEM24-mCherry vector resulted from PCR amplification of the coding sequence of TMEM24 (from TMEM24-EGFP) followed by cloning into pmCherry-N1 (BD Clontech) using XhoI and EcoRI sites. To assemble the TMEM24-CRY2-mCherry vector TMEM24, CRY2 and mCherry fragments were PCR amplified from TMEM24-EGFP, 5-phosphatase domain OCRL-CRY2 and pmCherry-C1, respectively. The amplification products were ligated by using the In-Fusion cloning Kit (BD Clontech). Theresulting TMEM24-CRY2-mCherryplasmid was used to generate ΔSMP-TMEM24-CRY2-mCherry vector by site directed mutagenesis (QuikChange II XL, Agilent Technology). pGP-CMV-GCaMP6s [Addgene plasmid # 40753 (55)], the untagged version of TMEM24 was from Origine (SC310158) and pSpCas9n(BB)-2A-Puro (PX462) V2.0 [Addgene plasmid # 62987 (56)]. ER-oxGFP, mRFP-Sec61b, ssHRP-KDEL, PH-PLCd-iRFP, P4M-iRFP, CAAX-GFP-CIBN and OCRL-CRY2 were previously described (24).
Cell cultures and transfection
HeLa, COS-7 and HEK cells were cultured at 37°C and 5% CO2 in DMEM medium (Life Technology) containing 10% fetal calf serum, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamax (Life Technology). N2A and PC12 cells were cultured in RPMI containing 10% fetal calf serum, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamax (Life Technology). INS1 cells (passage 30 to 50) were plated in RPMI-1640 with 11.1 mM D-Glucose and supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES-Na pH 7.4, 2 mM L-glutamine, 1 mM sodium pyruvate (Life Technologies), and 50 μM β-mercaptoethanol (Sigma Aldrich). 18 hours before experiments the standard medium with 11.1 mM glucose was replaced with fresh medium containing 5 mM glucose.
In all our experiments, plasmids were transfected by using Lipofectamine 2000 (Life Technology) and cells were cultured for 24 hours before analysis.
Immunofluorescence
Cells were plated and grown on 5 μg/ml human fibronectin (Millipore)-coated glass coverslips and fixed with 4% paraformaldehyde/4% sucrose in 0.1 M sodium phosphate buffer (pH 7.2) at room temperature. Coverslips were washed with 50 mM NH4Cl pH 7.2, then blocked and permeabilized in 0.1% Triton X-100 and 3% bovine serum albumin in PBS. Primary and secondary antibody incubations were performed using the same buffer. Coverslips were rinsed in 1X PBS and mounted in Prolong Gold + DAPI (Life Technology). Immunofluorescence data acquisition was performed using spinning disk confocal microscope (see below).
Live imaging
For live imaging, cells were incubated at 37°C in the following buffer: 136 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1.3 mM MgCl2, 3 mM D-glucose and 10 mM HEPES-Na pH 7.4. Spinning-disk confocal microscopy was performed using the Improvision UltraVIEW VoX system (Perkin-Elmer) built on a Nikon Ti-E inverted microscope, equipped with PlanApo objectives (60×1.45-NA) and controlled by Volocity (Improvision) software. TIRF microscopy was carried out on a Nikon TiE microscope equipped with 60× 1.49-NA and 100× 1.49-NA objectives. Excitation lights were provided by 488 nm (for GFP) and 561 nm (mCherry) diode-pumped solid-state lasers. An optical fiber coupled the lasers to the TIRF illuminator and an acousto-optic tunable filter controlled the output from the lasers. Fluorescent signals were detected with an EM-CCD camera (DU-887; Andor) and acquisition was controlled by Andor iQ software. Images typically were sampled at 0.2 Hz with exposure times in the 4 to 6 s range.
Electron microscopy
Cells were fixed in 1.3% glutaraldehyde-0.1M sodium cacodylate. To activate the HRP reaction, cells were incubated in 0.5 mg/mL 3′3′- Diaminobenzidine (DAB) plus 0.005% H2O2 in 0.1M ammonium phosphate. They were post-fixed with 1% OsO4 in 1.5% K4Fe(CN)6 and 0.1M sodium cacodylate, en bloc stained with 2% uranyl acetate in 50 mM sodium maleate buffer pH 5.2, dehydrated and embedded in Embed 812. Randomly selected cells were imaged at low magnification and cell perimeter was measured using iTEM by Olympus. Higher magnification images of selected cells were taken and ER-PM contact sites were counted and measured in length. All EM reagents were purchased from Electron Microscopy Sciences (Hatfield, PA).
Neuronal culture preparation
All experiments involving mice were performed in accordance with the Yale University Institutional Animal Care and Use Committee. Briefly, primary cultures of hippocampal neurons were dissected from P0-P2 brains. Hippocampal neurons were dissociated by papain treatment and trituration. Cells were counted in the presence of Trypan Blue and seeded at 8.3 × 104 cells/cm2 on poly-D-lysine-coated, glass-bottomed Petri dishes (MatTek). After two hours, the serum-based media was carefully removed and replaced with neuronal growth media [Neurobasal A supplemented with B27 and Glutamax (All reagents from Gibco)] and maintained for at least 21 days in vitro (DIV) at 37°C and 5% CO2.
All cultures were fed every 4 days with 10% cortical enriched media and 15% glial enriched media in freshly made neuronal growth media.
In vitro phosphorylation
In vitro TMEM24 phosphorylation by PKC was performed by incubating various concentrations (0–5 μM) of purified His12-TMEM24(36–706) with 2 units of commercial PKC (Promega) in PKC reaction buffer (200 mM HEPES pH 7.4, 16.7 mM CaCl2, 10 mM DTT, 100 mM MgCl2 and 0.6 mg/ml PS) in presence of 50 μM [γ-32P]-ATP, (1.5 μCi/sample). Samples were incubated at 37°C for 30 min and the reaction stopped by immediately adding Laemmli Stop buffer (57). Phosphorylation of TMEM24 was assessed by autoradiography of the SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel and densitometric analysis of the bands.
Optogenetic depletion of PI(4,5)P2 at the plasma membrane and light-induced recruitment of TMEM24-CRY2-mCherry
5′-dephosphorylation of plasma membrane PI(4,5)P2 by blue-light global illumination was performed as previously described (28, 58). Briefly, COS-7 cells were transfected with CRY2-5-ptaseOCRL, EGFP-CIBN-CAAX, TMEM24-mCherry and the PI(4,5)P2 reporter PH-PLCδ-iRFP. In transfected cells, 20–30 pulses of blue light (488 nm, 200 ms, 4 s apart) induce immediate binding of CRY2 to membrane-associated CIBN, thus bringing the 5-ptase to the plasma membrane. A similar approach was used to recruit TMEM24 to the plasma membrane to assess lipid transfer. Cells were transfected with TMEM24-CRY2-mCherry (or its nonfunctional variant ΔSMP-TMEM24-CRY2- mCHerry), EGFP-CIBN-CAAX, and the PI4P [P4M-iRFP (59)] or PI(4,5)P2 [iRFP- PH-PLCδ (60)] probe. Blue light illumination induced TMEM24 recruitment to the plasma membrane. TIRF microscopy was performed as described above, dimerization was induced by 50×100 ms 488 nm light pulses delivered through the evanescent field. Because of spectral overlap of EGFP with the absorption spectrum of CRY2 (61), EGFP-tagged CIBN expression and localization could only be imaged when dimerization was promoted.
Generation of TMEM24 KO INS1 cells by CRISPR/Cas9
TMEM24 KO INS1 cells were generated according to (56). Briefly, sgRNAs matching the first exon of rat TMEM24 were designed using an online CRISPR Design Tool (http://tools.genomeengineering.org Based on sequence analysis and quality score the oligos GE1 (CACCGCGGGGGTTCATTGCGCGAGC) and GE2 (CACCGTGCTGCGTTTGCGGGCGACC) were selected for the KO generation. After annealing, the oligonucleotides were cloned into the pSpCas9n(BB)-2A-Puro plasmid. Following sequence quality control the resulting vector was transfected into parental WT INS1 cells. After 24-hour transfection, cells were selected by using 1–3 μg/ml puromycin (Sigma) and incubated for a total of 72 hours. Resistant clones were selected and growth in standard medium. These clones were analyzed by PCR-genotyping and immunoblotting using an anti-TMEM24 antibody.
Insulin secretion assay
INS1 cell lines (WT or TMEM24 KO) were plated onto 24-well plates at a density of 800000 cells/well, transfected as described above and grown to 100% confluency before the secretion assay. Standard medium was replaced with fresh medium containing 5 mM glucose 18 hours before experiments. Stimulation of Insulin secretion was performed as described in (62). Briefly, cells were washed in 37°C warm HBSS (114 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.16 mM MgSO4, 20 mM HEPES pH 7.2, 2.5 mM CaCl2, 25.5 mM NaHCO3, and 0.2% bovine serum albumin) containing 3 mM glucose. After 1 hour at 37°C, cells were incubated in either basal HBSS or HBSS with 25 mM glucose (0.8 ml volume) for 1 hour at 37°C. Supernatants were collected and centrifuged at 2000 rpm for 10–15 min at 4°C to remove any cellular debris or floating cells. Cells were independently processed for the analysis of insulin content. Enzyme-linked immunosorbent assay (ELISA) was performed by using a rat insulin ELISA kit (Mecodia) according to manufacturer’s instructions.
Real Time Q-PCR
Total RNA was extracted from HeLa, COS-7, INS1 cells and cortical neuronal by using RNeasy MiniKit (QIAGEN). cDNA was obtained from 2 μg of RNA with the iScriptTM cDNA synthesis kit (Bio-Rad). The resulting DNA was subjected to real-time qPCR with SYBR green fast kit (Bio-Rad) and prevalidated primers (Bio-Rad) according to manufacturer’s instructions. Quantification results were expressed in terms of the cycle threshold (Ct). All real-time qPCR reactions were run in triplicate, and the Ct values were. Data were normalized to the internal controls: the reference gene S26 and HPRT. Differences between the mean Ct values of each gene and those of the reference gene were calculated as ΔCt = Ctgene Ctreference and represented as 2 ΔCt as shown in fig. S1B.
Protein expression and purification
TMEM24 fragment bacterial expression constructs were transformed into BL21(DE3) RIL Codon Plus (Agilent) or C43(DE3) (Lucigen) E. coli cells. LB precultures were diluted into large-scale expression cultures and grown at 37°C to an A600 of 0.6–0.8, then protein expression was induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) at 26°C for 12–18 hours with shaking. Selenomethionine-substituted TMEM24(76–260) was produced as previously described (63). Cells recovered by centrifugation were resuspended in TNT500 buffer (50 mM Tris-Cl, 500 mM NaCl, 0.5 mM TCEP, 1× complete EDTA-free protease inhibitor cocktail (Roche), pH 7.5), then lysed in an Emulsiflex-C5 cell disruptor (Avestin) at a pressure of 10,000–15,000 psi. Lysates were clarified by centrifugation at 27,000×g for 45 min. Soluble TMEM24 protein was bound to Glutathione Sepharose 4 Fast Flow (GE Healthcare) resin and washed with 3 × 10 bed volumes of TNT500 buffer. Protein was cleaved from the resin for 10–15 hours in TNT500 buffer with PreScission protease. Cleaved protein was eluted, concentrated in a 3 kDa NMWCO Amicon centrifugal filtration device, and loaded on a Superdex 200 16/60 column (GE Healthcare). Peak fractions were recovered and concentrated.
His12-TMEM24(36–706) was expressed in Expi293 cells (Invitrogen) according to manufacturer instructions for 72 hours. Cells recovered by centrifugation were resuspended in TNT500 buffer and lysed by four freeze-thaw cycles in liquid nitrogen and at 37°C, respectively. Insoluble material was pelleted at 15,000×g for 40 min. TMEM24 was recovered from the soluble fraction by incubation with Ni-NTA Agarose (Qiagen), followed by extensive washing. Protein was eluted with 200 mM imidazole, concentrated, and fractionated in TNT500 buffer on a Superdex 200 10/30 GL column (GE Healthcare).
Protein crystallization, structure determination, and refinement
Selenomethionine-derivatized TMEM24(76–260) at 3.5 mg/ml was crystallized by vapor diffusion using the sitting drop method in 100–200 mM MgCl2, 100 mM bis-tris propane, pH 6.5–8.0, 45% PEG 200, mixing the protein and well solution in equal volumes (1 μl each). Crystals were recovered in cryo-loops and flash frozen. All data were collected at the Advanced Photon Source (NE-CAT beamline 24-ID-E) at 0.9792 Å to maximize the selenium anomalous signal and data were processed in XDS (64). We used the single-wavelength anomalous dispersion method to obtain de novo phases. Selenium positions were identified using SHELX (65), and phases were calculated and further improved by solvent flattening and noncrystallographic 6-fold averaging, as implemented in Phenix (66). COOT (67) was used for model building. Phases calculated from partial models were iteratively combined with experimental phases to improve the maps in unmodeled regions. The model was refined to 2.96 Å using Phenix (66). Refinement was carried out using least squares, real space, TLS and individual B-factor, and torsion angle annealing options and was alternated with manual rebuilding in COOT. Figures were prepared using PyMol (v 1.7.2.0) software.
Lipid analysis by LC/MS
3xFLAG-TMEM24(76–260) was expressed in Expi293 cells according to the manufacturer’s instructions for 72 hours. Protein was immunoprecipitated, along with bound lipids, using anti-FLAG M2 affinity gel (Sigma), followed by elution with 3xFLAG peptide (Sigma) and size exclusion chromatography. Purified sample was sent to Avanti Polar Lipids, Inc. for analysis, where it was diluted in HPLC-grade water and extracted into 2:1 (v/v) chloroform:methanol. The chloroform layer was dried and reconstituted with an internal standard containing molecular species of PC, PE, PI, PS, PA, PG, and SM for quantitative measurement. The sample was injected onto a 100 × 2.1 mm i.d., 3.5 μm HILIC column under gradient conditions to resolve the phospholipids according to class and polarity. The molecular species for each class were detected by an AB Sciex 4000 QTrap using multiple reaction monitoring for ~250 compounds. These compounds were quantified above a signal to noise of 10:1 and summed by lipid class.
Native TMEM24-lipid complex analysis by LC/MS
3xFLAG-TMEM24(76–260) was expressed in Expi293 cells and purified as above. Purified protein sample was exchanged into 20 mM ammonium acetate pH 6.8 using a Vivaspin 0.5 mL filter. Native mass spectrometry analyses were performed in positive ion mode. The sample was directly injected at a flow of 1 μL/min into a quadrupole time-of-flight (QTOF) 6550 Agilent mass spectrometer equipped with an Agilent 1260 HPLC system through a SilicaTip nanospray emitter (New Objective, USA). The nanoelectrospray voltage was set to 1600 V (Vcap), temperature 280°C, drying gas flow 11 L/min and fragmentor 175 V. The samples were injected in 20 mM ammonium acetate pH 6.8 and spectra acquired at a scan rate of 1 spectrum/sec. Data were collected over a mass range of 700–8000 m/z.
Liposome preparation
Lipid solutions in chloroform were mixed in glass test tubes, evaporated to dryness under N2 gas, then dried 30 min under vacuum. For liposome sedimentation assays, lipid films were rehydrated under 20 mM Tris pH 7.5, 100 mM NaCl, 0.5 mM TCEP at 85°C for 1 hour, then vortexed to generate crude liposomes. Crude liposomes were frozen in liquid nitrogen and thawed at the rehydration temperature 7 times. Liposomes were used immediately or stored at −80°C. For lipid transfer assays, lipid films were rehydrated under 20 mM Tris pH 7.5, 100 mM NaCl (for light liposomes) or 20 mM Tris pH 7.5, 100 mM NaCl, 0.75 M sucrose (for heavy liposomes) at 37°C for 1 hour. Rehydrated lipids were vortexed to generate crude liposomes. Crude liposomes were frozen in liquid nitrogen and thawed at 37°C 5 times, then extruded through 100 nm (light lip.) or 400 nm (heavy lip.) pore size cellulose acetate filters in a mini-extruder (Avestin). Liposomes were clarified by centrifugation at 16,100×g for 15 min. Light liposome supernatants were transferred to a clean tube, while heavy liposome pellets were washed twice, then resuspended, all in sucrose-free buffer. Liposomes were stored at 4°C and used within 48 hours.
Liposome sedimentation assays
100 μg liposomes were incubated with 1 μg protein for 15 min at room temperature, then liposomes were pelleted at 16,100×g for 30 min at 4°C. Supernatants were saved and pellets resuspended in buffer. Equal proportions of supernatants and pellets were analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue. Destained gels were imaged electronically using a GE ImageQuant LAS 4000 and the appropriate bands were quantitated using ImageJ (ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij, 1997–2016) to determine the proportion of liposome-bound protein.
In vitro TMEM24 SMP binding to PI
Purified TMEM24 (76–260) was incubated 48 hours at 4°C with liposomes composed of 100% liver PI at a 10-fold molar excess over protein in 200 μl total reaction volume. Control reactions were prepared by replacing either protein or lipids with buffer. Protein and liposome mixtures were resolved on a Superdex 200 10/300 Increase column (GE Healthcare). Peak fractions were sampled for SDS-PAGE, then pooled. Lipids were acid-extracted as previously described (68). The organic phase was evaporated to dryness under nitrogen gas. Dried lipids were resuspended in 20 μl chloroform:methanol (1:1), spotted on a silica gel 60 TLC plate (EMD) pretreated with 50% methanol/1% potassium oxalate and resolved with freshly prepared mobile phase [water:acetic acid:methanol: acetone:chloroform (7:16:12:15:32)] against a liver PI standard. Plates were stained with iodine vapor and imaged.
Isolation of tritiated PI
COS-7 cells were incubated in inositol-free DMEM supplemented with dialyzed FBS and 20 μCi/ml 3H-myo-inositol (American Radiolabeled Chemicals) for 72 hours at 37°C. Cells were scraped after incubation of plate on ice in 0.6 ml 4.5% perchloric acid for 15 min., then pelleted and washed three times with ice-cold 0.1 M EDTA. Lipids were acid-extracted as described above, then separated by TLC, as above. Tritiated phosphoinositides on the plate were detected by autoradiography. Bands containing 3H-phosphatidylinositol were scraped from the TLC plate into a microcentrifuge tube and PI was acid-extracted as before.
Lipid transfer assays
100 μl of light and heavy liposomes (each at 1 mM final concentration) were incubated with rotation in the presence of 10 μl of 0.1 mg/ml protein or 10 μl of buffer for 15 (scint. assay) or 30 (HPLC assay) minutes, then quenched by the addition of 100 μl stop cocktail (600 mM imidazole, 10 mM EDTA, 0.1 mg/ml elastase, pH 8.0) and incubation for 60 min at room temperature. Heavy and light liposomes were separated by centrifugation at 16,100×g for 15 min at room temperature. Supernatants were reserved, while liposome pellets were washed once, then resuspended in sucrose-free buffer. For radioactive lipids, supernatant and pellet fractions were mixed with EcoScint scintillation fluid and measured by liquid scintillation counting. For unlabeled lipids, equal proportions of supernatant and pellet fractions were acid-extracted as described above. The organic phase was transferred to a clean tube and evaporated to dryness under nitrogen gas. Lipid content of each fraction was analyzed as described below.
Lipid head group analysis and HPLC quantitation
Extracted lipids were resuspended and deacylated in 40% methylamine:water:n-butanol:methanol (36:8:9:47) for 1 hour at 50°C and evaporated to dryness under vacuum. Head groups were dissolved and extracted into ultrapure water against n-butanol:petroleum ether:ethyl formate (20:40:1). The aqueous phase, containing the lipid head groups, was removed and evaporated to dryness under vacuum. Lipid head groups were then dissolved in ultrapure water and filtered through 0.22 μm cellulose acetate Spin-X filters (Corning). Head groups were resolved in a discontinuous KOH gradient on an IonPac AS11-HC (2 × 250 mm) analytical column using a Dionex ICS-3000 ion chromatography system. Each lipid class was separately quantitated in the Chromeleon software suite by integration of the corresponding chromatogram peak areas.
Quantification and statistical analysis
Fluorescent signals were quantified with Fiji Image J (http://fiji.sc/wiki/index.php/Fiji) or Volocity 3D Image Analysis software (Improvision). Immunoblots were analyzed by Fiji ImageJ or Image Studio (Odyssey). Graphs were made using Graph Pad Prism (Graph Pad Software).
Statistical analyses were performed by Graph Pad Prism using Student’s t test for independent samples, one-way (grouped samples) or two-way (two independent variables) ANOVA, followed by Tukey’s multiple comparison post hoc test.
Supplementary Material
Acknowledgments
We thank T. Levine (University College London, London, UK) for valuable scientific discussion. We also thank F. Wilson, L. Lucast, L. Liu, and C. Schauder for superb technical assistance. X-ray data were collected at Northeastern Collaborative Access Team beamline 24-ID-E, funded by National Institute of General Medical Sciences (P41 GM103403), and we are grateful to NE-CAT staff for their assistance. P.D.C. was supported in part by NIH (R37NS036251, DK45735, DA018343, and DK082700); K.M.R. was supported by NIH (R01GM80616); E.W.S. was supported in part by NIH 5T32GM007223-38/39; M.R.W. and F.T. were supported by grants from the National University of Singapore via the Life Sciences Institute (LSI) and the Biochemical Research Council (BMRC-SERC Diag-034). Data and coordinates for the TMEM24 SMP domain have been deposited in the Protein Data Bank (accession number 5TOD).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/355/6326/eaah6171/suppl/DC1
Author Contributions
References
REFERENCES AND NOTES
- 1.Tavassoli S, et al. Plasma membrane: Endoplasmic reticulum contact sites regulate phosphatidylcholine synthesis. EMBO Rep. 2013;14:434–440. doi: 10.1038/embor.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holthuis JCM, Levine TP. Lipid traffic: Floppy drives and a superhighway. Nat Rev Mol Cell Biol. 2005;6:209–220. doi: 10.1038/nrm1591. [DOI] [PubMed] [Google Scholar]
- 3.Stefan CJ, et al. Osh Proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 2011;144:389–401. doi: 10.1016/j.cell.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 4.Stefan CJ, Manford AG, Emr SD. ER-PM connections: Sites of information transfer and inter-organelle communication. Curr Opin Cell Biol. 2013;25:434–442. doi: 10.1016/j.ceb.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saheki Y, et al. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol. 2016;18:504–515. doi: 10.1038/ncb3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung J, et al. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science. 2015;349:428–432. doi: 10.1126/science.aab1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mesmin B, et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 8.Gatta AT, et al. A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. eLife. 2015;4 doi: 10.7554/eLife.07253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser von Filseck J, et al. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science. 2015;349:432–436. doi: 10.1126/science.aab1346. [DOI] [PubMed] [Google Scholar]
- 10.Manford AG, Stefan CJ, Yuan HL, Macgurn JA, Emr SD. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell. 2012;23:1129–1140. doi: 10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Toulmay A, Prinz WA. A conserved membrane-binding domain targets proteins to organelle contact sites. J Cell Sci. 2012;125:49–58. doi: 10.1242/jcs.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alva V, Lupas AN. The TULIP superfamily of eukaryotic lipid-binding proteins as a mediator of lipid sensing and transport. Biochim Biophys Acta. 2016;1861(8 Pt B):913–923. doi: 10.1016/j.bbalip.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Pottekat A, et al. Insulin biosynthetic interaction network component, TMEM24, facilitates insulin reserve pool release. Cell Rep. 2013;4:921–930. doi: 10.1016/j.celrep.2013.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou JC, Min L, Pessin JE. Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm. 2009;80:473–506. doi: 10.1016/S0083-67290800616-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 16.Spurlin BA, Thurmond DC. Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta-cells. Mol Endocrinol. 2006;20:183–193. doi: 10.1210/me.2005-0157. [DOI] [PubMed] [Google Scholar]
- 17.Daniel S, Noda M, Straub SG, Sharp GW. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes. 1999;48:1686–1690. doi: 10.2337/diabetes.48.9.1686. [DOI] [PubMed] [Google Scholar]
- 18.Bratanova-Tochkova TK, et al. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes. 2002;51(suppl 1):S83–S90. doi: 10.2337/diabetes.51.2007.S83. [DOI] [PubMed] [Google Scholar]
- 19.Wuttke A, Sågetorp J, Tengholm A. Distinct plasma-membrane PtdIns(4)P and PtdIns(4,5)P2 dynamics in secretagogue-stimulated beta-cells. J Cell Sci. 2010;123:1492–1502. doi: 10.1242/jcs.060525. [DOI] [PubMed] [Google Scholar]
- 20.Thore S, Wuttke A, Tengholm A. Rapid turnover of phosphatidylinositol-4,5-bisphosphate in insulin-secreting cells mediated by Ca2+ and the ATP-to-ADP ratio. Diabetes. 2007;56:818–826. doi: 10.2337/db06-0843. [DOI] [PubMed] [Google Scholar]
- 21.Martin TF. PI(4,5)P2-binding effector proteins for vesicle exocytosis. Biochim Biophys Acta. 2015;1851:785–793. doi: 10.1016/j.bbalip.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hille B, Dickson EJ, Kruse M, Vivas O, Suh BCC. Phosphoinositides regulate ion channels. Biochim Biophys Acta. 2015;1851:844–856. doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie B, et al. Plasma membrane phosphatidylinositol 4,5-bisphosphate regulates Ca2+-influx and insulin secretion from pancreatic β cells. Cell Chem Biol. 2016;23:816–826. doi: 10.1016/j.chembiol.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Giordano F, et al. PI(4,5)P(2)-dependent and Ca2+-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013;153:1494–1509. doi: 10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schikorski T, Young SM, Jr, Hu Y. Horseradish peroxidase cDNA as a marker for electron microscopy in neurons. J Neurosci Methods. 2007;165:210–215. doi: 10.1016/j.jneumeth.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Caberoy NB, Zhou Y, Alvarado G, Fan X, Li W. Efficient identification of phosphatidylserine-binding proteins by ORF phage display. Biochem Biophys Res Commun. 2009;386:197–201. doi: 10.1016/j.bbrc.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci USA. 2012;109:E2316–E2323. doi: 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy MJ, et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rizo J, Südhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 30.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 33.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toullec D, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 35.Liu J, et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-86749190124-H. [DOI] [PubMed] [Google Scholar]
- 36.Sumi M, et al. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291X9192031-E. [DOI] [PubMed] [Google Scholar]
- 37.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 38.Kameshita I, Ishida A, Fujisawa H. Phosphorylation and activation of Ca2+/calmodulin-dependent protein kinase phosphatase by Ca2+/calmodulin-dependent protein kinase II. FEBS Lett. 1999;456:249–252. doi: 10.1016/S0014-57939900958-8. [DOI] [PubMed] [Google Scholar]
- 39.Kopec KO, Alva V, Lupas AN. Homology of SMP domains to the TULIP superfamily of lipid-binding proteins provides a structural basis for lipid exchange between ER and mitochondria. Bioinformatics. 2010;26:1927–1931. doi: 10.1093/bioinformatics/btq326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schauder CM, et al. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature. 2014;510:552–555. doi: 10.1038/nature13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.AhYoung AP, et al. Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci USA. 2015;112:E3179–E3188. doi: 10.1073/pnas.1422363112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu H, et al. Extended synaptotagmins are Ca2+-dependent lipid transfer proteins at membrane contact sites. Proc Natl Acad Sci USA. 2016;113:4362–4367. doi: 10.1073/pnas.1517259113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maeda K, et al. Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature. 2013;501:257–261. doi: 10.1038/nature12430. [DOI] [PubMed] [Google Scholar]
- 44.Sampaio JL, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci USA. 2011;108:1903–1907. doi: 10.1073/pnas.1019267108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim YJ, Hernandez MLG, Balla T. Inositol lipid regulation of lipid transfer in specialized membrane domains. Trends Cell Biol. 2013;23:270–278. doi: 10.1016/j.tcb.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hay JC, Martin TF. Phosphatidylinositol transfer protein required for ATP-dependent priming of Ca2+-activated secretion. Nature. 1993;366:572–575. doi: 10.1038/366572a0. [DOI] [PubMed] [Google Scholar]
- 47.James DJ, Martin TFJ. CAPS and Munc13: CATCHRs that SNARE Vesicles. Front. Endocrinol (Lausanne) 2013;4(187) doi: 10.3389/fendo.2013.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang L, et al. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab. 2006;3:463–468. doi: 10.1016/j.cmet.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 49.Eberhard DA, Cooper CL, Low MG, Holz RW. Evidence that the inositol phospholipids are necessary for exocytosis. Loss of inositol phospholipids and inhibition of secretion in permeabilized cells caused by a bacterial phospholipase C and removal of ATP. Biochem J. 1990;268:15–25. doi: 10.1042/bj2680015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaspers LD, et al. Hormone-induced calcium oscillations depend on cross-coupling with inositol 1,4,5-trisphosphate oscillations. Cell Reports. 2014;9:1209–1218. doi: 10.1016/j.celrep.2014.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harootunian AT, Kao JP, Paranjape S, Tsien RY. Generation of calcium oscillations in fibroblasts by positive feedback between calcium and IP3. Science. 1991;251:75–78. doi: 10.1126/science.1986413. [DOI] [PubMed] [Google Scholar]
- 52.Kim YJ, Guzman-Hernandez MLL, Wisniewski E, Balla T. Phosphatidylinositol-phosphatidic acid exchange by Nir2 at ER-PM contact sites maintains phosphoinositide signaling competence. Dev Cell. 2015;33:549–561. doi: 10.1016/j.devcel.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang CLL, Liou J. Phosphatidylinositol 4,5-bisphosphate homeostasis regulated by Nir2 and Nir3 Proteins at endoplasmic reticulum-plasma membrane junctions. J Biol Chem. 2015;290:14289–14301. doi: 10.1074/jbc.M114.621375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong R, et al. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell. 2016;166:408–423. doi: 10.1016/j.cell.2016.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 58.Idevall-Hagren O, Decamilli P. Manipulation of plasma membrane phosphoinositides using photoinduced protein-protein interactions. Methods Mol Biol. 2014;1148:109–128. doi: 10.1007/978-1-4939-0470-9_8. [DOI] [PubMed] [Google Scholar]
- 59.Hammond GR, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol. 2014;205:113–126. doi: 10.1083/jcb.201312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/S0960-98229870135-6. [DOI] [PubMed] [Google Scholar]
- 61.Liu H, et al. Photoexcited CRY2 interacts with CIB1 to regulate transcription and floral initiation in Arabidopsis. Science. 2008;322:1535–1539. doi: 10.1126/science.1163927. [DOI] [PubMed] [Google Scholar]
- 62.Hohmeier HE, et al. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 63.Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. doi: 10.1016/S0076-68799776075-0. [DOI] [PubMed] [Google Scholar]
- 64.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 66.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nasuhoglu C, et al. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal Biochem. 2002;301:243–254. doi: 10.1006/abio.2001.5489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.