

Abstract
Liver fibrosis is a reversible wound-healing process that is protective in the short term, but prolonged fibrotic responses lead to excessive accumulation of extracellular matrix components that suppresses hepatocyte regeneration, resulting in permanent liver damage. Upon liver damage, nonparenchymal cells including immune cells and hepatic stellate cells (HSCs) have crucial roles in the progression and regression of liver fibrosis. Here, we report differential roles of the glucocorticoid receptor (GR), acting in immune cells and HSCs, in liver fibrosis. In the carbon tetrachloride hepatotoxin-induced fibrosis model, both steroidal and nonsteroidal GR ligands suppressed expression of fibrotic genes and decreased extracellular matrix deposition but also inhibited immune cell infiltration and exacerbated liver injury. These counteracting effects of GR ligands were dissociated in mice with conditional GR knockout in immune cells (GRLysM) or HSC (GRhGFAP): the impacts of dexamethasone on immune cell infiltration and liver injury were totally blunted in GRLysM mice, whereas the suppression of fibrotic gene expression was diminished in GRhGFAP mice. The effect of GR activation in HSC was further confirmed in the LX-2 HSC cell line, in which antifibrotic effects were mediated by GR ligand inhibition of Sma and mad-related protein 3 (SMAD3) expression. We conclude that GR has differential roles in immune cells and HSCs to modulate liver injury and liver fibrosis. Specific activation of HSC-GR without alteration of GR activity in immune cells provides a potential therapeutic approach to treatment of hepatic fibrosis.
Hepatic fibrosis is a wound-healing response in which excessive extracellular matrix (ECM) accumulation leads to chronic liver injury (1). It can be caused by various intrinsic and extrinsic stresses such as metabolic abnormalities, chronic inflammation, viral infection, alcohol consumption, vascular complications, and hepatotoxic drugs. After acute injury, limited ECM deposition protects parenchymal cells from further damages and helps them to regenerate. If the primary insult is removed, the transient changes are reversed (1). In contrast, if hepatic injury persists, the balance between pro- and antifibrogenic responses is disrupted and inflammatory signaling is increased. Repetitive hepatic damage results in extensive hepatic fibrosis, which causes necrosis/apoptosis of parenchymal cells and impaired liver regeneration. Finally, substitution of parenchymal cells with permanent scar tissue distorts liver architecture, leading to organ dysfunction (1).
In a fibrotic liver, there is complex cellular cross talk between nonparenchymal cells. Activation of hepatic stellate cells (HSCs) directly stimulate fibrosis progression in liver (2). Under normal conditions, quiescent HSCs act as retinoid (vitamin A) storage cells but they transdifferentiate into myofibroblast-like cells after liver injury. In the injured liver, activated HSCs can deposit large quantities of ECM components and also modulate inflammatory responses through the cross talk with immune cells (3, 4). Diverse immune cells, including endogenous Kupffer cells (KCs) as well as infiltrated monocytes and lymphocytes, can also contribute to liver fibrosis by responding to intracellular components released from damaged hepatocytes (HCs) and secreting a range of cytokines to promote inflammatory responses (5, 6). Immune cells are also important to fibrosis regression and HC regeneration by degrading scarring ECM proteins and enhancing liver progenitor cell proliferation (5, 7). However, how the integrated responses of these specialized cells contribute to control overall liver fibrosis and its molecular mechanisms remain unclear.
Nuclear receptors play many crucial roles in diverse processes including development, immune responses and energy homeostasis (8). Several nuclear receptors, including retinoid X receptor, peroxisome proliferator-activated receptors (PPARs), vitamin D receptor, and farnesoid X-activated receptor, have been reported to modulate hepatic fibrosis in various animal models (2). For example, PPARγ deletion in either immune cells or HSC accelerates inflammatory response and fibrosis progression, whereas PPARγ ligand treatment has antifibrotic effects through a decrease in platelet-derived growth factor-induced HSC proliferation and inhibition of α-smooth muscle actin expression (9, 10). Recently, activated vitamin D receptor was found to inhibit HSC activation and attenuate hepatic fibrosis through inhibitory cross talk with Sma and mad-related protein (SMAD) signaling (11). Thus, nuclear receptors are crucial regulators as well as potential therapeutic targets of liver fibrosis.
We confirmed earlier results indicating that the well-known nuclear receptor glucocorticoid receptor (GR; also known as NR3C1, nuclear receptor subfamily 3, group c, member 1) is highly expressed in nonparenchymal cells in liver (12). GR can be activated by endogenous glucocorticoids and orchestrates many biological roles in the regulation of stress responses, metabolic homeostasis, and inflammatory signaling (13). GR ligands have potent antiinflammatory and immunosuppressive effects that are mostly mediated by transrepression of Nuclear factor kappa-B (NF-κB) and Activator protein 1 (AP1). Thus, many synthetic GR ligands such as prednisolone, budesonide, and dexamethasone (DEX) are widely used to treat of immune-mediated diseases such as inflammatory bowel disease, autoimmune hepatitis and organ transplantation rejection (14). However, GR transactivation by these agonists is associated with deleterious side effects such as hyperglycemia and muscle breakdown. In liver, studies of GR function have focused on HC functions such as gluconeogenesis, fat accumulation, and the circadian clock (15–17), but potential nonparenchymal functions of hepatic GR remain largely unexplored.
We have found that GR activation suppresses fibrotic gene expression, but increases liver injury in carbon tetrachloride (CCl4)-treated animals. Very similar effects were observed with the nonsteroidal GR modulator Compound A (CpdA), which does not transactivate GR but retains NF-κB-mediated transrepression. Knocking out GR either in immune cells or HSC revealed differential effects on liver injury and fibrotic gene expression. Loss of GR in HSC blocked the ability of GR agonists to suppress fibrotic gene expression, whereas loss of GR in immune cells prevented their ability to inhibit immune cell infiltration and to exacerbate liver injury. The inhibitory effect of GR activation on fibrotic gene expression was confirmed in the LX-2 HSC cell line. Together, we conclude that GR activation can differentially modulate liver injury and hepatic fibrosis in immune cell and HSC, raising a possibility of novel therapeutic strategy for treating fibrotic liver disease by specifically targeting HSC.
Materials and Methods
Materials
DEX, DEX 21-phosphate disodium salt (DEX-NaPO4), prednisolone, and CCl4 were purchased from Sigma-Aldrich (D4902, D1159, P6004, and 319961, respectively). Budesonide was provided from Tocris (2671). CpdA was supplied from EMD Millipore (346110). Predesigned nontargeting small interfering RNA (siRNA) siRNA (NT siRNA) and GR-targeting siRNA (siGR) were purchased from Life Technologies (12935–400 and HSS104484, respectively).
Animal studies
The C57BL/6J wild-type mice, GRF/F mice (B6.129), LysM-cre/+ (B6.129) mice, and hGFAP-cre/+ (FVB) mice were obtained from the Jackson Laboratory (000664, 012914, 004781, and 004600). The immune cell-specific GR knockout mice (GRLysM) was generated by crossing GRF/F mice with a transgenic mice having cre recombinase inserted into the lysozyme 2 gene locus (LysM-cre/+). For HSC-specific GR knockout mice (GRhGFAP), GRF/F mice were crossed with hGFAP-cre/+ mice, which is a transgenic mice harboring cre recombinase gene fused to human glial fibrillary acidic protein (GFAP) promoter. In the knockout animal experiments, only male siblings at F3 generation were used to minimize mouse-to-mouse and generation-to-generation variation. To induce hepatic fibrosis, 10% CCl4 dissolved in corn oil were injected ip for 6 weeks (3 times per week, 0.5 mL/kg). For the treatment of DEX, mice received 1-mg/kg DEX by ip injection of an equimolar amount of DEX-NaPO4 (1.32 mg/kg) dissolved in sterile phosphate buffered saline. CpdA in dimethyl sulfoxide was further diluted in PBS and administrated to mice by ip injection (10 mg/kg). All mice were killed at 10 am to 12 pm to avoid circadian changes. Serum alanine transaminase (ALT) level was analyzed by Comparative Pathology Laboratory at Center for Comparative Medicine (Baylor College of Medicine). All animal studies and procedures described in this study were approved by the Institutional Animal Care and Use Committee of the Baylor College of Medicine.
Isolation of HCs, immune cells, and HSCs
Mouse HCs were isolated using a modified 2-step perfusion technique as previously described (18). After low-speed centrifugation, parenchymal cells were purified by 40% of Percoll gradient (P4937; Sigma-Aldrich). Nonparenchymal cell fraction (in suspension) was further separated into KC and HSC fractions using discontinuous Optiprep gradient (1114542; Axis-Shields) according to the manufacturer's protocols (application sheet C33 and C50).
Cell culture and siRNA transfection
The human immortalized stellate cell line LX-2 was maintained in DMEM (11965–092; Life Technologies) supplemented with 10% fetal bovine serum (F4135; Sigma-Aldrich). For transient transfection of siRNA, 25nM NT siRNA or siGR was transfected into LX-2 cells with Lipofectamine 2000 (11668–019; Life Technologies) according to the manufacturer's protocols. After 12 hours, transfected cells were treated with human TGFβ1 (5 ng/mL; 7754-BH-005/CF; R&D Systems) and DEX (2μM) for additional 24 hours.
Real-time quantitative PCR (Q-PCR)
Total RNA was isolated using either Quick-RNA MiniPrep kit (for LX-2 cells, 11–328; Zymo Research) or TRIzol reagent (for liver tissues, 15596; Life Technologies). Then, cDNA was prepared using amfiRivert cDNA synthesis Platinum Master Mix (5600; GenDEPOT). Gene expression level was determined by real-time PCR using LightCycler 480 Real-Time PCR System (Roche) with PerfeCTa SYBR Green FastMix (95072; Quanta Biosciences). Relative mRNA amounts were calculated by comparative Ct method (ΔΔCt method) with the normalization to the GAPDH gene. Primer information used in this study is listed in Supplemental Table 1.
Immunohistochemistry
Fresh liver tissues were fixed in 10% neutral buffered formalin (BDH0502; BDH Chemicals), embedded in paraffin, and sectioned at 5–7 μm. Hematoxylin/eosin staining and Sirius red staining were processed by Comparative Pathology Laboratory at Center for Comparative Medicine (Baylor College of Medicine) using Richard-Allan Scientific Histology Signature Series Stains (Thermo Scientific) and Picro-Sirius red stain kit (American MasterTech). Fibrotic area was quantified using ImageJ software on at least 3–5 nonoverlapped images at a low magnification (×40). GR staining in mouse liver section was performed according to the general guidelines of immunohistochemistry. Briefly, deparaffinized and antigen-retrieved tissue sections were blocked with 10% normal goat serum followed by incubated with 1/50 diluted anti-GR antibody (sc-8992; Santa Cruz Biotechnology, Inc). After binding of secondary antibody conjugated with biotin (BA-1000; Vector Laboratories), GR immunoreactivity was visualized by avidin-biotin complex method using VECTASTAIN Elite avidin-biotin complex reagent (PK-7100; Vector Laboratories) and VECTOR NovaRED Peroxidase (horseradish peroxidase, HRP) Substrate kit (SK-4800; Vector Laboratories). For fluorescent staining, anti-GR antibody (1:20, rabbit; sc-8992) were combined with either macrophage-specific F4/80 antibody (1:250, rat, MCA497; AbD Serotec) or HSC-specific anti-GFAP antibody (1:50, mouse, 3670P; Cell Signaling Technology). Then, Alexa Fluor 594 (antimouse IgG) and Alexa Fluor 488 (antirat IgG or antirabbit IgG) conjugated secondary antibodies (1:250) were applied to visualize GR, F4/80, and Gfap signal under fluorescent microscope (BX-50; Olympus). 4′,6′-Diamidino-2-phenylindole (DAPI) mounting medium (EverBrite Hardset Mounting Medium with DAPI, 23004; Biotium) was used for nuclear counterstain. All fluorescent images were taken at ×400 resolution and cropped. Images of GR expression patterns in human liver were provided by The Human Protein Atlas (available at http://www.proteinatlas.org) (19).
Statistical analysis
Comparison of gene expressions in HCs, immune cells, and HSCs was analyzed by one-way ANOVA followed by Bonferroni's post hoc test. Gene expressions in LX-2 cells were analyzed using the Student's t test. All the animal results are presented as mean ± SEM, and P values were calculated by 2-tailed Student's t test. Data were considered to be statistically significant at P < .05. In some cases, although P value was over 0.05, Cohen's d value was calculated to consider substantive significance. If d > 0.8 (large effect size), actual P values are indicated by with †.
Results
GR is highly expressed in nonparenchymal cells
To identify potential therapeutic targets of liver fibrosis, we compared mRNA expression of 49 murine nuclear receptors in HCs, KCs, and HSCs. Consistent with previous observations (8, 12), GR is among the most highly expressed nuclear receptors in both parenchymal and nonparenchymal cells (Supplemental Figure 1). In immunohistochemical analysis, GR immunoreactivity was observed in both parenchymal and nonparenchymal cells of mouse (Figure 1A) and human liver sections (Figure 1B) with modest enrichment in nonparenchymal cells (Figure 1B, lower panel). Direct analysis showed that GR mRNA expression was higher in immune cell and HSC fractions than in HCs (Figure 1C). Dual staining of GR and cell type specific marker also confirmed that GR is expressed in F4/80-positive macrophages and GFAP-positive HSCs (Figure 1, D and E), suggesting nonparenchymal functions of GR.
Figure 1. GR is highly expressed in KCs and HSC.

A, C57BL6/J wild-type livers were stained with anti-GR antibody (×200). Arrowheads indicate GR-positive signals in nonparenchymal cells. B, The human liver sections stained with 2 kinds of GR antibody (CAB010435 and HPA004248) provided by The Human Protein Atlas (available at http://www.proteinatlas.org). Arrowhead indicate GR-positive signals in nonparenchymal cells. C, Comparison of GR expression level in isolated HCs, KCs and HSCs. Purity of each cell fraction was confirmed by the expression levels of albumin (Alb), macrophage antigen (CD68), and Col1α1. Asterisks denote statistically significant differences (one-way ANOVA; *, P < .05; ***, P < .001). Normal livers were double-stained with (D) anti-GR antibody (red) and F4/80 antibody (green) or (E) anti-GR antibody (red) and anti-GFAP antibody (green). Nuclei were visualized by DAPI staining (blue). Arrowheads indicate colocalization of GR and cell-specific markers. CV, central vein.
GR activation blocks fibrotic gene expressions but increases liver injury
To test the impact of GR activation on fibrosis, we employed the standard CCl4-treated murine model of hepatotoxin-induced hepatitis. CCl4 was ip administrated for 6 weeks, and at the start of the sixth week, mice were coinjected with DEX followed by CCl4 (Supplemental Figure 2A). When we evaluated the histological impact of DEX treatment, perivascular infiltration of small mononuclear cells was diminished (Figure 2, A, upper arrowheads, and B), consistent with the expected antiinflammatory effect of GR activation. We also observed a significant reduction of Sirius red-positive fibrotic area (Figure 2, A lower and C), which was consistent with decreased expression of major fibrotic genes such as type I collagen, α-1 (Col1α1), Col1α2, Tgfβ1, and matrix metalloproteinase 2 (Figure 2E). A recent report indicated that immune cell depletion decreased liver regeneration and aggravated CCl4-induced liver injury (20). In accord with this, we also observed increased liver injury in the DEX-treated mice (Figure 2D), despite the decreased fibrosis.
Figure 2. DEX attenuates CCl4-induced hepatic fibrosis but exacerbates liver injury.
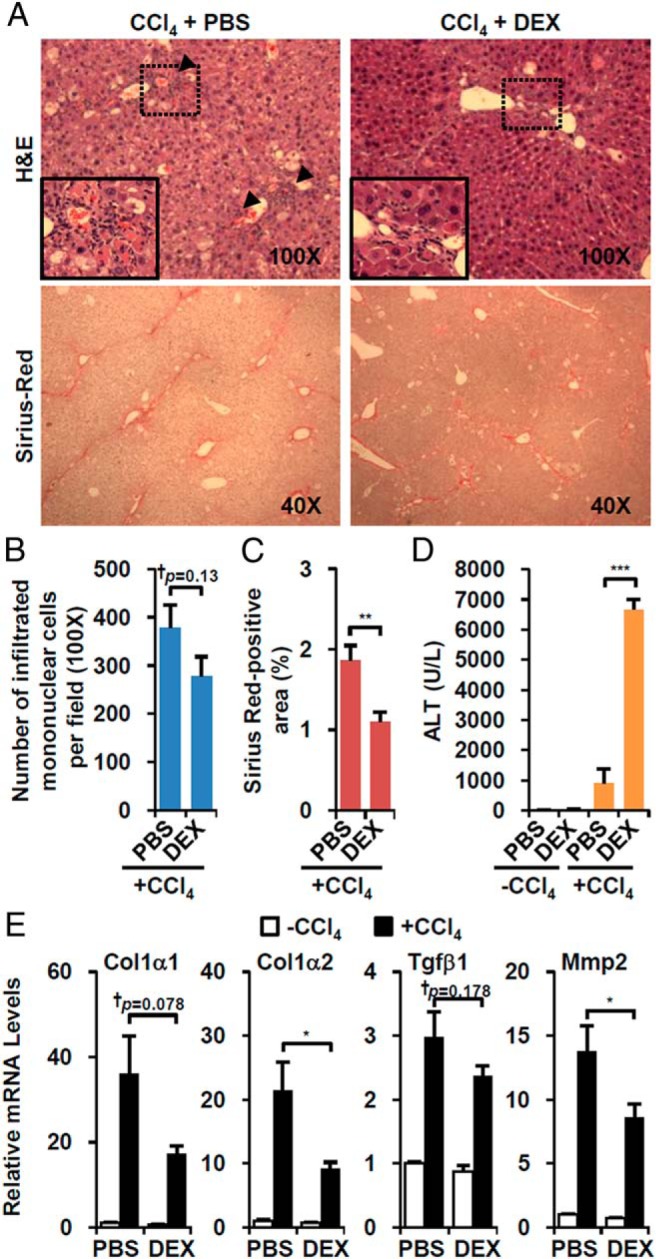
A, Wild-type mice were treated with CCl4 plus vehicle (CCl4+PBS, n = 7) or CCl4 plus water-soluble form of DEX (DEX-NaPO4; CCl4+DEX, n = 7). Liver sections were stained with hematoxylin/eosin (H&E) (×100, upper) and Sirius red (×40, lower). Arrowheads indicate areas of infiltrating mononuclear cells. B, The number of infiltrated cells were counted. C, The size of fibrotic area was quantified. D, Serum ALT level of DEX-treated mice. E, Expressions of fibrotic genes in the presence of CCl4 and/or DEX. Asterisks denote statistically significant differences (Student's t test; *, P < .05; **, P < .01; ***, P < .005; †, Cohen's d > 0.8).
CpdA is a dissociating nonsteroidal GR ligand isolated from the Namibian shrub Salsola tuberculatiformis Botschantzev (21). CpdA favors GR monomer formation and does not induce GRE-mediated transactivation of primary GR target gene expression but retains transrepression of NF-κB-mediated expression of proinflammatory genes (22, 23). Thus, CpdA has decreased side effects relative to classical steroid-based GR ligands and is a useful tool to dissect the molecular mechanisms of GR function. When CpdA was administrated to CCl4-treated mice (Supplemental Figure 2B), it decreased mononuclear immune cell infiltration (Figure 3, A upper and B) and dramatically decreased Sirius red-positive fibrotic area (Figure 3, A lower and C), accompanied by reduced expression of fibrotic genes (Figure 3E). As observed with DEX, CpdA also increased overall liver injury (Figure 3D). These results indicate that NF-κB-mediated transrepression mediated the suppression of overall hepatic fibrosis and also the decreased immune cell infiltration and increased liver injury.
Figure 3. GR modulator, CpdA, has comparable effects with DEX.
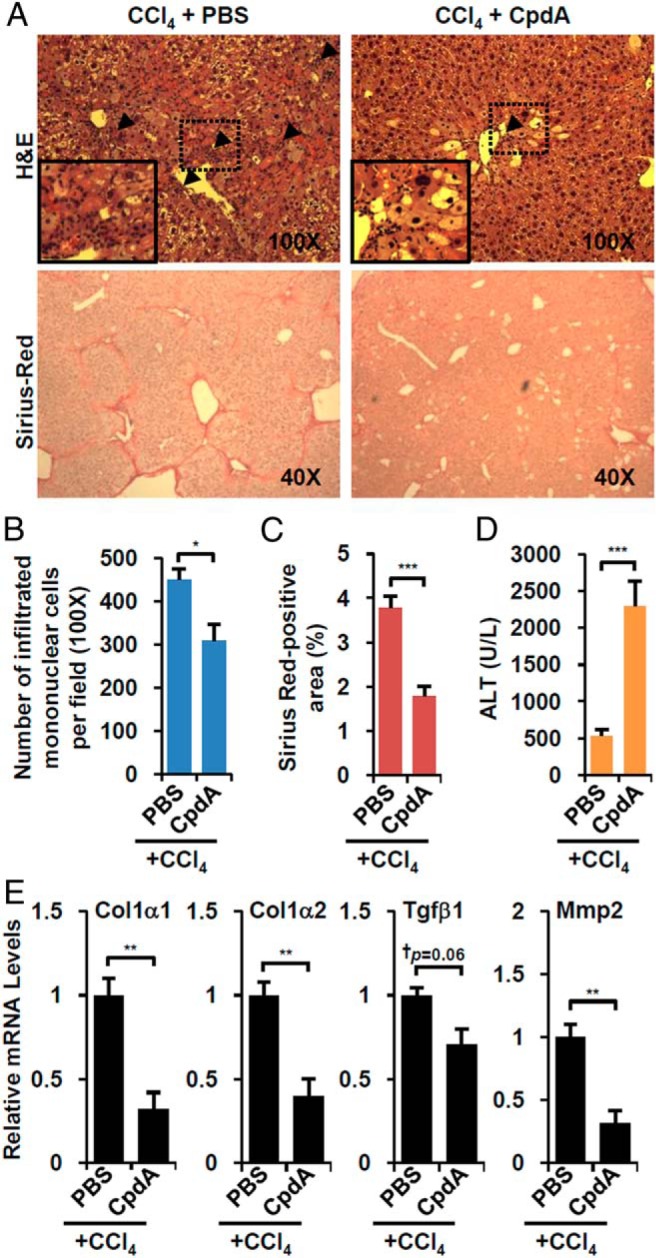
A, Wild-type mice were treated with CCl4 plus vehicle (CCl4+PBS, n = 5) or CCl4 plus CpdA (CCl4+CpdA, n = 5). Liver sections were stained with hematoxylin/eosin (H&E) (×100, upper) and Sirius red (×40, lower). Arrowheads indicate areas of infiltrating mononuclear cells. B, The number of infiltrated cells were counted. C, The percentage of fibrotic area was calculated. D, Serum ALT level of CpdA-treated mice. E, Fibrotic gene expression in CpdA-treated animals. Asterisks denote statistically significant differences (Student's t test; *, P < .05; **, P < .01; ***, P < .005; †, Cohen's d > 0.8).
GR activation in immune cell contributes to liver injury upon CCl4 treatment
To verify the role of GR in immune cell in vivo, we crossed GRF/F control mice with LysM-cre/+ mice (GRLysM) to deplete GR in immune cells. As expected, the combination of CCl4 and DEX treatment (Supplemental Figure 2A) significantly decreased the number of infiltrated mononuclear cells in GRF/F mice, but not in GRLysM mice (Figure 4, A upper and B). Without DEX, the basal level of fibrotic area determined by Sirius red staining was attenuated somewhat in GRLysM but it was further decreased by DEX treatment (Figure 4C), which is generally consistent with decreased expression of fibrotic genes (Figure 4E). Interestingly, the increased CCl4-induced liver injury in response to DEX in GRF/F mice was lost in GRLysM mice (Figure 4D).
Figure 4. Immune cell-specific GR deletion reversed DEX effect on immune cell infiltration and liver injury.

A, CCl4-tretaed immune cell-specific GR knockout mice (GRLysM) and control littermates (GRF/F) were administered with vehicle (PBS, n = 3 per each genotype) or DEX-NaPO4 (DEX, n = 3 per each genotype). Representative liver sections stained with hematoxylin/eosin (H&E) (×100, upper) and Sirius red (×40, lower) are shown. Arrowhead indicates areas of infiltrating mononuclear cells. The number of infiltrated mononuclear cells (B), size of fibrotic area (C), and serum ALT level (D) were analyzed. E, Fibrotic gene expressions were quantified by Q-PCR analysis. Asterisks denote statistically significant differences (Student's t test; *, P < .05; **, P < .01; ***, P < .005; †, Cohen's d > 0.8).
GR in HSC is crucial for the attenuation of fibrotic gene expression by GR ligand treatment
Because GR deletion in immune cells did not affect DEX-induced suppression of fibrotic gene expression (Figure 4E), we hypothesized that it is a direct effect of GR in HSC. To decrease GR function in HSC, we crossed a transgenic line expressing cre recombinase under the control of the human glial fibrillary acidic protein promoter (hGFAP-cre/+) with GRF/F mice (24) to generate GRhGFAP. Although several studies have shown that the hGFAP-cre transgene is expressed in brain (especially glial cells), cre expression was also detected in at least a subset of HSCs (Supplemental Figure 3) (25). In accord with this, GR expression was specifically attenuated in HSCs but not in HCs or KCs (Supplemental Figure 3), confirming partial knockout of GR in GRhGFAP mice (hGFAP-cre/+;GRF/F), which is consistent with previous reports (26, 27).
Without DEX treatment, control littermates (GRF/F) and GRhGFAP mice did not show a significant difference in responses to CCl4. As expected, GRhGFAP did not affect the impact of DEX on mononuclear cell infiltration and liver injury, which was comparably decreased in GRF/F and GRhGFAP livers (Figure 5, A upper, B, and D). However, the decrease of fibrotic area upon DEX treatment was blunted in GRhGFAP mice (Figure 5, A lower and C). In accord with this, DEX-induced suppression of fibrotic gene expression was also blunted in GRhGFAP mice (Figure 5E). Taken together, these results suggest that deletion of GR in HSC reversed the effect of GR activation on fibrotic responses without affecting immune cell infiltration and liver injury, supporting a crucial role for HSC GR in the control of hepatic fibrosis.
Figure 5. HSC-specific GR deletion abolishes DEX effect on fibrotic gene expression.

A, HSC-specific GR knockout mice (GRhGFAP) and control littermates (GRF/F) were injected with CCl4 in combination with vehicle (PBS, n = 3 per each genotype) or DEX-NaPO4 (DEX, n = 3 per each genotype). Representative liver sections stained with hematoxylin/eosin (H&E) (×100, upper) and Sirius red (×40, lower) are shown. Arrowhead areas of infiltrating mononuclear cells. B, Infiltrated mononuclear cells were counted. C, The size of fibrotic area were quantified. D, Serum ALT level was analyzed. E, The changes of fibrotic gene expression were determined by Q-PCR analysis. Asterisks denote statistically significant differences (Student's t test; *, P < .05; **, P < .01; †, Cohen's d > 0.8).
GR activation blunts fibrotic gene expression in HSC
To confirm the effect of GR activation in HSCs, we used LX-2 cells, which express most HSC markers and retain characteristics of primary HSCs (28). We assessed the effects of DEX and other steroidal GR ligands on fibrotic gene expression in the presence or absence of fibrogenic TGF (TGFβ1). TGFβ signaling is a potent stimulator of fibrotic gene expression through the activation of SMAD2/3 (29). As expected, TGFβ treatment significantly induced many fibrotic and inflammatory genes and this was blocked by the DEX (Figure 6A). Other GR agonists such as prednisolone and budesonide had comparable effects (Figure 6A). Like DEX, the nonsteroidal GR modulator CpdA also suppressed fibrotic gene expression in TGFβ-activated LX-2 cells (Figure 6B).
Figure 6. GR ligands suppress fibrotic gene expression in HSC.

LX-2 cells were pretreated with (A) Steroidal GR agonists (DEX; water-soluble DEX, DEX-NaPO4; prednisolone; budesonide; 2μM) and (B) GR modulator (CpdA; 2μM or 4μM) were pretreated. They were then treated with recombinant human TGFβ1 (5 ng/mL) for additional 24 hours, and Q-PCR analysis of fibrotic genes was carried out. Asterisks denote statistically significant differences compared with vehicle-treated sample (Student's t test; *, P < .05; **, P < .01; ***, P < .001). C, LX-2 cells were transfected with NT siRNA and siGR. After 12 hours, DEX (2μM) and TGFβ1 (5 ng/mL) were treated for 24 hours, and the fibrotic gene expression was analyzed by Q-PCR. In the presence of TGFβ1, the relative changes of fibrotic gene expression by DEX were denoted (%). Asterisks denote statistically significant differences compared with vehicle-treated sample (Student's t test; *, P < .05; **, P < .01; ***, P < .001).
We used siGR to confirm that the effects of GR ligands in LX-2 cells are GR dependent. Control NT siRNA had no effect on the impact of DEX on the induction of fibrotic gene expression by TGFβ treatment, as expected However, GR knockdown (siGR), which reduced the GR expression by approximately 70%, strongly attenuated the inhibitory functions of GR on fibrotic gene expression (Figure 6C).
Suppression of SMAD3 expression mediates antifibrotic effect of GR
To determine whether GR activation influences de novo transcription of fibrotic genes, we assessed COL1α1 gene transcription by measuring COL1α1 pre-mRNA using quantitative PCR with of COL1α1 intron primers (30–32). Similar to Figure 6A, COL1α1 pre-mRNA level was strongly blunted by GR ligand treatments (Figure 7A), indicating that GR activation directly suppresses de novo COL1α1 gene transcription.
Figure 7. Inhibition of SMAD3 expression mediates antifibrotic effect of GR.

A, LX-2 cells were pretreated with various GR ligands (2μM) for 2 hours then recombinant human (rh) TGFβ1 (5 ng/mL) was added. After 24 hours, expressions of COL1α1 pre-mRNA level were analyzed by Q-PCR. B, SMAD2 and SMAD3 expressions were determined by Q-PCR. C, LX2 cells were transfected with NT siRNA or siGR. After 12 hours, DEX and TGFβ1 were treated for additional 24 hours, and the SMAD2/3 expression was analyzed by Q-PCR. The relative changes by DEX treatment were denoted (%). Asterisks denote statistically significant differences compared with vehicle-treated sample (Student's t test; *, P < .05; **, P < .01; ***, P < .001). D, Summary of differential roles of GR in immune cell and HSC on liver fibrosis.
Because the GR modulator CpdA mimics the DEX effects in HSC and mouse liver (Figures 3 and 6B), we hypothesized that GR function in HSC is largely mediated by NF-κB transrepression. We analyzed GR cistrome data of mouse liver in a public database (GSM1111748 of GSE45674 at GEO dataset) to assess potential direct GR recruitment to the proximal region of fibrotic genes. In contrast to known target genes of GR (Pepck1 and Gilz), we did not observe recruitment of GR to fibrotic gene loci (Supplemental Figure 4), implying an indirect effect. Interestingly, however, a peak of GR occupancy was found in the promoter region of Smad3 (Supplemental Figure 4, arrowhead), a transcription factor that is an essential mediator of TGFβ signaling (11, 33), mediating profibrotic responses in HSC. In accord with this, we found that SMAD3 expression was markedly suppressed by DEX treatment of LX-2 cells regardless of TGFβ treatment (Figure 7B) and this suppression was abrogated by siRNA-mediated GR knockdown (Figure 7C). These results indicate that direct inhibition of SMAD3 expression by activated GR contributes to the attenuation of fibrotic gene expression in HSCs.
Discussion
We conclude that GR activation has quite different effects in immune cells and HSC in response to liver injury (Figure 7D). Our results are generally consistent with a previous report that GR activation decreases immune cell infiltration and exacerbates liver injury (20). Based on the loss of both responses in GRLysM mice, we conclude that they are direct effects of GR activation in immune cells (Figure 4). In parallel, GR activation is known to decrease fibrotic TGFβ signaling without affecting αSMA expression and differentiated phenotype in cultured HSCs (34–36), which is very similar to our observations (Figures 5 and 6). At the molecular level, we conclude that the inhibitory impact of GR on fibrotic gene expression is likely mediated by NF-κB transrepression (Figures 3 and 6B) and also by repression of the profibrotic transcription factor SMAD3 (Figure 7). Overall, these results suggest that the potential therapeutic utility of GR ligands in fibrosis would be improved by currently available strategies (37–39) that direct target HSC GR.
In injured liver, the combined effects of diverse immune cell subsets, including endogenous KCs and infiltrated monocytes and lymphocytes, modulate hepatic fibrosis (6). To try to clarify the impact of GR in these specific immune cell subtypes, we coinjected DEX with clodronate liposome (Clod), which specifically depletes macrophages, including endogenous KCs (40). Interestingly, Clod did not significantly alter CCl4-driven liver damage in either PBS or DEX-treated animals (Supplemental Figure 5A), suggesting that the ability of DEX to enhance liver damage is mediated by infiltrating immune cell types such as lymphocyte and monocyte rather than endogenous KCs or recruited macrophages. Consistent with a previous report using a different mouse model (41), fibrotic gene expression was partly attenuated by macrophage depletion. The inhibitory effect of DEX on fibrotic gene expression was retained in the CCl4/Clod-treated livers, as expected (Supplemental Figure 5B). However, the CCl4/Clod/DEX-treated mice showed significant toxicity, indicating that these conclusions remain to be clarified in further studies.
As with many other knockout models, the specificities of LysM-cre and hGFAP-cre are not absolute because these are also expressed in other types in a different tissue (24, 42). In particular, although the hGFAP-cre line has been successfully employed for studying lineage tracing of HSCs and HSC function in various animal models, it was originally designed to study astrocyte function in brain (26, 27, 43, 44). Therefore, we cannot rule out a possibility that other tissues (eg, brain) and/or other cell types could be involved in the impact of GR activation on hepatic fibrosis.
To validate the hGFAP-cre knockout strategy, we confirmed that Cre recombinase protein is expressed in a subset of GFAP-positive perisinusoidal and pericentral HSCs, and also in some cholangiocytes (Supplemental Figure 3B). However, our results are not consistent with a recent report that neither hGFAP-cre nor mGFAP-cre efficiently targets HSCs identified by desmin expression, which primarily detects periportal HSCs (3). As shown previously (45), GFAP protein is mainly detected in perisinusoidal/pericentral HSCs, but not in periportal HSCs (Supplemental Figure 6), which is consistent with results in the hGFAP-cre mice (24, 25). Thus, it is likely that the differential expression pattern of desmin and GFAP in distinct periportal and pericentral subsets of HSCs accounts for both the previous negative conclusion and our observation of partial deletion of GR in purified HSC (Supplemental Figure 3C).
The antifibrotic effect of GR is consistent with a similar effect observed in other tissues and cell types, such as skin fibroblasts and bronchial epithelial cells. For example, steroids have been widely used for treating keloid and hypertrophic scars to suppress abnormal deposition of ECM components (46). In lung fibrosis, the antiinflammatory properties of glucocorticoids have been well characterized, but GR also inhibits TGFβ signaling to ameliorate restrictive and obstructive lung disease (47).
A number of clinical trials have attempted to evaluate the efficacy of GR ligands such as prednisone, prednisolone and budesonide for treating hepatic fibrosis/cirrhosis. Some early reports suggested that prednisolone treatment improved short-term survival with better hepatic function in severe alcoholic hepatitis and primary biliary cirrhosis patients (48, 49). In addition, combined treatment with prednisolone and ursodeoxycholic acid showed higher efficacy than ursodeoxycholic acid alone in primary biliary cirrhosis (50). However, the efficacy of GR ligands is controversial and negative results have been obtained in various liver diseases such as viral/alcoholic hepatitis, biliary atresia, and primary biliary cirrhosis (51–56). Mixed results have also been reported on the impact of GR ligands in different animal models of hepatic fibrosis (20, 57–60).
We suggest that these apparently discrepant results are a reflection of the counteracting effects of GR in HSC and immune cells, which could result in quite different overall outcomes in distinct contexts where one or the other predominated. Thus, beneficial antifibrotic effects of GR activation in HSC are accompanied by decreased recruitment of immune cells, as expected from the well-known antiinflammatory effects of GR activation (Figures 2B and 3B), and also increased liver injury (Figures 2D and 3D). These deleterious effects are consistent with a recent report of linking negative consequences of immune cell depletion in injured liver to decreased regeneration and aggravated liver injury (20). Also, DEX conjugated with mannosylated albumin (Dexa(5)-Man(10)-HSA), which specifically targets immune cells, accelerated fibrogenesis through the induction of tissue inhibitor of metalloproteinase 1 (59). Decreased immune cell function may account for the observation that the overall antifibrotic effects of DEX decreased, rather than increased, when we extended the treatment of DEX from 1 to 2 weeks. It is apparent that multiple factors, including the mode-of-action of various GR ligands as well as duration and dose of treatment regimens could influence the overall therapeutic outcome of such treatments.
Collectively, we conclude that activation of GR in immune cells and HSC has quite different, opposing impacts on liver fibrosis. GR activation in immune cell suppresses immune cell infiltration, which is associated with exacerbated liver injury, whereas, in HSC, it inhibits fibrotic gene expressions through NF-κB transrepression and suppression of SMAD3 expression. These contrasting effects can explain why systemic administration of GR ligands can have both positive and negative effects, and suggest that currently available strategies for selective activation of GR in HSC (37–39) may be a novel therapeutic approach for treating hepatic fibrosis.
Acknowledgments
This work was supported by the R. P. Doherty Jr.-Welch Chair in Science Grant Q-0022.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the R. P. Doherty Jr.-Welch Chair in Science Grant Q-0022.
Footnotes
- ALT
- alanine transaminase
- AP1
- activator protein 1
- CCl4
- carbon tetrachloride
- Col1α1
- type I collagen, α-1
- CpdA
- Compound A
- DAPI
- 4′,6-diamidino-2-phenylindole
- DEX
- dexamethasone
- DEX-NaPO4
- DEX 21-phosphate disodium salt
- ECM
- extracellular matrix
- GFAP
- glial fibrillary acidic protein
- GR
- glucocorticoid receptor
- HC
- hepatocyte
- HSC
- hepatic stellate cell
- KC
- Kupffer cell
- NF-kB
- nuclear factor kappa-B
- NT siRNA
- nontargeting siRNA
- PPAR
- peroxisome proliferator-activated receptor
- Q-PCR
- real-time quantitative PCR
- siRNA
- small interfering RNA
- siGR
- GR-targeting siRNA
- SMAD
- Sma and mad-related protein.
References
- 1. Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: concept to treatment. J Hepatol. 2015;62:S15–S24. [DOI] [PubMed] [Google Scholar]
- 2. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Winau F, Hegasy G, Weiskirchen R, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. [DOI] [PubMed] [Google Scholar]
- 5. Wallace K, Burt AD, Wright MC. Liver fibrosis. Biochem J. 2008;411:1–18. [DOI] [PubMed] [Google Scholar]
- 6. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–194. [DOI] [PubMed] [Google Scholar]
- 7. Elsegood CL, Chan CW, Degli-Esposti MA, et al. Kupffer cell-monocyte communication is essential for initiating murine liver progenitor cell-mediated liver regeneration. Hepatology. 2015;62:1272–1284. [DOI] [PubMed] [Google Scholar]
- 8. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor γ modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478. [DOI] [PubMed] [Google Scholar]
- 10. Morán-Salvador E, Titos E, Rius B, et al. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J Hepatol. 2013;59:1045–1053. [DOI] [PubMed] [Google Scholar]
- 11. Ding N, Yu RT, Subramaniam N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raddatz D, Henneken M, Armbrust T, Ramadori G. Subcellular distribution of glucocorticoid receptor in cultured rat and human liver-derived cells and cell lines: influence of dexamethasone. Hepatology. 1996;24:928–933. [DOI] [PubMed] [Google Scholar]
- 13. Patel R, Williams-Dautovich J, Cummins CL. Minireview: new molecular mediators of glucocorticoid receptor activity in metabolic tissues. Mol Endocrinol. 2014;28:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev. 2009;30:830–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Opherk C, Tronche F, Kellendonk C, et al. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–1353. [DOI] [PubMed] [Google Scholar]
- 16. Shteyer E, Liao Y, Muglia LJ, Hruz PW, Rudnick DA. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40:1322–1332. [DOI] [PubMed] [Google Scholar]
- 17. Lamia KA, Papp SJ, Yu RT, et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. [DOI] [PubMed] [Google Scholar]
- 19. Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 20. Kwon HJ, Won YS, Park O, Feng D, Gao B. Opposing effects of prednisolone treatment on T/NKT cell- and hepatotoxin-mediated hepatitis in mice. Hepatology. 2014;59:1094–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Louw A, Swart P, de Kock SS, van der Merwe KJ. Mechanism for the stabilization in vivo of the aziridine precursor –(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride by serum proteins. Biochem Pharmacol. 1997;53:189–197. [DOI] [PubMed] [Google Scholar]
- 22. De Bosscher K, Vanden Berghe W, Beck IM, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci USA. 2005;102:15827–15832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Bosscher K, Beck IM, Dejager L, et al. Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-κB and AP-1. Cell Mol Life Sci. 2014;71:143–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. [DOI] [PubMed] [Google Scholar]
- 25. Yang L, Jung Y, Omenetti A, et al. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26:2104–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kocabayoglu P, Lade A, Lee YA, et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J Hepatol. 2015;63:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu L, Hui AY, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S. TGF-β/Smad signaling in the injured liver. Z Gastroenterol. 2006;44:57–66. [DOI] [PubMed] [Google Scholar]
- 30. Ponzio TA, Yue C, Gainer H. An intron-based real-time PCR method for measuring vasopressin gene transcription. J Neurosci Methods. 2007;164:149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gaidatzis D, Burger L, Stadler MB. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015;33:722–729. [DOI] [PubMed] [Google Scholar]
- 32. Madsen JG, Schmidt SF, Larsen BD, Loft A, Nielsen R, Mandrup S. iRNA-seq: computational method for genome-wide assessment of acute transcriptional regulation from total RNA-seq data. Nucleic Acids Res. 2015;43:e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshida K, Murata M, Yamaguchi T, Matsuzaki K. TGF-β/Smad signaling during hepatic fibro-carcinogenesis (review). Int J Oncol. 2014;45:1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bolkenius U, Hahn D, Gressner AM, Breitkopf K, Dooley S, Wickert L. Glucocorticoids decrease the bioavailability of TGF-β which leads to a reduced TGF-β signaling in hepatic stellate cells. Biochem Biophys Res Commun. 2004;325:1264–1270. [DOI] [PubMed] [Google Scholar]
- 35. Lindert S, Wickert L, Sawitza I, et al. Transdifferentiation-dependent expression of α-SMA in hepatic stellate cells does not involve TGF-β pathways leading to coinduction of collagen type I and thrombospondin-2. Matrix Biol. 2005;24:198–207. [DOI] [PubMed] [Google Scholar]
- 36. Weiner FR, Czaja MJ, Jefferson DM, et al. The effects of dexamethasone on in vitro collagen gene expression. J Biol Chem. 1987;262:6955–6958. [PubMed] [Google Scholar]
- 37. Li F, Li QH, Wang JY, Zhan CY, Xie C, Lu WY. Effects of interferon-γ liposomes targeted to platelet-derived growth factor receptor-β on hepatic fibrosis in rats. J Control Release. 2012;159:261–270. [DOI] [PubMed] [Google Scholar]
- 38. Sato Y, Murase K, Kato J, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26:431–442. [DOI] [PubMed] [Google Scholar]
- 39. van Beuge MM, Prakash J, Lacombe M, et al. Enhanced effectivity of an ALK5-inhibitor after cell-specific delivery to hepatic stellate cells in mice with liver injury. PLoS One. 2013;8:e56442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Biewenga J, van der Ende MB, Krist LF, Borst A, Ghufron M, van Rooijen N. Macrophage depletion in the rat after intraperitoneal administration of liposome-encapsulated clodronate: depletion kinetics and accelerated repopulation of peritoneal and omental macrophages by administration of Freund's adjuvant. Cell Tissue Res. 1995;280:189–196. [DOI] [PubMed] [Google Scholar]
- 41. Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orthgiess J, Gericke M, Immig K, et al. Neurons exhibit Lyz2 promoter activity in vivo: implications for using LysM-Cre mice in myeloid cell research. Eur J Immunol. 2016;46:1529–1532. [DOI] [PubMed] [Google Scholar]
- 43. Lujambio A, Akkari L, Simon J, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mochizuki A, Pace A, Rockwell CE, et al. Hepatic stellate cells orchestrate clearance of necrotic cells in a hypoxia-inducible factor-1α-dependent manner by modulating macrophage phenotype in mice. J Immunol. 2014;192:3847–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niki T, De Bleser PJ, Xu G, Van Den Berg K, Wisse E, Geerts A. Comparison of glial fibrillary acidic protein and desmin staining in normal and CCl4-induced fibrotic rat livers. Hepatology. 1996;23:1538–1545. [DOI] [PubMed] [Google Scholar]
- 46. Roques C, Téot L. The use of corticosteroids to treat keloids: a review. Int J Low Extrem Wounds. 2008;7:137–145. [DOI] [PubMed] [Google Scholar]
- 47. Schwartze JT, Becker S, Sakkas E, et al. Glucocorticoids recruit Tgfbr3 and Smad1 to shift transforming growth factor-β signaling from the Tgfbr1/Smad2/3 axis to the Acvrl1/Smad1 axis in lung fibroblasts. J Biol Chem. 2014;289:3262–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mitchison HC, Palmer JM, Bassendine MF, Watson AJ, Record CO, James OF. A controlled trial of prednisolone treatment in primary biliary cirrhosis. Three-year results. J Hepatol. 1992;15:336–344. [DOI] [PubMed] [Google Scholar]
- 49. Ramond MJ, Poynard T, Rueff B, et al. A randomized trial of prednisolone in patients with severe alcoholic hepatitis. N Engl J Med. 1992;326:507–512. [DOI] [PubMed] [Google Scholar]
- 50. Leuschner M, Maier KP, Schlichting J, et al. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double-blind trial. Gastroenterology. 1999;117:918–925. [DOI] [PubMed] [Google Scholar]
- 51. Porter HP, Simon FR, Pope CE 2nd, Volwiler W, Fenster LF. Corticosteroid therapy in severe alcoholic hepatitis. A double-blind drug trial. N Engl J Med. 1971;284:1350–1355. [DOI] [PubMed] [Google Scholar]
- 52. The Copenhagen Study Group for Liver Diseases. Sex, ascites and alcoholism in survival of patients with cirrhosis. Effect of prednisone. N Engl J Med. 1974;291:271–273. [DOI] [PubMed] [Google Scholar]
- 53. Gregory PB, Knauer CM, Kempson RL, Miller R. Steroid therapy in severe viral hepatitis. A double-blind, randomized trial of methyl-prednisolone versus placebo. N Engl J Med. 1976;294:681–687. [DOI] [PubMed] [Google Scholar]
- 54. Prince M, Christensen E, Gluud C. Glucocorticosteroids for primary biliary cirrhosis. Cochrane Database Syst Rev. 2005;CD003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sokol RJ. Corticosteroid treatment in biliary atresia: tonic or toast? Hepatology. 2007;46:1675–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bezerra JA, Spino C, Magee JC, et al. Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA. 2014;311:1750–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin JC, Peng YJ, Wang SY, Young TH, Salter DM, Lee HS. Role of the sympathetic nervous system in carbon tetrachloride-induced hepatotoxicity and systemic inflammation. PLoS One. 2015;10:e0121365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Swain MG, Appleyard C, Wallace J, Wong H, Le T. Endogenous glucocorticoids released during acute toxic liver injury enhance hepatic IL-10 synthesis and release. Am J Physiol. 1999;276:G199–G205. [DOI] [PubMed] [Google Scholar]
- 59. Melgert BN, Olinga P, Van Der Laan JM, et al. Targeting dexamethasone to Kupffer cells: effects on liver inflammation and fibrosis in rats. Hepatology. 2001;34:719–728. [DOI] [PubMed] [Google Scholar]
- 60. Eken H, Ozturk H, Ozturk H, Buyukbayram H. Dose-related effects of dexamethasone on liver damage due to bile duct ligation in rats. World J Gastroenterol. 2006;12:5379–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]