

Abstract
Purpose of review
To describe the role of D-type cyclins and CDKs 4 and 6 in breast cancer, and to discuss potential biomarkers for sensitivity or resistance to CDK4/6 inhibitors.
Recent findings
A small number of preclinical and clinical studies have explored potential mechanisms of CDK4/6 inhibitor response and resistance in breast cancer. Putative markers of response include ER-positivity, luminal patterns of gene expression, high cyclin D1 levels, and low p16 levels. Possible resistance mechanisms include loss of Rb function, overexpression/amplification of cyclin E, and CDK6 amplification. Most these remain speculative and have not been validated in clinical specimens.
Summary
If early successes with CDK4/6 inhibitors are to be capitalized upon, it is critical that our understanding of CDK4/6 biology in breast cancer extends beyond its current rudimentary state. Only then we will be able to develop rational therapeutic combinations that further enhance the efficacy of these agents.
Keywords: breast cancer, CDK4/6, cyclin, drug resistance, estrogen receptor
INTRODUCTION
In order for a healthy cell to divide, it must pass through each stage of the cell cycle in a sequential and tightly orchestrated fashion. The control of cellular proliferation is governed by a vast array of molecular players, most important of which are the cyclins and their partner kinases, the cyclin-dependent kinases (CDKs). Given the fundamental role of dysregulated cellular proliferation in cancers, it is not surprising, therefore, that the development of drugs that inhibit the cyclin/CDK axes in tumor cells has been the subject of investigation for many years.
The cell cycle machinery is frequently dysregulated in cancer through a variety of mechanisms. For example, certain tumors harbor amplification of genes encoding particular cyclins and CDKs, hence increasing their levels within tumor cells(1). In other cases, genes for endogenous CDK inhibitors are deleted, facilitating unconstrained CDK activity(2). Most commonly, activation of upstream mitogenic pathways (e.g. PI3K-AKT-mTOR or Ras-Raf-MEK-ERK) leads to increased levels of particular cyclins(3). The D-type cyclins and their partner kinases, CDKs 4 and 6, play a particularly important role in breast cancer. In mouse models, mammary tumorigenesis often requires the presence of cyclin D1 and CDK4, and both are also needed for the growth of certain established mammary tumors(4–6). As detailed below, the cyclin D-CDK4/6 complexes are critical regulators of cellular transition through the G1 to the S phase of the cell cycle.
Despite the knowledge that cyclin D-CDK4/6 complexes are very important for breast cancer growth, pharmacologic targeting of CDK4 and 6 has remained an elusive goal until recently. Older generations of CDK inhibitors lacked specificity, potency, or both, and yielded disappointing clinical trial results. Recently, three relatively selective inhibitors of CDKs 4 and 6 have entered clinical development (palbociclib, ribociclib, and abemaciclib)(7). Each of these is a potent inhibitor of CDK4 and 6, and abemaciclib may also possess inhibitory activity against other kinases, including CDK9(8). Early phase clinical trials in breast cancer have shown extremely promising results(9, 10) and as a testament to the activity of these agents, they have progressed rapidly to phase 3 trials. One (palbociclib) has received U.S. Food and Drug Administration (FDA) approval as a treatment for advanced, estrogen receptor-positive (ER-positive) breast cancer at the time of writing(11, 12). Although outside the scope of this article, it is worth noting that these three inhibitors show differential relative potency for inhibition of CDK4 and CDK6, as well as distinct pharmacokinetic and toxicity profiles.
For reasons discussed below, the main development path for CDK4/6 inhibitors in breast cancer has been in the setting of ER-positive breast cancer. Other pharmacologic agents have also been studied in this disease subtype, including inhibitors of mTOR(13) and PI3-Kinase (particularly for tumors harboring PIK3CA mutations)(14–16). Each has shown evidence of efficacy, testament to the multitude of pathways that serve to mediate resistance to standard endocrine therapy. In this review, we focus on the mechanisms behind CDK4/6 inhibitor efficacy, and also discuss potential biomarkers of both response and resistance to these agents.
CELL CYCLE REGULATION: The role of CDKs 4 and 6 in the G1-S transition
The mammalian cell cycle is composed of the interphase during which DNA is replicated and repaired (G1, S and G2) followed by the mitotic phase in which chromosome segregation and cell division occur (G2 and M)(17). In order for a cell to proceed through the cell cycle from G1 into S phase, it must pass through a “restriction point” which is tightly regulated by the retinoblastoma tumor suppressor protein (Rb)(18). In particular, the phosphorylation of Rb is a critical trigger for passage through the restriction point (see Figure 1).
Figure 1.
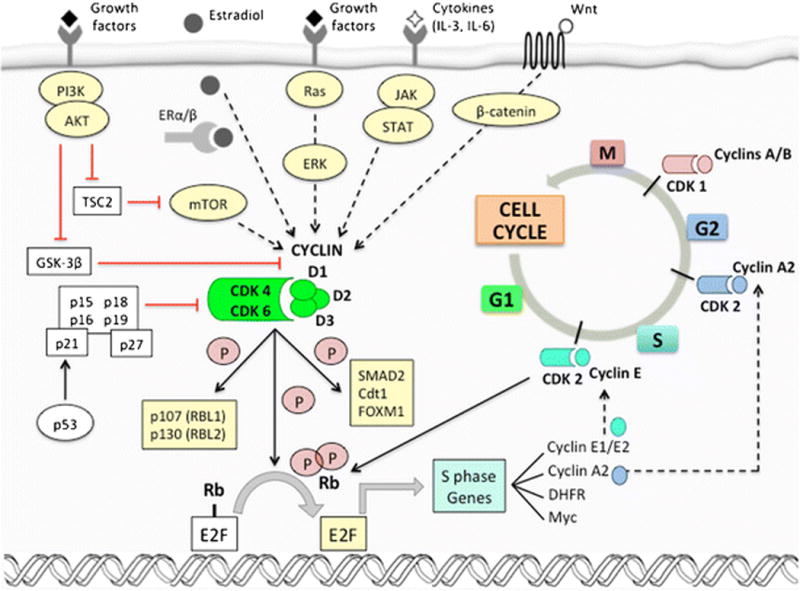
The role of cyclins/cyclin-dependent kinases (CDK) in cell-cycle progression and the crosstalk with oncogenic signaling pathways.
Classically, the first event triggering Rb phosphorylation is a proliferative stimulus. Stimulation can arise after exposure to extracellular mitogens and growth factors, or due to dysregulation of proliferative signaling pathways within cancer cells. Collectively, these elevate intracellular D-type cyclin levels (cyclins D1, D2, and D3). The amount of D-type cyclin within the cellular nucleus is controlled at the levels of transcription, nuclear retention, and protein stability, and all are governed by mitogenic signaling(19, 20). Once present, D-type cyclins preferentially associate with CDK4 and CDK6, and the holoenzyme phosphorylates Rb in addition to other Rb family members known as “pocket proteins” (RBL1, also known as p107, and RBL2, also known as p130). This phosphorylation of Rb partially uncouples it from the E2F transcription factors, in turn enabling the expression of E-type cyclins. CDK2-cyclin E complexes then act to further phosphorylate and completely inactivate Rb and the pocket proteins. This ultimately results in a more complete derepression of E2F transcription factor activity, facilitating transcription of genes promoting transition into S phase(21).
The catalytic function of CDKs 4 and 6 is regulated by several mechanisms(22). Their activation is mainly controlled by binding to cyclins, which show a cyclical pattern of synthesis and degradation. CDK4/6 activation also requires a second step – the phosphorylation of the Thr160 residue of the CDK activation loop by CDK-activating kinase (CAK). The Cdc25A phosphatase also assists in CDK4 activation by removing inhibitory phosphate groups from various tyrosine residues. In addition to decreasing levels of D-type cyclins as cells progress through S phase, endogenous inhibition of CDK4/6 is also enabled by two families of CDK inhibitors: the INK4 family (p16INK4A, p15INK4B, p18INK4C, and p19INK4D) and the Cip/Kip family (p21, p27, and p57). The INK4 family is composed of 15–20 kDa proteins with repeated ankyrin motifs that facilitate binding to CDK4 and CDK6 and inhibit the construction of CDK4/6-cyclin D complexes. Notably, INK4A and INK4D require the presence of functional Rb to induce cell cycle arrest, as demonstrated by the lack of growth arrest observed with overexpression of p16INK4A and p19INK4D in Rb-deficient cells(23). On the other hand, Cip/Kip family members bind to all cell cycle-related CDKs and have more complex positive or negative regulatory functions. Of note, p21 and p27 can bind to cyclin D-CDK4 complexes in G1, stabilizing these complexes. Their sequestration in these complexes, in turn, relieves inhibition cyclin E-CDK2 complexes(19, 22, 24).
THE ROLE OF CYCLIN D-CDK4/6 IN BREAST CANCER
Given the role that D-type cyclins and CDKs 4 and 6 play in regulating cell cycle progression, it is not surprising that aberrant upregulation of their activity is a common feature in cancer(25). Notably, the cyclin D1-CDK4 axis plays a particularly important role in mammary tissue, and in breast cancer. Although mice lacking cyclin D1 are viable and show few organ-specific deficits, they demonstrate specific defects in the development and proliferation of mammary tissue during pregnancy(26). Conversely, transgenic mice engineered to overexpress CCND1 in the mammary glands demonstrate abnormal mammary proliferation and, in some instances, develop mammary adenocarcinoma (27). Although it remains unclear as to why cyclin D1/CDK4 are of particular importance in the mammary epithelium, these observations do suggest that mammary epithelial cells might show particular sensitivity to the inhibition of CDK4/6(28).
Preclinical data further supports the notion that cyclin D1 and CDK4 are critical players in some breast cancers(29). In an elegant study, Yu and colleagues demonstrated that cyclin D1 knockout mice are completely resistant to the formation of breast cancers induced either by the Erbb2 or Ras oncogenes(4). This demonstrates a critical role for cyclin D1 in tumor initiation. Notably, cyclin D1 deficiency did not protect against formation of c-Myc or Wnt-1 driven tumors, suggesting that the role of cyclin D1 in mammary carcinoma is pathway-specific. Indeed, additional analyses in vitro and in vivo demonstrated that Ras and Erbb2 are dependent on cyclin D1 for malignant transformation of mammary epithelial cells and that these oncogenic pathways act through regulatory elements located within CCND1 promoter, as opposed to Wnt-1- and Myc that may be connected to the cell cycle machinery of breast tumors through other targets. A subsequent study confirmed that the role of cyclin D1 in this context is dependent upon its capacity to activate CDK4, as genetic ablation of Cdk4 in Erbb2 models also protected mice from the development of mammary tumors(5).
Transgenic animal studies also show that cyclin D1/CDK4 are important for the maintained growth of already established mammary tumors. Acute and global genetic ablation of Ccnd1 in adult female mice halts progression of Erbb2-driven mammary carcinomas, accompanied by cessation of cell proliferation and induction of a senescent-like phenotype in tumor cells(6). Similar results are seen after pharmacologic inhibition of CDK4/6 activity with palbociclib or abemaciclib(6, 30). Treatment with CDK4/6 inhibitors in these tumors inhibits Rb phosphorylation, reduces expression of E2F target genes, and reduces expression and phosphorylation of the FOXM1 transcription factor, all of which contribute to a senescent-like state in cancer cells(31).
In addition to this empirical evidence, there is a strong theoretical rationale for the importance of the cyclin D-CDK4/6 axis in breast cancer. This rationale primarily relates to the presence of significant crosstalk between the cell cycle machinery and oncogenic signaling pathways in breast cancer (Figure 1). First, approximately 75% of all breast cancers express ER and show estrogen-dependent growth, which is specifically dependent upon cyclin D1. Estrogenic steroids promote cell cycle progression in G1-arrested MCF-7 cells by increasing CCND1 transcription, and CCND1 is indeed a known ER target gene(32). Furthermore, estrogens also induce Cdc25A expression. Finally, cyclin D1 can also directly bind to ERα and induce ER-mediated transcription, even in the absence of estradiol(33).
Second, approximately 15 percent of breast cancers harbor amplification of ERBB2, which encodes the transmembrane growth factor receptor HER2, or activating mutations in PIK3CA (approximately 35 percent), which encodes for the p110-catalytic subunit of PI3-kinase. Heightened activity of the HER2-PI3K-AKT axis increases cyclin D1 levels in cells through a variety of mechanisms: (i) HER2-EGFR heterodimers can trigger MAP-kinase pathway signaling which directly activates CCND1 transcription(19, 34–36); (ii) AKT phosphorylates glycogen synthase kinase-3β (GSK-3β) in a site-specific manner, reducing cyclin D1 phosphorylation and hence preventing its nuclear export and proteosomal degradation(37).
Finally, studies of human breast cancers have revealed genomic aberrations that might be expected to predict increased cyclin D-CDK4/6 activity. Luminal A, luminal B and HER2-enriched tumors often harbor amplification of CCND1 (29%, 58% and 38%, respectively), and/or CDK4 gain (14%, 25% and 24%, respectively) (38). In addition, luminal cancers (particularly luminal A tumors) show high levels of Rb protein and mRNA expression.
BIOMARKERS OF RESPONSE AND RESISTANCE TO CDK4/6 INHIBITORS
Clinical trials of CDK4/6 inhibitors in breast cancer have, in general, shown very encouraging results. In concordance with their mechanism of action, CDK4/6 inhibitors have resulted in prolongation of stable disease in patients with advanced ER-positive breast cancer, both when given as monotherapy or in combination with endocrine therapy(9, 10, 39). Intriguingly, a small proportion of patients with ER-positive breast cancer have also experienced regression of metastatic disease in response to CDK4/6 inhibitor monotherapy(9, 10, 40). Given that ER-positive breast cancer cells have not been shown to undergo apoptosis in response to CDK4/6 inhibition, the mechanisms behind true tumor responses remain elusive.
Larger randomized trials have confirmed that CDK4/6 inhibitors have significant activity in ER-positive disease, as shown in Table 1(41–46). All such trials have restricted study entry to patients with ER-positive disease, based on preclinical evidence that luminal breast cancer cells (largely ER-positive) retain higher levels of Rb and show greater sensitivity to CDK4/6 inhibition(47). Notably, not all patients in these studies derived benefit from CDK4/6 inhibition. Similarly, there is speculation that breast cancers in other clinical subgroups (e.g. HER2-positive or triple-negative cancers) might also be candidates for therapeutic CDK4/6 inhibition in particular circumstances(30, 48). For these reasons, it is critical to identify biomarkers that accurately predict response and resistance to CDK4/6 inhibitors.
Table 1.
Results of phase II/III clinical trials with CDK4/6 inhibitors in advanced breast cancer
| PALBOCICLIB | RIBOCICLIB | ABEMACICLIB | |||
|---|---|---|---|---|---|
| PALOMA-1/TRIO-18 | PALOMA-2 | PALOMA-3 | MONALEESA-2 | MONARCH-1 | |
| Study arms | Palbociclib + Letrozole vs. Letrozole | Palbociclib + Letrozole vs. Placebo + Letrozole | Palbociclib + Fulvestrant vs. Placebo + Fulvestrant (+/− Goserelin) | Ribociclib + Letrozole vs. Placebo + Letrozole | Abemaciclib |
| Trial design | Phase II Open-label | Phase III Double-blind | Phase III Double-blind | Phase III Double-blind | Phase II Single-arm |
| Menopause status inclusion criteria | Postmenopausal | Postmenopausal | All | Postmenopausal | All |
| Prior ET allowed for advanced BC | No | No | Yes | No | Yes |
| Pts, n | 165 | 666 | 521 | 668 | 132 |
| Dose CDK4/6 Inhibitor | 125mg QD 3w on/1w off | 125mg QD 3w on/1w off | 125mg QD 3w on/1w off | 600mg QD 3w on/1w off | 200mg BID continuously |
| Median PFS (experimental arm vs. control arm), mo. | 20.2 vs. 10.2 | 24.8 vs. 14.5 | 9.5 vs. 4.6 | NR vs. 14.7 | 6.0 |
| HR PFS (95% CI) | 0.49 (0.32–0.75) | 0.58 (0.46–0.72) | 0.46 (0.36–0.59) | 0.56 (0.43–0.72) | NA |
| Median OS (experimental arm vs. control arm), mo. | 37.5 vs. 33.3 | NA | NA | NA | 17.7 |
| HR OS (95% CI) | 0.81 (0.49–1.35) | NA | NA | NA | NA |
| ORR (ITT; measurable disease), % | 43 vs. 33; 55 vs. 39 | 42 vs. 35; 55 vs. 44 | 19 vs. 9; 25 vs. 11 | 41 vs. 28; 53 vs. 37 | 20; NA |
| CBR (ITT), % | 81 vs. 58 | 85 vs. 70 | 67 vs. 40 | 80 vs. 73 | 42 |
| Any grade 3–4 AE | 76 vs. 21 | 76 vs. 24 | 69 vs. 18 | 81 vs. 33 | NA |
| Grade 3–4 neutropenia, % | 54 vs. 1 | 67 vs. 1 | 65 vs. 1 | 59 vs. 1 | 27 |
| Grade 3–4 diarrhea, % | 4 vs. 0 | 1 vs. 1 | 0 vs. 0 | 1 vs. 1 | 20 |
ET: endocrine therapy; BC: breast cancer; w: weeks; mo: months; pts: patients; n: number; NA: not available/not applicable; NR: not reached; PFS: progression-free survival; OS: overall survival; HR: hazard ratio; ORR: overall response rate; CBR: clinical benefit rate; ITT: intention-to-treat; AE: adverse event
Biomarkers of Response
Estrogen-receptor positivity. For reasons described above, all randomized clinical trials have explored the use of CDK4/6 inhibitors in patients with ER-positive disease. Although the positive results from these trials do not confirm the utility of ER-positivity as a biomarker for drug efficacy, early phase clinical data does support this notion. For example, in a phase 1 study, abemaciclib monotherapy was administered to 47 patients with metastatic breast cancer, 36 of which were ER-positive(9). All eleven responses were seen in patients with ER-positive tumors. It is worth noting that in an analysis of primary tumor specimens from the randomized PALOMA-2 study (letrozole vs. letrozole plus palbociclib in advanced ER-positive breast cancer), the degree of benefit from palbociclib did not differ by the level of ER-protein expression as measured by immunohistochemistry(49). Similar observations were made in the randomized PALOMA-3 trial that explored fulvestrant in combination with palbociclib in patients with pretreated metastatic disease(43). However, given the biologic rationale for using these agents in ER-positive tumors (ER-driven cyclin D1 expression and higher Rb levels), it is reasonable at this time to state that ER-positivity is a useful clinical marker to identify potential candidates for CDK4/6 inhibitor therapy. Although FDA approval has only been granted to date in the setting of ER-positive disease, it still remains unclear as to whether this is the only subgroup of tumors that derive benefit from these agents.
Luminal pattern of gene expression. In a study determining the effects of palbociclib in a large number of human breast cancer cell lines, Finn et al. noted that almost all sensitive cell lines showed a luminal pattern of gene expression. These cell lines also showed higher levels of RB1 and CCND1 mRNA compared to the resistant group. Interestingly, some of the sensitive cell lines were ER-negative(47). Based on these data, it is appealing to speculate that human breast cancers with a luminal gene expression pattern (for example, as assessed by the PAM50 gene set)(50) might show sensitivity to CDK4/6 inhibition, irrespective of ER status. This hypothesis remains to be tested in clinical trial samples, however.
-
CCND1 amplification and/or loss of p16. Both basic biology and the preclinical data of Finn suggested that tumors with higher levels of cyclin D1 or lower levels of p16INK4a might be more sensitive to CDK4/6 inhibitors(47). These hypotheses have been tested in numerous clinical samples through assessment of gene copy number and protein expression. First, in a phase II study of single-agent palbociclib in 37 patients with Rb-positive breast cancer, neither p16INK4A nuclear expression nor CCND1 amplification were predictive of clinical benefit or prolonged progression-free survival (10).
These findings have been upheld in two randomized cohorts. In the phase II PALOMA-1 study, 165 postmenopausal women with previously untreated ER-positive, HER2-negative advanced breast cancer were randomized to the combination of letrozole and palbociclib or letrozole alone(39). Two sequential cohorts were designed in an attempt to identify biomarkers of response. In the first cohort, only ER-positivity and HER2-negative status were considered for enrolment. In the second, central confirmation of CCND1 amplification and/or p16INK4A loss in the tumor specimen was required. Ultimately, patient selection based on CCND1 amplification or p16INK4A loss, compared to ER/HER2 status alone, did not predict for benefit from palbociclib(39). Similarly, immunohistochemical staining for cyclin D1 and p16INK4A on the PALOMA-2 specimens did not show differential benefit from palbociclib in patients with tumors with different degrees of expression for either of these proteins(49).
Importantly, CCND1 amplification is only one reason for heightened cyclin D1 levels within tumor cells (as discussed above), and immunohistochemistry can be an imprecise measure. Assessment of CCND1 mRNA levels within tumors has not been studied as a predictor of CDK4/6 inhibitor benefit, and might provide more meaningful insights than those we currently have.
Tumor proliferative capacity. In vitro, CDK4/6 inhibition has shown significant activity against breast cancer cell lines with widely varying doubling times, and there is no evidence to suggest that cells with higher or lower proliferation rates show preferential sensitivity to CDK4/6 inhibitors. Consistent with this, primary tumor Ki-67 levels were not predictive of palbociclib benefit in the PALOMA-2 study(49).
Biomarkers of Resistance
-
Loss of Rb function. Given that CDK4/6 inhibitors function primarily by suppressing Rb phosphorylation, it would seem intuitive that tumors lacking functional Rb (and thus showing unconstrained E2F transcription factor activity) are resistant to these agents. Indeed cell line experiments suggest that cells with low Rb mRNA levels are less sensitive to CDK4/6 inhibitors(47). A challenge with validating this hypothesis in clinical samples is determining the appropriate method to measure Rb status, and the appropriate “cut-off” that separates Rb-proficient from Rb-deficient tumors. Immunohistochemistry is convenient, but it is not known whether all Rb detected with this method is functional. Furthermore, results vary considerably between labs. Alternate methods include measurement of gene expression signatures of Rb loss-of-function and genomic sequencing to look for RB1 mutation or gene loss, although the latter may markedly underestimate the frequency of Rb dysfunction.
In the laboratory, chronic loss of Rb has been associated with the development of a CDK4/6-inhibitor resistant state in breast cancer cell lines(51). The same was true in explants derived from human breast tumors(48). Recently, Malorni et al. interrogated data from The Cancer Genome Atlas to develop an 87-gene expression signature of Rb loss (RBsig)(52). The RBsig confirmed the previously established notion that tumors with impaired Rb function are associated with worse prognosis. Importantly, the Rbsig was able to predict with reasonable accuracy the likelihood of breast cancer cell lines being sensitive to palbociclib (high RBsig correlated with relative palbociclib resistance)(52).
Notably, the RBsig scores were highest in luminal A breast cancers and lowest in basal like breast cancers, in keeping with the notion that ER-positive tumors in general retain greater Rb functionality(52). This begs the question of whether the most meaningful and accurate biomarker for CDK4/6 inhibitor-resistance might in fact be loss of Rb function rather than ER-negativity. This is an important distinction, as a subgroup of triple-negative breast cancers also retains Rb expression (e.g. luminal androgen receptor positive tumors)(53), and such tumors have been found in the laboratory to be sensitive to CDK4/6 inhibition(53, 54). In addition, although the majority of ER-positive primary breast cancers show Rb expression, the rate of Rb functional loss in the metastatic setting is unknown.
Hyperactivity of the cyclin E-CDK2 axis. Aside from CDKs 4 and 6, CDK2 is the kinase that also has the capacity to phosphorylate Rb, and the presence of sustained CDK4/6 inhibition, CDK2 can substitute for CDK4/6, providing a mechanism of escape from cell cycle arrest(51). In vitro studies in ER-positive breast cancer cell lines have shown that chronic exposure to palbociclib can lead to sustained expression of CDK2 and/or its binding partner cyclin E. In cell lines, this can be mediated by CCNE1 amplification, which may play an important role in early adaptation and acquired resistance to palbociclib(55). Interestingly, CDK4/6 inhibition can also increase tumor cell AKT phosphorylation(30, 55), which increases cyclin D1 levels. In this state, cyclin D1 is able to bind to and activate CDK2 in a non-canonical manner(55). Of note, inhibition of the PI3K-AKT-mTOR axis is able to prevent this phenomenon, and heightened CDK2 activity is potentially amenable to pharmacological inhibition, with CDK2 inhibitors currently in early phases of clinical development.
Increased CDK6 activity. Although mutations in the kinase domains of CDK4 or 6 have not been reported as mechanisms of resistance to CDK4/6 inhibitors, a recent report describes in vitro evidence of CDK6 amplification in breast cancer cell lines with acquired abemaciclib resistance(56). This amplification was sufficient to promote resistance. It is not known as yet whether this phenomenon is observed in human tumors.
Other genomic aberrations (ESR1, PIK3CA, TP53). Investigators have interrogated clinical trial samples in an attempt to uncover other genomic predictors for resistance to CDK4/6 inhibitors. For example, the PALOMA-3 study was analyzed to determine the predictive role of mutations in the ER gene ESR1, which often occur in the setting of resistance to aromatase inhibitor therapy(57, 58). ESR1 mutations in ctDNA were analyzed in baseline blood samples of 360 patients, and found in approximately 25% of patients. Although an ESR1 mutation was associated with a worse outcome compared to ESR1 wild-type status in the overall population, ESR1 mutant and wild-type patients derived similar PFS benefit with palbociclib (HR 0.43 and 0.49, respectively)(59). Similarly, mutations in PIK3CA in plasma were also not associated with palbociclib treatment effect(43). Finally, it is notable that in the abemaciclib phase 1 study, TP53 mutations were more common in non-responding tumors(9). Given the role of TP53 in mediating cellular senescence, there is a biologic rationale behind this observation, but it awaits confirmation in larger randomized datasets.
CONCLUSIONS
Cell cycle regulation through CDK-targeted drugs has revolutionized the treatment of metastatic breast cancer. Dramatic improvements in clinical outcomes have positioned CDK4/6 inhibitors as a new standard of care for patients with ER-positive disease, in combination with endocrine therapies. This has encouraged widespread interest in exploring the mechanisms of response and failure to these drugs, although many questions remain unanswered.
Moving forward, three priorities clearly exist. The first is to interrogate samples from existing randomized studies in more depth. Ideally, trials in metastatic disease should utilize metastatic tissue when ever possible, given its possible discordance with primary archival tissue. Moreover, a number of biopsy-rich neoadjuvant studies have now been completed, and the tissue from these studies (obtained before and after CDK4/6 inhibitor therapy) is an invaluable resource. Critically, assays performed on clinical samples must be carefully considered before they are performed. For example, consideration must be given to the best way to assess Rb functionality in tumors.
The second priority is to better comprehend the biological mechanisms underlying CDK4/6 inhibitor activity in cancer. It is clear that these effects extend well beyond cell cycle arrest, and include development of a senescent phenotype, changes in tumor cell kinase signaling, and altered metabolism(30, 31, 60). Until these phenomena are better understood, our understanding of response and resistance mechanisms will remain rudimentary.
Third, it is critical that we understand whether CDK4/6 inhibitors show any activity in tumors that have developed “resistance” to these agents. Anecdotal reports from laboratory studies suggest that breast cancers that respond to CDK4/6 inhibitors and subsequently develop resistance might in fact be re-sensitized to these agents after a short drug holiday. This begs the clinical question of whether using CDK4/6 inhibitors beyond progression might be a useful strategy. It is important to understand the biological basis for such observations before large-scale clinical trials, which are expensive and expose patients to drug toxicity, are begun.
Acknowledgments
This work was supported by a Career Development Award provided through the Dana-Farber/Harvard Cancer Center SPORE in Breast Cancer (NIH 2015 P50 CA) to Shom Goel.
Footnotes
Disclosures
Shom Goel has served as a paid scientific advisor to Eli Lilly, and conducts laboratory research funded by Eli Lilly.
Conflict of Interest
Ana C. Garrido-Castro declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Section Editors: Tari King, Elizabeth A. Mittendorf
References
- 1.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18(22):2699–711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer discovery. 2016;6(4):353–67. doi: 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 5.Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer cell. 2006;9(1):23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, et al. The requirement for cyclin D function in tumor maintenance. Cancer cell. 2012;22(4):438–51. doi: 10.1016/j.ccr.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–30. doi: 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- 8.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32(5):825–37. doi: 10.1007/s10637-014-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer discovery. 2016;6(7):740–53. doi: 10.1158/2159-8290.CD-16-0095. [DOI] [PubMed] [Google Scholar]
- 10.DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(5):995–1001. doi: 10.1158/1078-0432.CCR-14-2258. [DOI] [PubMed] [Google Scholar]
- 11.Walker AJ, Wedam S, Amiri-Kordestani L, Bloomquist E, Tang S, Sridhara R, et al. FDA Approval of Palbociclib in Combination with Fulvestrant for the Treatment of Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0493. [DOI] [PubMed] [Google Scholar]
- 12.Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(21):4760–6. doi: 10.1158/1078-0432.CCR-15-1185. [DOI] [PubMed] [Google Scholar]
- 13.Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England journal of medicine. 2012;366(6):520–9. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baselga J, Im S-A, Iwata H, Clemons M, Ito Y, Awada A, et al. PIK3CA Status in Circulating Tumor DNA Predicts Efficacy of Buparlisib Plus Fulvestrant in Postmenopausal Women with Endocrine-resistant HR+/HER2-Advanced Breast Cancer: First results from the randomized, phase III BELLE-2 trial. Cancer Research. 2016;76(4 suppl):S6–01. [Google Scholar]
- 15.Saura C, Sachdev J, Patel MR, Cervantes A, Juric D, Infante JR, et al. Phase Ib study of the PI3K inhibitor taselisib (GDC-0032) in combination with letrozole in patients with hormone receptor-positive advanced breast cancer. Cancer Res. 75(9 suppl):PD5–2. [Google Scholar]
- 16.Dickler M, Saura C, Richards D, Krop I, Cervantes A, Bedard PL, et al. A phase II study of the PI3K inhibitor taselisib (GDC-0032) combined with fulvestrant (F) in patients (pts) with HER2-negative (HER2-), hormone receptor-positive (HR+) advanced breast cancer (BC) J Clin Oncol. 2016;34(suppl) doi: 10.1158/1078-0432.CCR-18-0613. Abstract 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24(17):2909–15. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 18.Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1(2):103–10. [PubMed] [Google Scholar]
- 19.Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121(Pt 23):3853–7. doi: 10.1242/jcs.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220(2):292–6. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23(36):9408–21. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- 22.Donjerkovic D, Scott DW. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. 2000;10(1):1–16. doi: 10.1038/sj.cr.7290031. [DOI] [PubMed] [Google Scholar]
- 23.Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O’Keefe CL, et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8(24):2939–52. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 24.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18(6):1571–83. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nature reviews Cancer. 2001;1(3):222–31. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 26.Sicinski P, Weinberg RA. A specific role for cyclin D1 in mammary gland development. J Mammary Gland Biol Neoplasia. 1997;2(4):335–42. doi: 10.1023/a:1026391128117. [DOI] [PubMed] [Google Scholar]
- 27.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369(6482):669–71. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 28.Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast cancer research: BCR. 2016;18(1):17. doi: 10.1186/s13058-015-0661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutherland RL, Musgrove EA. Cyclin D1 and mammary carcinoma: new insights from transgenic mouse models. Breast cancer research: BCR. 2002;4(1):14–7. doi: 10.1186/bcr411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer cell. 2016;29(3):255–69. doi: 10.1016/j.ccell.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer cell. 2011;20(5):620–34. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foster JS, Henley DC, Bukovsky A, Seth P, Wimalasena J. Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function. Mol Cell Biol. 2001;21(3):794–810. doi: 10.1128/MCB.21.3.794-810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88(3):405–15. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
- 34.Winston JT, Coats SR, Wang YZ, Pledger WJ. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996;12(1):127–34. [PubMed] [Google Scholar]
- 35.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J. 1997;326(Pt 1):61–8. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proceedings of the National Academy of Sciences of the United States of America. 1998;95(3):1091–6. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. The Lancet Oncology. 2015;16(1):25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 40.Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, et al. A Phase I Study of the Cyclin-Dependent Kinase4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. The New England journal of medicine. 2015;373(3):209–19. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 42.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. The New England journal of medicine. 2016;375(18):1738–48. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 43.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. The Lancet Oncology. 2016;17(4):425–39. doi: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- 44.Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon KA, et al., editors. PALOMA-2: Primary results from a Phase 3 trial of Palbociclib plus Letrozole compared with Placebo plus letrozole in postmenopausal women with ER+/HER2-advanced breast cancer. 2016 American Society of Clinical Oncology (ASCO) Annual Meeting; June 3–7, 2016; Chicago, IL (USA) J Clin Oncol. 2016;34(suppl) abstr 507. [Google Scholar]
- 45.Dickler M, Tolaney SM, Rugo HS, Cortes J, Dieras V, Patt DA, et al., editors. MONARCH-1: Results from a phase 2 study of abemaciclib, a CDK4 and CDK6 inhibitor, as monotherapy, in patients with HR+/HER2-breast cancer, after chemotherapy for metastatic disease. 2016 American Society of Clinical Oncology (ASCO) Annual Meeting; June 3–7, 2016; Chicago, IL (USA) J Clin Oncol. 2016;34(suppl) abstr 510. [Google Scholar]
- 46.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and Letrozole in Advanced Breast Cancer. The New England journal of medicine. 2016;375(20):1925–36. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 47.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012;11(14):2756–61. doi: 10.4161/cc.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finn RS, Jiang Y, Rugo H, Moulder SL, Im S, Gelmon KA, et al. Biomarker analyses from the phase 3 PALOMA-2 trial of palbociclib with letrozole compared with placebo plus letrozole in postmenopausal women with ER+/HER2-advanced breast cancer. Annals of Oncology. 2016;27(7):1–36. [Google Scholar]
- 50.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27(8):1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29(28):4018–32. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 52.Malorni L, Piazza S, Ciani Y, Guarducci C, Bonechi M, Biagioni C, et al. A gene expression signature of Retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget. 2016;7(42):68012–22. doi: 10.18632/oncotarget.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011;121(7):2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asghar U, Herrera-Abreu MT, Cutts R, Babina I, Pearson A, Turner NC. Identification of subtypes of triple negative breast cancer (TNBC) that are sensitive to CDK4/6 inhibition. J Clin Oncol. 2015;33(suppl) Abstract 11098. [Google Scholar]
- 55.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer research. 2016;76(8):2301–13. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2016 doi: 10.1038/onc.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clin Cancer Res. 2016;22(5):1130–7. doi: 10.1158/1078-0432.CCR-15-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–67. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2016 doi: 10.1200/JCO.2016.67.3061. [DOI] [PubMed] [Google Scholar]
- 60.Franco J, Balaji U, Freinkman E, Witkiewicz AK, Knudsen ES. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell reports. 2016;14(5):979–90. doi: 10.1016/j.celrep.2015.12.094. [DOI] [PMC free article] [PubMed] [Google Scholar]