

Abstract
Isoforms of flavin-containing monooxygenase (FMO) are involved in xenobiotic metabolism but have also been implicated in the regulation of glucose and lipid homeostasis and in the development of atherosclerosis. However, we have recently shown that improved insulin action is associated with increased FMO expression in livers of protein kinase C-deficient mice. Here, we investigated whether FMO3 expression affected insulin signaling, glucose metabolism, and endoplasmic reticulum (ER) stress in hepatocytes. HepG2 and IHH hepatocytes were transfected with FMO3 cDNA for overexpression, or small interfering RNA for knockdown. Cells were treated with palmitate to induce insulin resistance and insulin signaling, phosphoenolpyruvate carboxykinase (PEPCK) gene expression and ER stress markers were examined by immunoblotting and RT-PCR. Glycogen synthesis was measured using [14C]glucose. Palmitate treatment reduced insulin signaling at the level of Akt phosphorylation and glycogen synthesis, which were little affected by FMO3 overexpression. However, the fatty acid also increased the levels of several ER stress markers and activation of caspase 3, which were counteracted by FMO3 overexpression and exacerbated by FMO3 knockdown. Although FMO3 expression did not reverse lipid effects on protein thiol redox in hepatocytes, it did prevent up-regulation of the gluconeogenic enzyme PEPCK by pharmacological ER stress inducers or by palmitate. ER stress and PEPCK levels were also reduced in livers of fat-fed protein kinase Cδ-deficient mice. Our data indicate that FMO3 can contribute to the regulation of glucose metabolism in the liver by reducing lipid-induced ER stress and the expression of PEPCK, independently of insulin signal transduction.
The development of insulin resistance is strongly correlated to fat oversupply and lipid accumulation in insulin target tissues such as liver (1). This can involve the actions of one or more isoforms of the lipid-activated protein kinase C (PKC) family, which are proposed to disrupt insulin signal transduction through serine phosphorylation of insulin receptor substrate-1 (IRS-1) (2). We and others have previously shown that both PKCδ and PKCϵ, which are activated by the fatty acid intermediate diacylglycerol, play an inhibitory role in glucose homeostasis. Ablation of either isoform resulted in improvement of glucose tolerance and insulin sensitivity in mice fed a high-fat diet (HFD) (3–6). However, the molecular mechanisms underlying the beneficial effects of PKC deletion may not be limited to effects on proximal insulin signaling. For example, PKCδ deletion has been shown to promote a reduction in hepatic lipid accumulation which may also promote insulin sensitivity (4, 6).
To examine additional mechanisms, we recently conducted quantitative proteomics analyses of the livers from fat-fed wild-type (WT) and PKCδ and PKCϵ knockout (KO) mice, to compare protein expression profiles and to identify novel PKC-dependent proteins (7). We showed that certain flavin-containing monooxygenase (FMO) isoforms were up-regulated in livers of fat-fed PKC-deficient mice. PKCδ deletion resulted in up-regulation of FMO3, whereas deletion of PKCϵ caused up-regulation of FMO1 and FMO5. This suggested that FMOs may play a role in the protection of glucose intolerance observed in both lines of PKC KO mice.
FMO isoforms have been well characterized in terms of their ability to metabolize a broad range of xenobiotics and therapeutic agents in detoxification processes (8). There are 5 functional human and mouse FMO proteins (FMO1–FMO5), FMO3 being the best-characterized and most common isoform in adult human liver (9). FMOs are located in the endoplasmic reticulum (ER), and their role in the oxygenation of endogenous nucleophilic compounds such as cysteine derivatives has now also been highlighted (10). Furthermore, FMO in yeast is involved in the unfolded protein response (UPR) (11), which is linked to ER stress. Indeed, FMO3 activity generates trimethylamine N-oxide (TMAO) (8), an cellular osmolyte that is known for its chemical chaperone properties (12). Overall, this is consistent with a potential beneficial role for FMO isoforms in regulating hepatic insulin sensitivity through the amelioration of lipid-induced ER stress. ER stress has been implicated in obesity-associated insulin resistance in animals and humans (13, 14).
ER stress results from insults, including lipid oversupply, that cause accumulation of misfolded protein in the ER lumen. At first, the UPR is initiated to relieve ER stress, through dissociation of an ER chaperone, binding immunoglobulin protein (BiP) (GRP-78/HSPA5), from specific ER transmembrane proteins (15). Downstream events include phosphorylation of eukaryotic translation initiation factor 2 α-subunit (eIF2α), production of activating transcription factor 4 (ATF4), and splicing of X-box-binding protein 1 (XBP-1s) (16). This results in both a reduction in the protein load of the ER and up-regulation of chaperones and isomerases to improve protein folding. Unresolved ER stress results in the activation of apoptotic pathways, involving the up-regulation of CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) (17), cleavage of caspase 3, and phosphorylation of Jun N-terminal kinase (JNK) (18). Several of these events lead to dysregulation of insulin action at the levels of glucose and lipid homeostasis, through further effects of JNK, CHOP, and other transcription factors (14).
Although a role for FMO activity in the modulation of ER stress in mammalian cells remains to be determined, recent studies have suggested that FMO3 and its product TMAO have a negative impact, promoting hyperglycaemia, hyperlipidemia, and atherosclerosis (19–22). It is therefore important to clarify its effects on liver ER function.
Here, we investigated the role of FMO3 in the modulation of hepatic insulin signaling, ER stress, and glucose metabolism. By overexpression or knockdown of FMO3, we showed that FMO3 does not significantly modulate insulin signal transduction, but identified a novel function in the amelioration of lipid-induced ER stress and gluconeogenesis in both human hepatocytes and mouse liver.
Materials and Methods
Cell treatments
Hepatocytes were cultured in MEM (HepG2 and Huh-7 cells) or DMEM-F12 (IHH cells) supplemented with 10% fetal bovine serum, 100-U/mL penicillin and 100-μg/mL streptomycin, at 37°C, 5% CO2. HepG2 cells and IHH cells in 12-well plates were transfected with 1 μg/well mouse FMO3 cDNA in pENTR223.1 (MGC173248; Life Technologies), using Lipofectamine LTX with Plus Reagent (Invitrogen) and NanoJuice transfection reagent (Novagen), respectively, according to the manufacturer's instructions. IHH cells were transfected with NanoJuice Core Transfection Reagent:NanoJuice Transfection Booster:plasmid DNA (2:1:1). For FMO3 knockdown studies, IHH cells were transfected with 50nM ON-TARGETplus SMARTpool Fmo3 or control small interfering RNA (siRNA) (Dharmacon) using DharmaFECT4 according to the manufacturer's instructions. Treatments were commenced 24 hours after transfection. For lipid treatment, transfected cells were incubated in the presence of either BSA or BSA coupled to 0.75 mmol/L palmitate for 18 hours (23). For ER stress induction, cells were incubated with thapsigargin (Tg) (50nM), tunicamycin (Tm) (500 ng/mL), or dimethylsulfoxide vehicle alone (0.1%) for 16 hours. Where stated, cells were stimulated with 100 nmol/L insulin for 10 minutes for investigation of insulin signaling or 1 hour for measurement of glycogen synthesis. The experimental design is summarized in Supplemental Figure 1. After treatments, cells were washed twice with ice-cold PBS and extracts subjected to immunoblotting or real time RT-PCR as described below.
Animals
Ethical approval for mouse studies was granted by the Garvan Institute/St Vincent's Hospital Animal Ethics Committee. Mice were maintained on a hybrid 129/Sv x C57BL/6 background using either PKCδ or PKCϵ heterozygous breeding pairs. Age-matched male WT and PKC KO littermates were used for experiments and were fed either a chow or a HFD for 1 week as described previously (4, 5). Mice were killed after a 6 hour fast and livers rapidly removed and frozen. Samples were subjected to immunoblotting and RT-PCR (5).
RNA isolation and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from hepatocytes and mouse liver using the RNeasy minikit, and cDNA was synthesized using the QuantiTect Reverse Transcription kit (QIAGEN). qRT-PCR was performed with Power SYBR Green PCR Master Mix and gene-specific primers (Table 1) on a 7900HT Real-Time PCR System (Applied Biosystems). Results were normalized to 40S ribosomal protein S9 for hepatocyte extracts and cyclophilin A for liver extracts and quantification carried out using the ΔΔCt method.
Table 1.
Sequences of Oligonucleotide Primers Used for qRT-PCR
| Gene Symbol | 5′ Oligonucleotide | 3′ Oligonucleotide |
|---|---|---|
| Human | ||
| ATF4 | ATCCAGCAAAGCCCCACAAC | CAAGCCATCATCCATAGCCG |
| DDIT3 (Chop) | TTCACTACTCTTGACCCTGCGTC | CACTGACCACTCTGTTTCCGTTTC |
| EDEM1 | GCAATGAAGGAGAAGGAGACCC | TAGAAGGCGTGTAGGCAGATGG |
| FKBP11 | ACACGCTCCACATACACTACACGG | ATGACTGCTCTTCGCTTCTCTCCC |
| FMO1 | ACCTGGCGGAAAAGGTGT | CATGTTCTGAAAGCGTGTCAT |
| FMO3 | TGGCCCTTGTAGTCCCTACCAG | GGCTTCTGAAGTCTCCCGACC |
| FMO5 | GGCCTGAAGCCTAAACACAG | ACGATTTGGCAGGTCATCAT |
| PEPCK | TGCATGAAAGGTCGCACCA | CACAGAATGGAGGCATTTGACA |
| PHLDA1 | CCAGGACAGATGCTACTTGG | GACTACATAACCTAGCAGTGG |
| HSPA5 (BiP) | AGGACAAGAAGGAGGATGTGGG | ACCGAAGGGTCATTCCAAGTG |
| PDIA4 (Erp72) | AGTCAAGGTGGTGGTGGGAAAG | TGGGAGCAAAATAGATGGTAGGG |
| RPS9 | ATCCCGTCCTTCATTGTCCG | TGGCATTCTTCCTCTTCACGC |
| Mouse | ||
| FMO1 | TGTCTCTGGACAGTGGGAAGT | CATTCCAACTACAAGGACTCG |
| FMO3 | CACCACTGAAAAGCACGGTA | GTTTAAAGGCACCAAACCATAG |
| FMO5 | ATCACACGGATGCTCACCTG | GCTTGCCTACACGGTTCAAG |
| PEPCK | CAGCCAGTGCCCCATTATTG | AGGTATTTGCCGAAGTTGTAGCC |
Aliases of gene symbols given in parentheses.
Promoter reporter assays
HepG2 cells were transfected as above with an dual reporter clone expressing secreted Gaussia Luciferase under control of the FMO3 promoter and secreted alkaline phosphatase as internal control (pEZX-PG04; GeneCopoeia). After 24 hours, cells were treated with vehicle (0.1% dimethyl sulfoxide) or 100nM 12-O-tetradecanoylphorbol-13-acetate (TPA), in the presence or absence of palmitate as above. After a further 24 hours, media were assayed for Gaussia Luciferase expression, which was corrected for secreted alkaline phosphatase activity (24).
Immunoblotting
Cells were extracted in 50 mmol/L Tris (pH 7.5), 150 mM NaCl, 1% nonidet-P40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 10 mM sodium pyrophosphate and Complete Protease Inhibitor Cocktail (Roche), and centrifuged for 10 minutes at 17 000g. Supernatant samples of equal protein concentration were resolved by SDS-PAGE and subjected to immunoblotting as previously (7). To detect endogenous FMO3 in hepatocytes, protein extracts were concentrated by acetone precipitation (25). Primary antibodies were from Proteintech Group (59001-1-1g, FMO3, detected overexpressed protein only), Abcam (Ab126711, FMO3, detected endogenous protein), Sigma-Aldrich (I12658, phospho-Tyr612-IRS-1); Cell Signaling Technology (# 3194, total IRS-1; # 2966, total Akt; # 9271, phospho-Ser473-Akt; # 9252, total Jnk; # 9251, phospho-Thr183, Tyr185-Jnk; # 9721, phospho-Ser51-eIF2α; and # 9664, cleaved caspase 3), or Santa Cruz Biotechnology, Inc (sc-7160, XBP-1s; sc-11386, total eIF2α; sc-575, CHOP; and sc-1657, pan 14-3-3).
Glycogen synthesis
HepG2 cells in 6-well plates were incubated for 1 hour in MEM supplemented with 25mM HEPES containing D-[U-14C]glucose (2 μCi/mL) (PerkinElmer). Glycogen production was determined as previously (23).
Cell viability
Cells were treated as above and subjected to 2H-tetrazolium, 2-(4,5-dimethyl-2-thiazolyl)-3,5-diphenyl bromide cell viability assay (M6494; Life Technologies) according to the manufacturer's instructions.
ER redox potential
For technical reasons HEK293 cells were more amenable to fluorescence lifetime imaging employed here and were cultured and transfected as described for HepG2 cells. Cells were transfected with 1-μg FMO3 cDNA or empty vector and 0.2-μg ERroGFPiE cDNA, an ER-tuned fluorescent redox-responsive probe, in pCDNA3.1 (26). After 24 hours, cells were incubated for 48 hours in the absence or presence of palmitate as above. Fluorescence lifetime imaging microscopy was performed using a 63x NA 1.2 HC PL APO CS2 motCorr water immersion objective on a DMI6000 SP8 confocal microscope equipped with HyD detectors in the nondescanned (NDD/RLD) position (Leica Microsystems GmbH). The excitation source was a Chameleon Vision II Ti:Sapphire femtosecond pulsed laser (80 MHz) tuned to 890nm (Coherent) and Chroma dichroic (495lpxt) and bandpass (525/50) filters separated the required emission. Fluorescence excited state was measured via the NDD HyD detector in counting mode using PicoHarp300 time correlated single photon counting equipment and the data were analyzed, including instrument response function and dual exponential fitting, with Symphotime software (both PicoQuant GmbH). 12–15 individual cells, carefully maintained at 37°C with CO2 to ensure media buffering and expressing only low levels of ERroGFPiE, were analyzed for each condition in each experiment using a cell defined region of interest from a typically approximately 37 × 37-μm field of 448 × 448 pixels at 200 MHz (2.78-μs dwell time) with count rate typically approximately 500 kCounts/s until a max of 250 photons at the brightest pixel were acquired.
Reactive oxygen species (ROS)
Lipid-treated cells were incubated with 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (Invitrogen), and ROS determined according to the manufacturer's instructions. Control cells were transfected with empty vector rather than enhanced green fluorescent protein (eGFP) cDNA to avoid assay interference.
Statistical analysis
Data are presented as mean ± SEM. Multiple comparisons were made using two-way ANOVA. Single comparisons were made using a 2-tailed Student's t test. Analyses were performed using GraphPad Prism 5 (GraphPad Software) or STATA/SE9.2 (STATA) software. Differences were considered significant at P < .05.
Results
Palmitate reduces expression levels of FMO isoforms in liver cells
Previously, we showed that protein expression levels of FMO1, FMO3, and FMO5 were up-regulated 1.3- to 2-fold in livers from fat-fed PKCδ- and PKCϵ-deficient mice compared with WT mice, in association with protection against glucose intolerance caused by the diet (7). We now investigated the effect of lipid overload on FMO mRNA expression in cultured hepatocytes. We treated human hepatocellular carcinoma cell lines, HepG2 and Huh-7, as well as the immortalized human hepatocyte IHH cell line, with the saturated fatty acid palmitate. In all 3 liver cell lines, palmitate treatment induced a 40%–70% reduction in the endogenous mRNA levels of FMO1, FMO3, and FMO5 isoforms (Figure 1, A–C). In agreement, palmitate treatment also reduced FMO3 protein in IHH cells (Figure 1D).
Figure 1. Effect of palmitate on the expression of FMO isoform mRNA and in human hepatocyte cell lines.
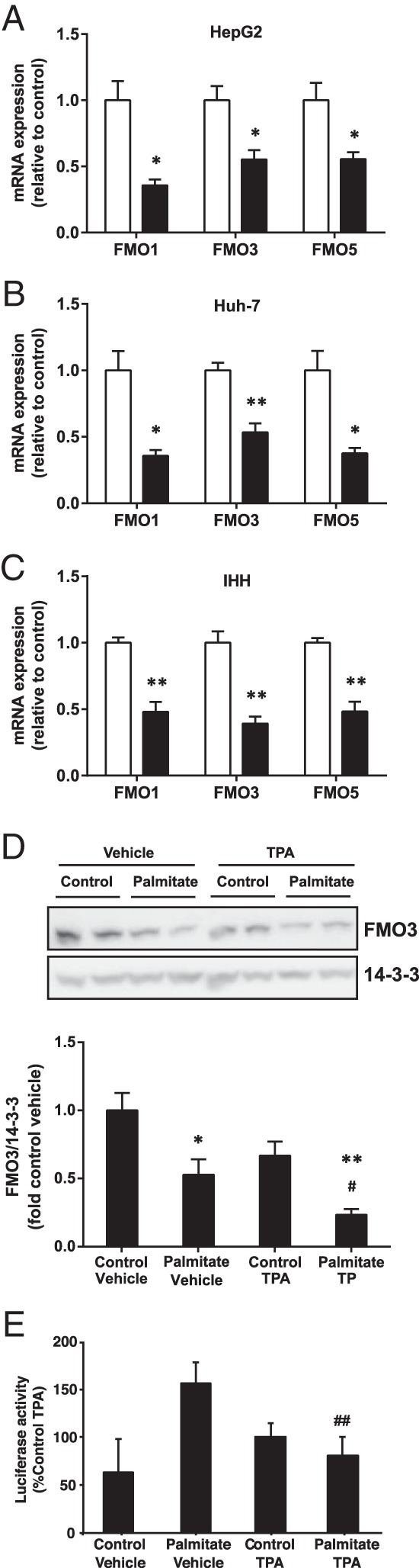
The mRNA levels of FMO1, FMO3, and FMO5 were measured in (A) HepG2, (B) Huh-7, and (C) IHH cells by qRT-PCR after treatment without (open bars) or with (filled bars) 0.75mM palmitate for 18 hours. Results are shown as mean ± SEM. Student's t test; *, P < .05; **, P < .01 vs untreated cells. D, FMO3 protein was measured by immunoblotting in IHH cells incubated with palmitate and TPA or vehicle as indicated and normalized to 14-3-3 protein levels. Results are mean ± SEM from 3 experiments. Student's t test; *, P < .05; **, P < .01 vs untreated cells; #, P < .05 vs palmitate-treated cells treated with vehicle. E, FMO3 promoter activity was measured by luciferase reporter assay in cells incubated with palmitate and TPA or vehicle as indicated. Results are mean ± SEM from 4 experiments. Student's t test; ##, P < .025 vs palmitate-treated cells treated with vehicle.
PKC activation in the presence of lipid oversupply reduces FMO3 promoter activity
To examine the role of PKC, we performed reporter assays to measure FMO3 promoter activity in palmitate-treated HepG2 cells also stimulated with the PKC activator TPA. Although not correlating directly with mRNA levels, which represent the net effect of transcription and degradation, FMO3 promoter activity was reduced upon PKC activation in the presence of the fatty acid (Figure 1E), consistent with enhanced FMO3 protein expression in fat-fed PKC-deficient mice (7), and with reduced FMO3 protein in palmitate- and TPA-treated cells (Figure 1D).
Overexpression of FMO3 has minimal effects on insulin signaling and glycogen synthesis
To examine whether changes in FMO3 expression contribute to alterations in insulin action, we first transfected HepG2 cells with FMO3 or eGFP cDNA. This liver cell line was the most insulin-responsive at the level of proximal insulin signaling (data not shown), and we examined the effect of FMO3 overexpression on the phosphorylation of IRS-1 and Akt in lipid-treated cells. After 18 hours of palmitate treatment, HepG2 cells exhibited a 60% decrease in insulin-stimulated serine phosphorylation of Akt, without changes in IRS-1 tyrosine phosphorylation (Figure 2, A–C). However, there was no difference between control and FMO3-overexpressing cells.
Figure 2. The effects of FMO3 overexpression on insulin signaling and glycogen synthesis in lipid-treated human hepatocytes.
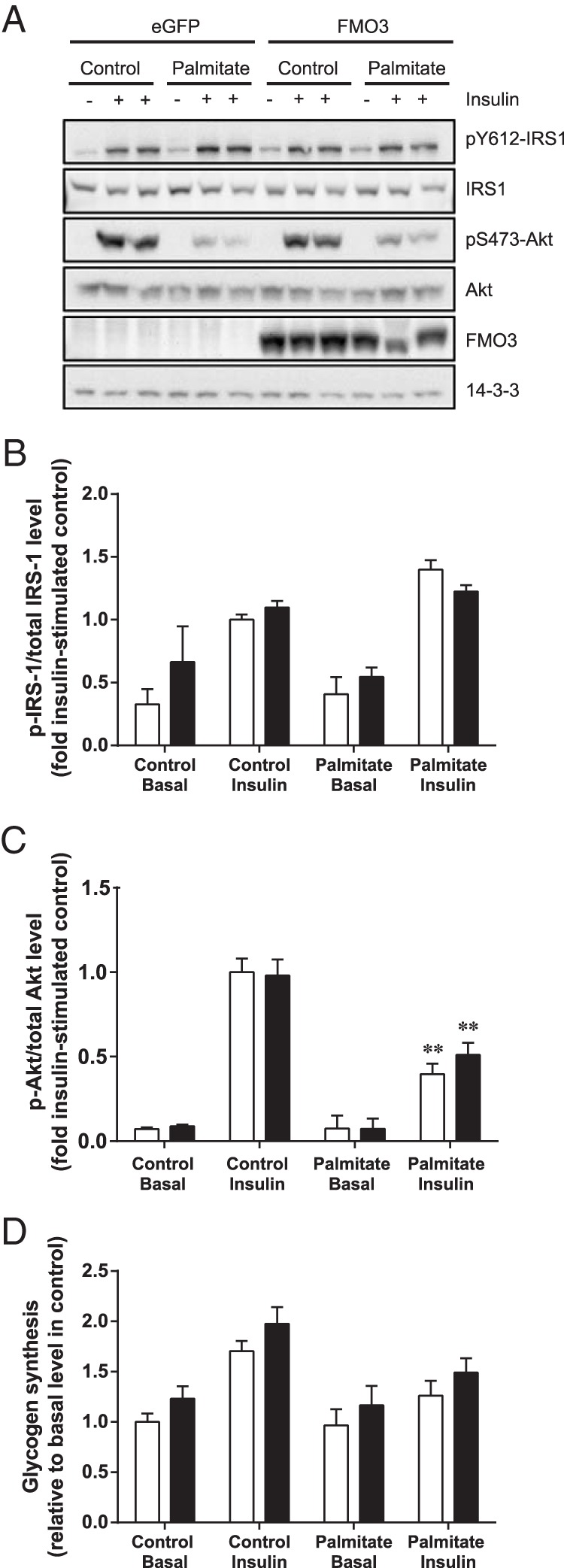
HepG2 cells were transfected with FMO3 cDNA (black bars), or eGFP cDNA (white bars) as control. Cells were incubated in the absence or presence of 0.75mM palmitate before stimulation with 100nM insulin. Cell lysates were subjected to immunoblotting to determine changes in IRS-1 and Akt phosphorylation and FMO3 expression. 14-3-3 was used as a loading control. Representative immunoblots from 4 independent experiments (A) and the mean ± SEM of densitometric analysis (B and C) are shown. Student's t test; **, P < .01 vs untreated cells. D, Glycogen synthesis was determined from the incorporation of [14C]glucose. Mean ± SEM from 5 independent experiments are shown. ANOVA, P < .02 for effect of plamitate in insulin-stimulated cells; P < .05 for effect of FMO3 overexpression.
We also assessed glycogen synthesis under these conditions. As expected, palmitate treatment impaired the insulin-stimulated increase of glycogen synthesis (Figure 2D). Hepatocytes overexpressing FMO3 exhibited slightly more glycogen synthesis under all conditions compared with the control cells. Taken together, these data indicate that FMO3 overexpression has little impact on proximal insulin signaling, and only a subtle effect on downstream glycogen metabolism.
FMO3 overexpression protects against palmitate-mediated ER stress
Enzymes involved in other aspects of hepatic glucose metabolism including gluconeogenesis can be affected independently of insulin signaling upon the induction of ER stress (27, 28). To study the role FMO3 plays in the modulation of fatty acid-induced ER stress in hepatocytes, we next examined the effects of FMO3 overexpression on markers of the UPR and downstream proapoptotic signals induced by ER stress. Palmitate treatment of HepG2 cells increased the phosphorylation of eIF2α and the levels of XBP-1s, markers of the adaptive UPR, and also elevated cleaved caspase 3 and JNK1/2 phosphorylation, indicative of unresolved ER stress (Figure 3, A–E). Palmitate-treated FMO3-overexpressing hepatocytes were greatly protected against the increase in each of these signals (Figure 3, A–E).
Figure 3. The effects of FMO3 overexpression on markers of ER stress in lipid-treated hepatocytes.

HepG2 cells were transfected with FMO3 cDNA (black bars), or eGFP cDNA (white bars) as control. Cells were incubated in the absence or presence of 0.75mM palmitate. Cell lysates were subjected to immunoblotting to determine FMO3 expression and changes in eIF2α phosphorylation, XBP-1s, JNK1/2 phosphorylation, and cleaved caspase 3 levels. Representative immunoblots from 4 independent experiments (A) and the mean ± SEM of densitometric analysis are shown (B–E). Alternatively, gene expression was analyzed by qRT-PCR, and mRNA levels expressed as mean fold change of the level in untreated control cells ± SEM (F–L). Changes in cell viability in response to palmitate treatment and FMO3 expression were determined by 2H-tetrazolium, 2-(4,5-dimethyl-2-thiazolyl)-3,5-diphenyl bromide (MTT) assay (M). Student's t test; *, P < .05; **, P < .01; ***, P < .001 vs untreated hepatocytes; ##, P < .01; ###, P < .001 vs palmitate-treated eGFP-expressing hepatocytes.
We also measured the mRNA expression levels of further ER stress markers under these conditions. Palmitate induced the expression of EDEM1, HSPA5 (BiP), PDIA4 (Erp72), and FKBP11, protein chaperones and isomerases located in the ER, consistent with activation of the UPR (Figure 3, F–I). In addition, the fatty acid increased mRNA levels of the phospho-eIF2α-dependent genes ATF4 and pleckstrin homology-like domain family A member 1 (PHLDA1), as well of the downstream proapoptotic transcription factor CHOP (Figure 3, J–L). Overexpression of FMO3 was generally protective against these elevations in ER stress markers, especially ATF4, PHLDA1, and CHOP. In agreement with the modulation of caspase 3 and CHOP, palmitate treatment moderately reduced cell viability, but this was no longer significant when FMO3 was overexpressed (Figure 3M). Taken together, these data indicate that FMO3 acts to attenuate ER stress, reducing the UPR as well as downstream events such as caspase 3 activation.
FMO3 knockdown up-regulates ER stress-regulated genes
We also used siRNA to determine whether knockdown FMO3 had reciprocal effects on ER stress in hepatocytes compared with overexpression. For these experiments, we employed IHH cells because in comparison with HepG2 cells they express higher endogenous levels of FMO3 mRNA (data not shown) and are thus more comparable with human liver as well as amenable to knockdown studies. FMO3 siRNA transfection led to a 70% reduction in FMO3 mRNA expression compared with control siRNA-transfected cells (Figure 4A), which was reflected in the reduced FMO3 protein levels also observed (Figure 4I). In contrast to the effects of FMO3 overexpression, this resulted in further augmentation of palmitate-induced mRNA expression levels of EDEM1, HSPA5, PDIA4, and FKBP11 (Figure 4, B–E). The expressions of ATF4, PHLDA1, and CHOP were also significantly increased in palmitate-treated IHH cells transfected with FMO3 siRNA (Figure 4, F–H). In agreement, CHOP protein levels were similarly increased (Figure 4I). However, we were unable to detect significant changes in the palmitate-induced phosphorylation of eIF2α and JNK1/2 and elevation of cleaved caspase 3 (data not shown). XBP-1s protein levels were below the limit of detection in IHH cells (data not shown). These data indicate that endogenous FMO3 plays a significant role in the regulation of the ER stress response in hepatocytes.
Figure 4. The effects of FMO3 knockdown on palmitate-induced ER stress in hepatocytes.
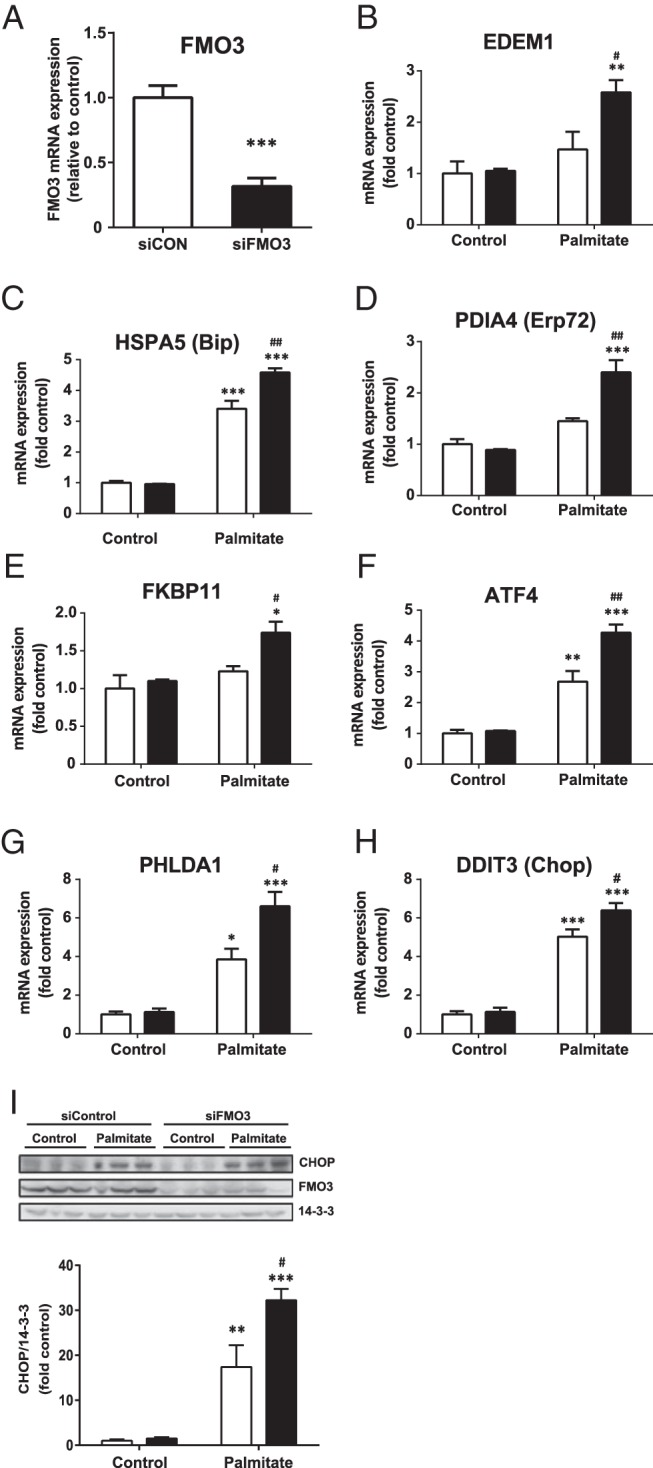
IHH cells were transfected with control siRNA (white bars) or siRNA directed against FMO3 (black bars). Cells were treated with or without 0.75mM palmitate for 18 hours, and the mRNA expression levels of FMO3 (A) and ER stress markers (B–H) were analyzed by qRT-PCR. mRNA levels were expressed as fold change of the level in untreated control cells ± SEM. Changes in the protein levels of CHOP and FMO3 were determined by immunoblotting (I). Student's t test; *, P < .05; **, P < .01; ***, P < .001 vs untreated hepatocytes; #, P < .05; ##, P < .01 vs palmitate-treated hepatocytes transfected with control siRNA.
FMO3 increases ROS in palmitate-treated hepatocytes
FMO activity is induced by alterations in redox balance in yeast, promoting protein folding in the ER by providing oxidizing equivalents necessary for disulphide bond formation (11). However, the production of ROS is associated with the detrimental effects of unresolved ER stress in mammalian cells (29). We therefore examined how alterations in FMO3 expression in palmitate-induced hepatocytes affect the levels of ROS. IHH cells were transfected with FMO3 or control cDNA and subjected to 18 hours of palmitate treatment. Palmitate elevated ROS production by over 2-fold in control cells, whereas FMO3 overexpressing hepatocytes exhibited a further increase in ROS production (Figure 5A). Conversely, knocking down FMO3 in IHH cells reduced basal and palmitate-induced ROS levels by 20% compared with cells transfected with control siRNA (Figure 5B). These data indicate that the protective effects of FMO3 are associated with increased rather than diminished ROS production in hepatocytes, which may promote protein folding in the ER.
Figure 5. Effect of FMO3 expression on the generation of ROS in palmitate-treated hepatocytes.
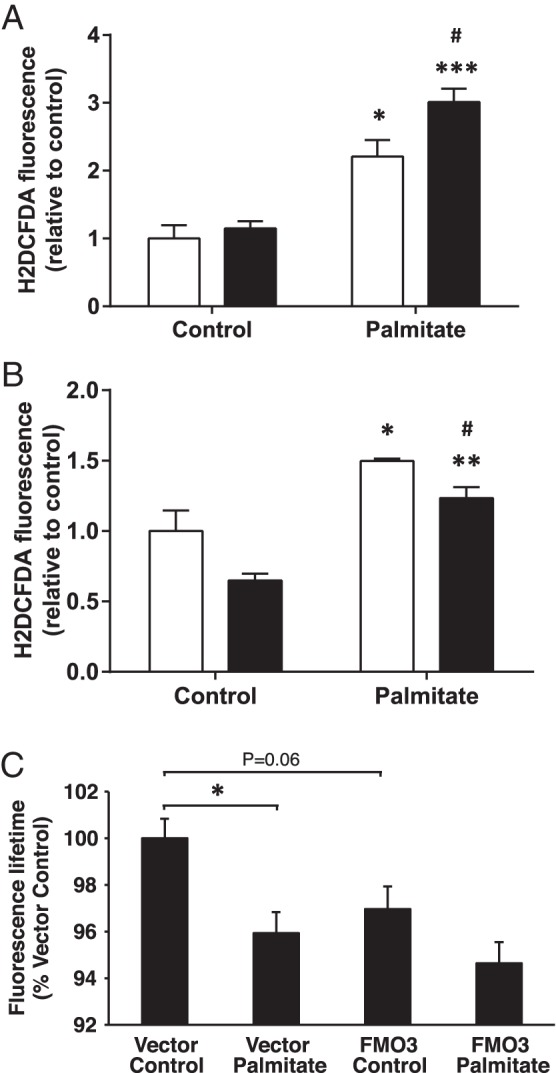
ROS were determined in hepatocytes, after incubation without or with palmitate (white bars, control hepatocytes; black bars, FMO3 overexpression (A) or knockdown (B)). Student's t test; *, P < .05; **, P < .01; ***, P < .001 vs untreated hepatocytes; #, P < .05 vs control siRNA hepatocytes. C, Fluorescence lifetime measurements were performed as described in experimental procedures on cells expressing ERroGFPiE, an ER-localized redox sensor (Avezov et al [26]). Cells under control conditions exhibited a mean ERroGFPiE lifetime of 1.52 ns in 4 independent experiments. Reduced ERroGFPiE lifetime is indicative of reduced disulphide formation due to a diminished oxidative environment. Data shown are mean ± SEM. Student's t test; *, P < .05.
Palmitate-induced reduction in ER redox balance is not prevented by FMO3 overexpression
We hypothesized that lipid oversupply specifically reduces ER redox potential, diminishing disulphide bond formation leading to incorrect protein folding and ER stress, and that this is prevented by enhanced FMO3 expression. Using an ER-targeted fluorescent protein thiol redox probe (26), we demonstrated that palmitate treatment did indeed reduce ER thiol redox (Figure 5C). Unexpectedly, however, FMO3 overexpression reduced redox balance similarly to lipid treatment and did not reverse the effects of the fatty acid (Figure 5C). The beneficial effects of FMO3 on ER stress are thus mediated independently of ER protein thiol redox.
FMO3 modulates phosphoenolpyruvate carboxykinase (PEPCK) gene expression
To assess whether the protection against ER stress by FMO3 affects the regulation of gluconeogenesis, we first examined the effect of the known ER stress inducers Tg and Tm on a marker of gluconeogenesis in IHH cells overexpressing FMO3 or control cDNA. Upon treatment with Tg or Tm, the expression of the gluconeogenic gene PEPCK was increased by 9- and 7-fold, respectively, compared with vehicle control. This increase in PEPCK mRNA levels was greatly reduced in cells overexpressing FMO3 (Figure 6A), although FMO3 knockdown was unable to significantly increase PEPCK mRNA beyond the effects of the pharmacological agents (Figure 6B). Next, we measured FMO3-dependent alterations PEPCK mRNA in hepatocytes treated without or with palmitate, a less potent physiological inducer of ER stress. Treatment with the fatty acid led to a more than 2-fold increase in PEPCK mRNA levels in eGFP-expressing control hepatocytes, whereas the expression of the gluconeogenic gene was unaffected by palmitate in the FMO3-overexpressing cells (Figure 6C), indicating complete protection by FMO3 against this effect of the lipid. Conversely, when we transfected cells with FMO3 siRNA (siFMO3) we observed a 70% increase in PEPCK mRNA levels upon palmitate treatment relative to cells transfected with control siRNA (Figure 6D). Taken together with the previous experiments, these data support the idea that FMO3 protects against the lipid-induced induction of gluconeogenic gene expression by reducing ER stress.
Figure 6. Effect of ER stress and FMO3 expression on PEPCK mRNA levels.
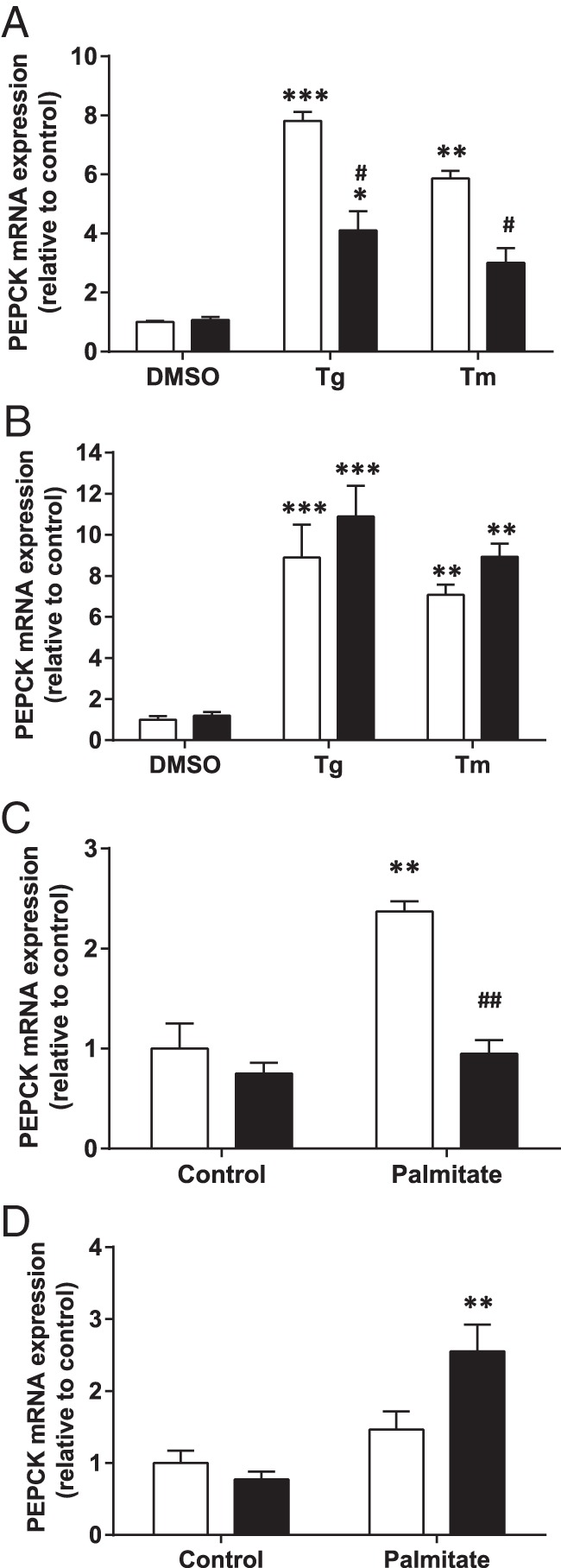
IHH hepatocytes either overexpressing FMO3 (A and C) or subjected to siRNA-mediated knockdown of FMO3 (B and D), were treated for 18 hours either with Tg, Tm, or vehicle to directly induce ER stress (A and B), or incubated for 18 hours without or with palmitate (C and D). In A–D, white bars represent control hepatocytes; black bars represent FMO3 overexpression or knockdown. In each case, PEPCK mRNA levels were determined by qRT-PCR. Means from 4 independent experiments are shown in each panel. Student's t test; **, P < .01; ***, P < .001 vs control; ##, P < .001 vs Tg-, Tm-, or palmitate-treated control hepatocytes.
Livers of PKCδ KO mice exhibit reduced HFD-induced ER stress and PEPCK expression
Previously we showed that fat-fed PKCδ KO mice exhibit increased hepatic FMO3 protein levels, and we now investigated whether an association between FMO3 expression, ER stress, and gluconeogenic gene mRNA levels can also be observed in vivo. We measured the levels of XBP-1s and CHOP in livers from WT and PKCδ KO mice fed either a chow or a HFD for 1 week (Figure 7, A and B). XBP-1s protein was increased by 1.5-fold in livers from WT mice by fat-feeding but this was not observed in livers from fat-fed PKCδ KO mice (Figure 7, A and B). Similarly, CHOP protein levels were elevated 1.4-fold by the fat diet in WT mice, whereas PKCδ deletion reduced CHOP expression (Figure 7, A and C). We also measured the relative PEPCK mRNA expression levels in these animals. PEPCK expression was not significantly affected by a 1 week HFD in WT mice; however, a reduction was observed in the livers from fat-fed PKCδ KO mice (Figure 7D). These results are consistent with a role for increased FMO3 expression in the protection of glucose tolerance in fat-fed PKCδ KO mice through a reduction in ER stress and hence in gluconeogenic gene expression.
Figure 7. Effect of PKCδ deletion on ER stress markers and PEPCK gene expression in livers of fat-fed mice.
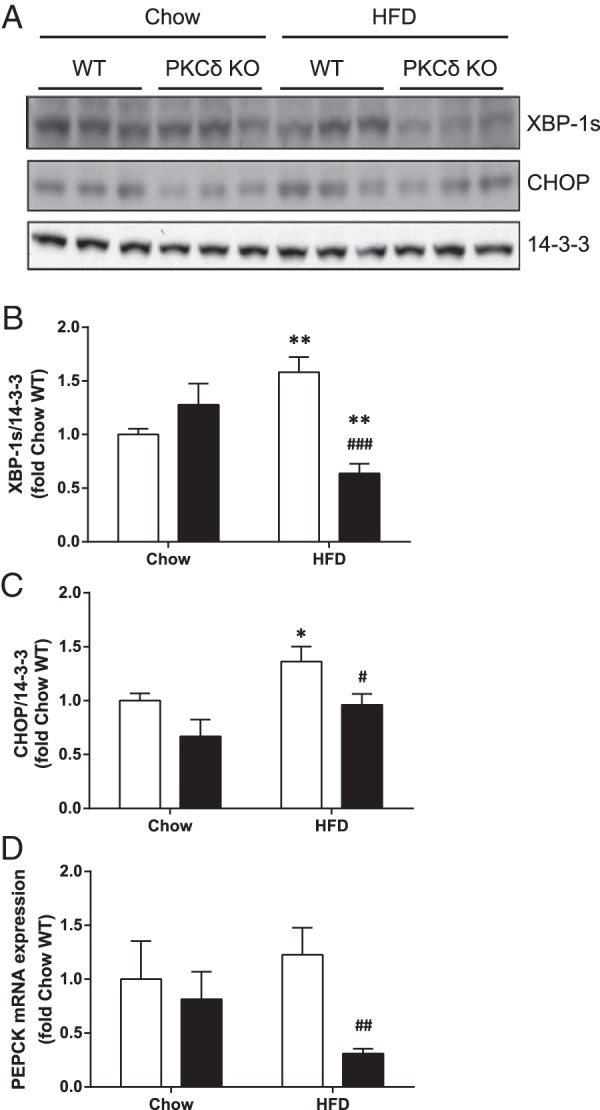
Liver lysates from chow or fat-fed WT (white bars) and PKCδ KO mice (black bars; n = 6 in each group) were subjected to immunoblotting to detect changes in XBP-1s and CHOP expression. Representative immunoblots (A) and mean ± SEM from densitometry (B and C) are shown. D, PEPCK mRNA expression in livers was analyzed by qRT-PCR. Student's t test; *, P < .05; **, P < .01 vs chow-fed mice; #, P < .05; ##, P < .01; ###, P < .001 vs HFD-fed WT mice.
Discussion
Previously, we showed that FMO isoforms were up-regulated in livers from fat-fed PKCδ and PKCϵ KO mice (7). Here, we investigated mechanisms by which FMO3 activity may contribute to the improvement in hepatic insulin action observed in PKC-deficient mice. FMO3 overexpression had minimal effects on insulin signaling and glycogen synthesis in hepatocytes. However, overexpression of the enzyme protected against palmitate-induced ER stress and elevation of PEPCK mRNA in vitro, whereas knockdown had reciprocal effects, consistent with observations in mouse liver. This is the first study to demonstrate a protective function of FMO3 in the regulation of glucose metabolism via modulation of ER stress.
Alterations in the abundance of FMO isoforms have been demonstrated in genomic and proteomic studies of livers from type 2 diabetes patients as well as rodent models of insulin resistance and diabetes (30–32). Furthermore, deletion of FMO1, which is highly expressed in liver, kidney, and adipose tissue, has been linked to a reduction in body weight and triglyceride storage in mice (33). FMO3 expression was shown to be up-regulated by a high-fat, cholesterol-containing diet supplemented with the bile acid cholic acid which also resulted in increased atherosclerosis (19). This was linked to elevated plasma levels of TMAO, the natural osmolyte generated by FMO3 activity. Similarly, FMO3 knockdown or deletion in vivo has recently been shown to reduce atherosclerosis (21, 22). Taken together, those studies could be interpreted to indicate detrimental effects of FMO activity in obesity and T2D. In contrast, however, increased FMO3 expression has been linked to the beneficial effects of calorie restriction in rodents (34). TMAO has been used as a chemical chaperone to improve protein folding and reduce ER stress (12), which has been associated with insulin resistance (13), and TMAO reverses insulin resistance in lipid-treated hepatocytes (35). Our data is thus consistent with a protective effect of FMO3 against ER stress, possibly through the generation of TMAO, although we were unable to detect an improvement in insulin signal transduction, suggesting a more distal site of action.
Alternatively, our measurements of ROS in lipid-treated cells suggested that FMO3 expression may improve protein folding in the ER by mediating changes in redox balance, which has been reported previously in yeast cells (11). Partial uncoupling of the NADPH-dependent reaction catalyzed by FMO enzymes can generate H2O2 (8), and we confirmed that FMO3 expression was associated with increased ROS. Although chronic ROS production by nutrient overload has been linked to the generation of insulin resistance, a beneficial role in insulin action for H2O2 production by NADPH oxidases has also been proposed (36). Different compartmentalization and roles for the ROS produced by palmitate metabolism and by FMO3 action in hepatocytes would reconcile the increased ROS levels observed in association with reduced ER stress in cells overexpressing FMO3. However, we also demonstrated that increased FMO3 expression was not associated with a prevention of lipid-induced changes in ER protein thiol redox balance.
Although FMO3 overexpression was able to reduce palmitate-mediated ER stress in hepatocytes, and also block the increase of PEPCK expression induced either by pharmacological ER stress agents or by the fatty acid, it did not reverse effects on insulin signaling at the level of Akt phosphorylation. This suggests that mechanisms other than phosphorylation of the key transcription factor Foxo1 by Akt may contribute to the reduction in PEPCK mRNA mediated by the oxygenase. Additional transcription factors also regulate PEPCK expression, including ATF2, cAMP-response element-binding protein and C/EBPβ. Furthermore, ER stress has been shown to modulate the expression of PEPCK by effects on cAMP-response element-binding protein and C/EBPβ (28, 37, 38), so it is possible that the effects of FMO3 are mediated through reversal of such changes instead of through the Akt/Foxo1 pathway.
Our findings made in vitro using cultured hepatocytes have relevance to changes occurring in livers from fat-fed PKC-deficient mice. Our proteomic studies demonstrated that livers from fat-fed PKC KO mice exhibit up-regulation of FMO isoforms, and here we have shown that the ER stress markers XBP-1s and CHOP are reduced upon PKCδ deletion, opposing the effects of a HFD. We did not detect a significant fat diet-dependent increase in PEPCK mRNA expression, in contrast to the effects of palmitate in hepatocytes. Importantly, however, PKCδ deletion resulted in greatly reduced expression of PEPCK mRNA compared with fat-fed WT mice. Increased FMO3 expression and a reduction of ER stress can therefore be correlated with reduced transcription of PEPCK. In this case, however, additional levels of regulation occurring in vivo are indicated by the fact that the ER stress associated with fat feeding was not sufficient to promote PEPCK expression in WT mice, at least after 1 week.
We have previously shown that FMO3 protein expression in liver of fat-fed mice is increased by PKC deletion (7), and we now show that this is mediated in part at the level of transcription. Lipid treatment alone reduced FMO3 mRNA and protein levels in hepatocyte cell lines. Importantly, PKC activation coupled with lipid-treatment resulted in reduced FMO3 promoter activity and protein expression, consistent with our observations in fat-fed WT and PKC-deficient mice. However, indirect effects of PKC deletion may play a role. FMO3 has been reported to be induced by bile acids acting on the hepatic farnesoid X receptor (19), and we have also observed PKC-dependent alterations in metabolic pathways of cholesterol and bile acid synthesis (7). Thus, although FMO3 levels may normally be responsive to dietary bile acid (19), PKC deletion may promote the expression of the enzyme by affecting de novo bile acid production. Interestingly, the action of bile acids on farnesoid X receptor is strongly associated with the regulation of lipid metabolism and the maintenance of insulin sensitivity (39). Whether such a mechanism explains the increased expression of each of the FMO isoforms up-regulated in PKC KO mice, or whether PKC activity plays a more direct role, remains to be determined.
In summary, we have extended our findings from livers of PKC KO mice to examine the potential role of FMO action in the amelioration of glucose intolerance caused by lipid oversupply. We have shown that FMO isoforms are down-regulated by lipid treatment in hepatocytes, whereas FMO3 overexpression protects against ER stress due to fatty acid treatment, with reciprocal effects of FMO3 knockdown. This can be linked to a modulation gluconeogenesis through changes in PEPCK gene expression, also observed in vivo. These mechanisms are summarized in a model (Supplemental Figure 2). Our studies therefore highlight a positive role for FMO3, and possibly other FMO isoforms, in the regulation of glucose homeostasis. This is an important consideration because a reduction in FMO3 activity has been proposed as a therapy for diabetes-associated atherosclerosis (22).
Acknowledgments
We thank the expert technical assistance of the Garvan Institute Biological Testing Facility. We also thank Professor Trevor Biden, Associate Professor Ross Laybutt, Dr Jeng Yie Chan, and Dr Mohammed Bensellam, Diabetes and Metabolism Division, Garvan Institute, for helpful advice and critical reading of the manuscript.
This work was supported by the National Health and Medical Research Council of Australia Grant 535917 (to C.S.-P.) and the Diabetes Australia Research Trust and an Australian Postgraduate Award (B.M.L.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the National Health and Medical Research Council of Australia Grant 535917 (to C.S.-P.) and the Diabetes Australia Research Trust and an Australian Postgraduate Award (B.M.L.).
Footnotes
- ATF4
- activating transcription factor 4
- BiP
- binding immunoglobulin protein
- C/EBP
- CCAAT/enhancer-binding protein
- CHOP
- C/EBP homologous protein
- eGFP
- enhanced green fluorescent protein
- eIF2α
- eukaryotic translation initiation factor 2 α-subunit
- ER
- endoplasmic reticulum
- FMO
- flavin-containing monooxygenase
- HFD
- high-fat diet
- IRS-1
- insulin receptor substrate-1
- JNK
- Jun N-terminal kinase
- KO
- knockout
- PEPCK
- phosphoenolpyruvate carboxykinase
- PHLDA1
- pleckstrin homology-like domain family A member 1
- PKC
- protein kinase C
- qRT-PCR
- quantitative RT-PCR
- ROS
- reactive oxygen species
- siRNA
- small interfering RNA
- Tg
- thapsigargin
- Tm
- tunicamycin
- TMAO
- trimethylamine N-oxide
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- UPR
- unfolded protein response
- WT
- wild type
- XBP-1s
- splicing of X-box-binding protein 1.
References
- 1. Hocking S, Samocha-Bonet D, Milner KL, Greenfield JR, Chisholm DJ. Adiposity and insulin resistance in humans: the role of the different tissue and cellular lipid depots. Endocr Rev. 2013;34:463–500. [DOI] [PubMed] [Google Scholar]
- 2. Schmitz-Peiffer C, Biden TJ. Protein kinase C function in muscle, liver, and β-cells and its therapeutic implications for type 2 diabetes. Diabetes. 2008;57:1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Samuel VT, Liu ZX, Wang A, et al. . Inhibition of protein kinase Cϵ prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frangioudakis G, Burchfield JG, Narasimhan S, et al. . Diverse roles for protein kinase C δ and protein kinase C ϵ in the generation of high-fat-diet-induced glucose intolerance in mice: regulation of lipogenesis by protein kinase C δ. Diabetologia. 2009;52:2616–2620. [DOI] [PubMed] [Google Scholar]
- 5. Raddatz K, Turner N, Frangioudakis G, et al. . Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia. 2011;54:1447–1456. [DOI] [PubMed] [Google Scholar]
- 6. Bezy O, Tran TT, Pihlajamäki J, et al. . PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J Clin Invest. 2011;121:2504–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liao BM, Raddatz K, Zhong L, Parker BL, Raftery MJ, Schmitz-Peiffer C. Proteomic analysis of livers from fat-fed mice deficient in either PKCδ or PKCϵ identifies Htatip2 as a regulator of lipid metabolism. Proteomics. 2014;14:2578–2587. [DOI] [PubMed] [Google Scholar]
- 8. Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106:357–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koukouritaki SB, Simpson P, Yeung CK, Rettie AE, Hines RN. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr Res. 2002;51:236–243. [DOI] [PubMed] [Google Scholar]
- 10. Elfarra AA. Potential role of the flavin-containing monooxygenases in the metabolism of endogenous compounds. Chem Biol Interact. 1995;96:47–55. [DOI] [PubMed] [Google Scholar]
- 11. Suh JK, Robertus JD. Yeast flavin-containing monooxygenase is induced by the unfolded protein response. Proc Nat Acad Sci USA. 2000;97:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones. 1996;1:109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15:623–634. [DOI] [PubMed] [Google Scholar]
- 15. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. [DOI] [PubMed] [Google Scholar]
- 16. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- 17. Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. [DOI] [PubMed] [Google Scholar]
- 18. Urano F, Wang X, Bertolotti A, et al. . Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. [DOI] [PubMed] [Google Scholar]
- 19. Bennett BJ, de Aguiar Vallim TQ, Wang Z, et al. . Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao X, Liu X, Xu J, Xue C, Xue Y, Wang Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J Biosci Bioeng. 2014;118:476–481. [DOI] [PubMed] [Google Scholar]
- 21. Shih DM, Wang Z, Lee R, et al. . Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J Lipid Res. 2015;56:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miao J, Ling AV, Manthena PV, et al. . Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat Commun. 2015;6:6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274:24202–24210. [DOI] [PubMed] [Google Scholar]
- 24. Tannous BA. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc. 2009;4:582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmitz-Peiffer C, Browne CL, Biden TJ. Characterization of two forms of protein kinase C α, with different substrate specificities, from skeletal muscle. Biochem J. 1996;320:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Avezov E, Cross BC, Kaminski Schierle GS, et al. . Lifetime imaging of a fluorescent protein sensor reveals surprising stability of ER thiol redox. J Cell Biol. 2013;201:337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deng Y, Wang ZV, Tao C, et al. . The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J Clin Invest. 2013;123:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choudhury M, Qadri I, Rahman SM, Schroeder-Gloeckler J, Janssen RC, Friedman JE. C/EBPβ is AMP kinase sensitive and up-regulates PEPCK in response to ER stress in hepatoma cells. Mol Cell Endocrinol. 2011;331:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. [DOI] [PubMed] [Google Scholar]
- 30. Takamura T, Sakurai M, Ota T, Ando H, Honda M, Kaneko S. Genes for systemic vascular complications are differentially expressed in the livers of type 2 diabetic patients. Diabetologia. 2004;47:638–647. [DOI] [PubMed] [Google Scholar]
- 31. Liang CP, Tall AR. Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in ob/ob mouse liver. J Biol Chem. 2001;276:49066–49076. [DOI] [PubMed] [Google Scholar]
- 32. Borbás T, Benko B, Dalmadi B, Szabó I, Tihanyi K. Insulin in flavin-containing monooxygenase regulation. Flavin-containing monooxygenase and cytochrome P450 activities in experimental diabetes. Eur J Pharm Sci. 2006;28:51–58. [DOI] [PubMed] [Google Scholar]
- 33. Veeravalli S, Omar BA, Houseman L, et al. . The phenotype of a flavin-containing monooyxgenase knockout mouse implicates the drug-metabolizing enzyme FMO1 as a novel regulator of energy balance. Biochem Pharmacol. 2014;90:88–95. [DOI] [PubMed] [Google Scholar]
- 34. Fu ZD, Klaassen CD. Short-term calorie restriction feminizes the mRNA profiles of drug metabolizing enzymes and transporters in livers of mice. Toxicol Appl Pharmacol. 2014;274:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Achard CS, Laybutt DR. Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: adaptation with enhanced insulin signaling or insulin resistance. Endocrinology. 2012;153:2164–2177. [DOI] [PubMed] [Google Scholar]
- 36. Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci. 2011;32:82–89. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lim JH, Lee HJ, Ho Jung M, Song J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell Signal. 2009;21:169–177. [DOI] [PubMed] [Google Scholar]
- 39. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. [DOI] [PubMed] [Google Scholar]