

Abstract
Estrogen receptor (ER)α is a well-characterized ligand-dependent transcription factor. However, the global picture of its nongenomic functions remains to be illustrated. Here, we demonstrate a novel function of ERα during mitosis that facilitates estrogen-dependent cell proliferation. An E3 ubiquitin ligase, UBE3C, was identified in an ERα complex from estrogen-treated MCF-7 breast cancer cells arrested at mitosis. UBE3C interacts with ERα during mitosis in an estrogen-dependent manner. In vitro, estrogen dramatically stimulates the E3 activity of UBE3C in the presence of ERα. This effect was inhibited by the estrogen antagonist tamoxifen. Importantly, estrogen enhances the ubiquitination of cyclin B1 (CCNB1) and destabilizes CCNB1 during mitosis in a manner dependent on endogenous UBE3C. ERα, UBE3C, and CCNB1 colocalize in prophase nuclei and at metaphase spindles before CCNB1 is degraded in anaphase. Depletion of UBE3C attenuates estrogen-dependent cell proliferation without affecting the transactivation function of ERα. Collectively, these results demonstrate a novel ligand-dependent action of ERα that stimulates the activity of an E3 ligase. The mitotic role of estrogen may contribute to its effects on proliferation in addition to its roles in target gene expression.
Estrogens play an essential role in growth, differentiation, female development and reproductive processes. They function in a broad range of target tissues in mammalian organisms and are also important in regulating the progression of breast and endometrial cancers. Estrogen receptor (ER)α, a member of the nuclear receptor (NR) superfamily, exerts vital effects on cellular functions upon binding to the ligand estrogen. Ligand-bound ERαs dimerize and are recruited to the cis-regulatory elements of target genes through direct interaction with estrogen-response elements (EREs). Once bound to DNA, ERα subsequently recruits a number of proteins in complexes that include general transcription factors, coactivators, and corepressors to either activate or repress the target gene.
In addition to this classical role of ERα as a transcription factor, accumulated evidence has demonstrated a diverse range of functions, including those mediated by nongenomic actions. ERα signaling mechanisms could be divided into 4 categories (1): 1) the classical mechanism of ligand- and ERE-dependent transcriptional activation (2); 2) ligand-dependent but ERE-independent genomic action, in which ERαs are indirectly recruited via protein-protein interactions with transcription factors (3–5); 3) ligand-independent but ERE-dependent genomic action, in which phosphorylation of ERα via growth factor-activated protein kinase cascades promotes the activation of ERα at EREs (6); and 4) ligand-dependent nongenomic action, in which ERα mediates rapid nongenomic response to estrogen through interactions with adaptor molecules, G proteins and kinases (7). Thus, the functions of ERα are not limited to transcriptional regulation or to genomic functions but encompass a broader scope of unknown cellular functions. In particular, the ligand-dependent nongenomic action of ERα has been intensively researched over the past decade and much has been revealed for its role in kinase activation. That include activation of endothelial nitric oxide synthase (eNOS) by plasma membrane-associated ERα through activating multiple kinases, including the phosphatidylinositol-3-kinase (PI3K), protein kinase B (AKT), and ERK1/2 (8–15). However, activation of other enzymes, for instance E3 ubiquitin ligase, through the ligand-dependent nongenomic action of ERα has not been elucidated. Furthermore, whether ERα functions during mitosis has not been addressed.
In this manuscript, we report a novel nongenomic function of ERα during mitosis beyond the above categories. We demonstrate ligand-dependent action of ERα to stimulate an E3 ubiquitin ligase, UBE3C. Cyclin B1 (CCNB1) was identified as a substrate of UBE3C that is destabilized by estrogen during mitosis. Depletion of UBE3C attenuates estrogen-dependent cell proliferation. Because the stimulation of UBE3C by liganded ERα is inhibited by the estrogen antagonist tamoxifen, this mechanism could be a novel target in clinical applications, including cancer treatment.
Materials and Methods
Cell lines and culture conditions
MCF-7 and HEK-293T cells were purchased from American Type Culture Collection and grown in low glucose DMEM (Wako), 10% fetal bovine serum (FBS), and 1% antibiotic-antimycotic agent (Life Technologies). Cell lines were not further tested or authenticated. The day before the assays, MCF-7 cells were cultured in phenol red-free medium supplemented with 10% charcoal-stripped FBS (GIBCO). MCF-7 cells stably expressing FLAG-tagged UBE3C or ERE-TATA-Luc were established by transient transfection with pcDNA3-FLAG-UBE3C using Lipofectamine 2000 reagent (Life Technology) and selected with 1-mg/mL G418. HEK-293T cells were transfected using the SuperFect Transfection Reagent (QIAGEN). MCF-7 cells were arrested in S phase or M phase by treatment with 2.5mM thymidine or 1μM nocodazole for 24 hours. For cell cycle synchronization by a double thymidine block, MCF-7 cells cultured in phenol red-containing medium were treated with 2.5mM thymidine for 18 hours, released for 8 hours, retreated with the thymidine for 16 hours, and released to the phenol red-free/charcoal-stripped FBS-containing medium with or without 10nM 17β estradiol for the indicated time length.
Plasmids
The cDNA for full-length human UBE3C was amplified by PCR from an MCF-7 cell cDNA library and subcloned into the vector pcDNA3 in-frame with an N-terminal FLAG tag. A UBE3C C1051A mutant was generated by site-directed mutagenesis. A pRL-CMV Renilla luciferase reporter plasmid (pCMV-Rluc) was purchased from Promega. The pGL3–3xERE-TATA-luciferase reporter plasmid (3xERE-TATA-Luc), pcDNA3-Flag-tagged ERα, pcDNA3-AR were kindly provided by Dr Fumiaki-Ohtake.
RNA interference
siRNA oligonucleotides targeting UBE3C (5′-GAGAAUGCUUGAAGUAUUUUU-3′, sense strand), ERα (5′-GAAUGUGCCUGGCUAGAGAUU-3′), and nontargeting control (4390844) were purchased (Ambion). Cells were transfected with RNA duplexes (final concentration 10nM) using Lipofectamine RNAiMAX or Lipofectamine 2000 reagent (Invitrogen) and analyzed 72 hours after transfection.
Antibodies
The next antibodies were used: rabbit polyclonal antibodies to ERα (HC-20), AR (N-20), GST (B-14) (Santa Cruz Biotechnology, Inc), FLAG (Sigma), and ubiquitin (Dako); and mouse monoclonal antibodies to ERα (for immunoprecipitation; B10, Merck Millipore), α-tubulin (DMIA, Neomarkers), β-actin (Abcam), CCNB1 (GNS, Santa Cruz Biotechnology, Inc), and control IgG2a (Abcam). The rabbit anti-UBE3C polyclonal antibody was raised against a synthetic peptide (EGDFKTRPKVSLGGASRC) and affinity purified.
Cell extracts, immunoprecipitation, and Western blotting
Immunoprecipitation and immunoblotting were performed as described (16) with TNE lysis buffer containing 45mM Tris-HCl (pH 7.8), 150mM NaCl, 2mM MgCl2, 0.1% NP-40, 1mM EDTA, 1mM DTT, protease inhibitor cocktail set III (Calbiochem) and Protein A Dynabeads (Life Technology). For straight immunoblotting, cells were lysed, clarified, adjusted for protein concentration and subjected to western blotting. For immunoprecipitation of in vivo ubiquitinated CCNB1, cells were lysed in radioimmunoprecipitation assay buffer (25mM Tris-HCl [pH 7.8], 150mM NaCl, 2mM MgCl2, 2mM EDTA, 0.1% SDS, 1% sodium deoxycholate, 50mM NaF, and protease inhibitor cocktail set III).
Purification of ERα interactants and mass spectrometry
Proteins immunoprecipitated from HeLa cells with Protein A chemically cross-linked to either anti-ERα or control IgG2a were subjected to SDS-PAGE and stained with Silver Quest (Life Technology). Proteins were excised from the gel and in-gel digested by trypsin as previously described (16). Peptides extracted from the gel were subjected to MALDI-TOF/MS analysis (Bruker Daltonics).
Purified proteins
Ubiquitin (Boston Biochem), rabbit E1, UBE2E1, UBE2E2, and UBE2R1 (Calbiochem) were purchased commercially. Other E2s/UBE2Ds and GST-CCNB1-His were purified from Rosetta 2 (DE3) bacterial cells (Merck Millipore) with IPTG induction. FLAG-ERα and FLAG-UBE3C were prepared using a baculovirus expression system (Invitrogen) and purified using FLAG M2 agarose beads.
In vitro Ub ligation assay
Purified FLAG-UBE3C was subjected to in vitro reaction with ubiquitin, E1, and E2s as previously described (16) in the presence or absence of 13.3μM FLAG-UBE3C, 26.6μM FLAG-ERα, and the indicated amount of 17β-estradiol. For substrate ubiquitination, 25 ng of GST-CCNB1-His were added to the reaction. In some experiments, UBE3C immune complexes immobilized on Protein G Sepharose beads prepared from MCF-7 cells arrested in mitosis with nocodazole and treated with/without 10nM 17β-estradiol were used instead of the purified FLAG-UBE3C and FLAG-ERα. MG132 (10μM) was added to the cell lysate and the reaction where indicated. The reaction was subjected to western blotting with anti-CCNB1 antibody.
Surface plasmon resonance (SPR) analysis
Purified FLAG-UBE3C peptides were immobilized on a CM5 sensor chip using an amine coupling kit, and SPR analysis with FLAG-ERα as the analyte was performed as previously described (17).
Immunofluorescence microscopy
Proliferating cells were fixed with 4% paraformaldehyde in 1mM EGTA/PBS for 20 minutes and permeabilized with 0.3% Triton X-100 for 15 minutes. Cells were washed with PBS twice, blocked with 0.3% normal goat serum in PBS-T (0.1% Tween 20), and stained with the indicated antibodies. Primary antibodies were diluted in blocking buffer at the next dilutions: anti-UBE3C, 1:4000; anti-α-tubulin, 1:4000; anti-ERα, 1:100; and anti-CCNB1, 1:100. Goat antirabbit Alexa Fluor 488 or goat mouse Alexa Fluor 594 secondary antibodies (Life Technology) were used at a dilution of 1:1000. The cells were then mounted with Prolong Gold with DAPI (Life Technology) and examined with a confocal laser scanning microscope (LSM 510; Carl Zeiss).
Luciferase assay
MCF-7 cells were first transected with siRNA. After 44 hours, the cells were then transfected with 250 ng of 3xERE-TATA-Luc vector per 5 × 105 cells in a 12-well plate. The vector pCMV-Rluc (1 ng; Promega) was also transfected as an internal control for transfection efficiency. Vehicle ethanol or 10nM 17β-estradiol was added 4 hours after the transfection, and the cells were further incubated for 24 hours before harvest. The cells were then subjected to the dual-luciferase reporter assay system (Promega). Each reaction was performed in triplicate, and the data were normalized to an internal control in each sample.
Quantitative real-time RT-PCR (qPCR)
Total RNA from each sample was prepared using the RNeasy plus mini kit (QIAGEN). A 500-ng aliquot of the total RNA was reverse transcribed using PrimeScript RT Master Mix (Perfect Real Tim3) (Takara). The gene expression level was determined by qPCR based on the StepOnePlus Real-Time PCR system with Power SYBR Green PCR Master Mix (Applied Biosystems). Each reaction was performed in triplicate. The data were analyzed according to the comparative Ct method and normalized to hGAPDH expression in each sample. The primers used are listed in the supplemental information below.
Results
Identification of UBE3C as an ERα-interacting protein
Previous studies suggested that the roles of ERα are not limited to genomic functions but contribute to broader cellular actions than expected. Because the known functions of ERα are limited to its role in interphase of the cell cycle, we investigated a possible role of ERα during mitosis. We first analyzed the transcriptional activity of ERα in mitosis. qPCR assays demonstrated that the transcription level of ERα-target genes, including FOS, NRIP1, TFF1, and MYC, in cells arrested in mitosis was only minimally affected by 17β-estradiol, whereas significant transactivation was observed in asynchronous cells or cells arrested in S phase (Supplemental Figure 1). These results are consistent with the global protection of transcription by chromosome condensation during mitosis.
We next sought to investigate a nongenomic role of ERα during mitosis. We first screened proteins in complex with ERα in estrogen-stimulated mitotic MCF-7 cells. Cells were arrested at mitosis by nocodazole and stimulated by 17β-estradiol. We analyzed the proteins that coprecipitated with an anti-ERα antibody by MALDI-TOF/MS and identified several of interest (Figure 1A). We selected UBE3C, a HECT family E3 ubiquitin ligase, for further analysis because its protein sequence contains significant similarity to UBE3A/E6-AP, which has been implicated in ERα function (18–20). To verify the interaction, we raised a rabbit polyclonal antibody specific to UBE3C (Supplemental Figure 2). The association of endogenous ERα and UBE3C in MCF-7 cells arrested at mitosis was observed by anti-ERα immunoprecipitation followed by anti-UBE3C immunoblotting (Figure 1B). Importantly, the detected interaction was dependent on 17β-estradiol treatment.
Figure 1. Identification of UBE3C as an ERα-binding protein from mitotic cells treated with estrogen.

A, Identification of UBE3C in the ERα immunocomplex. Exponentially growing MCF-7 cells were incubated with nocodazole for 24 hours and in the presence of 17β-estradiol (10nM) for the final 1.5 hours before harvest. Cell lysates were immunoprecipitated (IP) with either control IgG or anti-ERα antibody, resolved by SDS-PAGE, and subjected to silver staining (upper panel) or anti-ERα immunoblotting (lower panel). ERα-interacting proteins were identified by mass spectrometry and are indicated. *, IgG. B, Estrogen-induced interaction of endogenous ERα with UBE3C in mitosis. MCF-7 cells were cultured with nocodazole for 24 hours with or without 10nM 17β-estradiol for the indicated time before harvest. Cell lysates were IP with control IgG or anti-ERα antibody followed by immunoblotting with the indicated antibodies. Inputs (0.5%) were also loaded. C, Estrogen-induced interaction of exogenous ERα with UBE3C in mitosis. HEK-293T cells were transfected with ERα and FLAG-UBE3C as indicated and treated as in B with or without 2 hours of exposure to 17β-estradiol. Cell lysates were IP followed by immunoblotting as in B. *, nonspecific products. D, SPR analysis with ERα peptides (250nM) injected over immobilized FLAG-UBE3C peptides.
To obtain further evidence, HEK-293T cells were transfected with ERα and FLAG-tagged UBE3C and arrested at mitosis. ERα was only faintly detected in the anti-UBE3C immunoprecipitate in the absence of 17β-estradiol, and 17β-estradiol treatment dramatically enhanced the detection of ERα (Figure 1C). To determine whether ERα directly interacts with UBE3C, we purified recombinant FLAG-ERα and FLAG-UBE3C proteins (Supplemental Figure 3A). SPR analysis revealed the direct interaction of ERα with UBE3C (Figure 1D). These results suggest that ERα interacts with UBE3C during mitosis in the presence of estrogen. We then analyzed the mitotic interaction between ERα and UBE3C in more physiological condition using a double thymidine block. Progression through the cell cycle was monitored by flow cytometry, and the interaction was examined by anti-ERα immunoprecipitation followed by anti-UBE3C immunoblotting. Cells in G2/M phases were enriched during 9–14 hours after release from the double thymidine block (Figure 2A). The cell cycle progression was also corroborated by western blot for the expression of Geminin (a marker for S, G2, and early M phases), CCNA (S and G2 phases), CCNB1 (late S, G2, and M phases) and phospho-S126 CCNB1 (M phase) (Figure 2B). Significantly, the interaction between ERα and UBE3C was most evident during late M phase (12 h) to M/G1 transition (14 h) accompanied by the expression of phospho-S126 CCNB1.
Figure 2. Interaction of UBE3C and ERα during cell cycle.

MCF-7 cells were treated with double thymidine block, released to the medium containing 10nM 17β-estradiol for the indicated time length, and subjected to flow cytometry to monitor the cell cycle progression (A) or to immunoprecipitation (IP) with anti-ERα antibody followed by immunoblotting with anti-UBE3C and anti-ERα antibodies (B). Inputs were also loaded and immunoblotted with the indicated antibodies.
Estrogen stimulates the E3 activity of UBE3C in the presence of ER
We next examined whether the interaction of UBE3C with ERα affects the E3 activity of UBE3C. For the purpose, we examined the in vitro E2 activation with UBE3C in the absence of a substrate. We first identified the E2 ubiquitin-conjugating enzyme (E2) that provides the maximum activity with UBE3C in vitro. Eleven representative recombinant E2s were purified (Supplemental Figure 3B). The highest activity was observed with UBE2D3 (UbcH5c), and the other 2 UBE2Ds also exhibited activity with UBE3C (Figure 3A). This E2 specificity for UBE3C is consistent with previous observations (21, 22). Polyubiquitin formation with UBE2D3 was UBE3C-dependent (Figure 3B). Using UBE2D3 as an E2, we next analyzed the effect of ERα and estrogen on the reaction. Different concentrations of ERα and 17β-estradiol were added to the reaction containing constant concentration of UBE2D3 and UBE3C. Significantly, UBE3C-induced in vitro polyubiquitin formation was dramatically enhanced by ERα and 17β-estradiol in a dose-dependent manner (Figure 3C). We further tested whether the effect of ERα and 17β-estradiol was inhibited by the estrogen antagonist tamoxifen. Importantly, the ERα- and 17β-estradiol-dependent enhancement of polyubiquitination by UBE3C was completely blocked by tamoxifen, supporting the specificity of the effect of the liganded ERα (Figure 3D). Together with the interaction experiments, the results indicate that estrogen increases the E3 activity of ERα-bound UBE3C.
Figure 3. Estrogen enhances the E3 ligase activity of UBE3C in the presence of ERα.

A, Recombinant FLAG-UBE3C was incubated with ubiquitin, E1, and the indicated E2s (UBE2s, 83nM each) in the presence of ATP. The reactions were immunoblotted with antiubiquitin antibody. B, The ubiquitin ligation reaction was performed as in A with UBE2D3 and in the absence or presence of an increased amount of UBE3C. C, The ubiquitin ligation reaction was performed as in A with UBE2D3, UBE3C (13.3μM), and the indicated amount of ERα (shown as mole ratios to UBE3C) and 17β-estradiol (lanes 2, 5, and 8: 10nM; lanes 3, 6, and 9: 10μM). The reactions were immunoblotted with the indicated antibodies. *, nonspecific product from ERα preparation. D, The ubiquitin ligation reaction was performed as in C with or without 10nM 17β-estradiol and an increased amount of 4-hydroxytamoxifen (4OHT) (lanes 3 and 6, 100nM; lanes 4 and 7, 1μM; and lanes 5 and 8, 10μM). *, nonspecific product.
UBE3C ubiquitinates and destabilizes CCNB1 in an estrogen- and ERα-dependent manner
The observed enhancement of E3 activity suggested an allosteric effect of liganded ERα on UBE3C. However, it is also possible that liganded ERα is only served as a substrate that stimulates the reaction. To validate the allosteric effect, we searched for candidate substrate(s). Coincidently during the course of our investigation, it was reported that in the metazoan Caenorhabditis elegans, the UBE3C ortholog ETC-1 ubiquitinates and degrades the CCNB1 ortholog CYB-1 (23). Therefore, we tested the interaction of CCNB1 with UBE3C and ERα. Recombinant GST-CCNB1 was purified (Supplemental Figure 3A) and incubated with FLAG-UBE3C and FLAG-ERα. GST pull-down demonstrated that UBE3C effectively interacts with GST-CCNB1 but not with GST alone (Figure 4A). By contrast, no interaction of ERα with GST-CCNB1 was detected.
Figure 4. Estrogen enhances UBE3C-mediated ubiquitination and degradation of CCNB1.

A, Interaction of UBE3C with CCNB1 in vitro. The indicated recombinant proteins and 17β-estradiol were mixed and subjected to GST pull-down. Protein interactions were evaluated by immunoblotting with the indicated antibodies. Inputs (0.5%) were also loaded. Note that the faint amount of ERα detected in the GST-CCNB1 pull-downs is likely nonspecific, because a similar amount of ERα was detected in the control GST pull-down. B, ERα and 17β-estradiol enhance the ubiquitination of CCNB1 by UBE3C in vitro. Ubiquitin, E1, UBE2D3, and GST-CCNB1-His were incubated for the indicated time with or without FLAG-UBE3C, FLAG-ERα, and 17β-estradiol. The reactions were immunoblotted with anti-CCNB1 antibody. C, UBE3C from cells treated with estrogen is capable of ubiquitinating CCNB1 in vitro. Anti-UBE3C immunoprecipitates from mitotic MCF-7 cells treated with or without 17β-estradiol for 2 hours were mixed with ubiquitin, E1, UBE2D3, and GST-CCNB1-His and left untreated (right panel) or incubated (left panel) with ATP. MG132 was added to the cell lysate and the reaction where indicated. The reaction was immunoblotted with anti-CCNB1 antibody. D, Estrogen enhances the ubiquitination of CCNB1 in vivo. MCF-7 cells were treated with nocodazole for 24 hours. MG132 and 17β-estradiol were added as indicated during the last 4 hours of the incubation. Cell lysates were immunoprecipitated with anti-CCNB1 antibody or control IgG under denaturing conditions and subjected to immunoblotting with an anti-CCNB1 antibody. E, UBE3C-dependent destabilization of CCNB1 by estrogen during mitosis. MCF-7 cells transfected with the indicated siRNAs were treated with nocodazole for 24 hours. MG132 and 17β-estradiol were added as indicated during the last 4 hours of the incubation. Steady-state levels of CCNB1, UBE3C, and actin were analyzed by immunoblot. F, CCNB1 mRNA expression levels in cells treated as in E were analyzed by qPCR. Relative ratios normalized to GAPDH mRNA expression are shown.
We next examined whether ERα and 17β-estradiol affect the UBE3C-mediated ubiquitination of CCNB1. GST-CCNB1 was incubated with ubiquitin, E1, UBE2D3, and UBE3C in the presence or absence of ERα and 17β-estradiol. Anti-CCNB1 immunoblotting demonstrated the UBE3C-dependent ubiquitination of CCNB1, which was enhanced by ERα and 17β-estradiol (Figure 4B). We also examined the in vitro ubiquitination of CCNB1 using an anti-UBE3C immunoprecipitate obtained from mitotic MCF-7 cells treated or not with 17β-estradiol (Figure 4C). CCNB1 ubiquitination was not detected after incubation with the UBE3C immunocomplex regardless of the addition of 17β-estradiol to the cell culture. Interestingly, however, the CCNB1 ubiquitination was detected when proteasome inhibitor (MG132) was added to the cell lysate only when 17β-estradiol was added to the cell culture (Figure 4C, lane 4). Because UBE3C is known to interact with the proteasome (24–28), we speculate that coprecipitated proteasomes in the anti-UBE3C immunocomplex may suppress the CCNB1 ubiquitination in vitro without MG132.
We further analyzed the estrogen-induced ubiquitination of UBE3C under physiological conditions in vivo. MCF-7 cells were arrested at mitosis, and CCNB1 was precipitated with an anti-CCNB1 antibody from the cell lysate. Anti-CCNB1 immunoblotting revealed a ladder at approximately 100–120 kDa in the presence of 17β-estradiol (Figure 4D, lane 2). Addition of MG132 enhanced the ladder, suggesting the 17β-estradiol-induced polyubiquitination of CCNB1 (lane 4). The MG132 dependence observed in Figure 4, C and D, suggests that this ubiquitination is the signal for proteasomal degradation, which is generally mediated by Lys11- or Lys48-linked polyubiquitin chains (29, 30). UBE3C catalyzes the polyubiquitination of Lys29- and Lys48-linked chains (22). We therefore analyzed the chains catalyzed by UBE3C in combination with ERα and 17β-estradiol with mutant ubiquitins, in which all but 1 Lys residue were substituted with Arg. Specifically, UBE3C with ERα and 17β-estradiol was able to catalyze only chains with Lys48-ubiquitin (Supplemental Figure 4A). In addition, quantitative mass spectrometric analysis demonstrated that UBE3C-mediated Lys48-linked chains were significantly enhanced by 17β-estradiol in the presence of ERα (Supplemental Figure 4B).
We further examined the 17β-estradiol- and UBE3C-dependent degradation of CCNB1. MCF-7 cells were transfected with either control or UBE3C-specific siRNA and arrested in mitosis with nocodazole. UBE3C expression was effectively inhibited by the siRNA (Figure 4E, middle panel). Incubation with 17β-estradiol dramatically decreased the steady-state level of CCNB1 in control cells (upper panel, lane 2). Importantly, the steady-state level was restored by treatment with MG132 or inhibition of UBE3C. 17β-estradiol treatment did not suppress CCNB1 at the mRNA level, and inhibition of UBE3C did not increase it (Figure 4F). Together, these results suggest that UBE3C and liganded ERα coordinately destabilize CCNB1 during mitosis.
Colocalization of ERα, UBE3C, and CCNB1 during mitosis
Although ERα localizes in the nucleus in interphase cells, its localization during mitosis has not been clarified. Therefore, we analyzed the subcellular localization of ERα during each phase of mitosis (Figure 5A). ERα predominantly accumulated in the nucleus in prophase. It then localized to the mitotic spindle during metaphase and anaphase and to the polar microtubules during telophase. A competing peptide for the epitope of the ERα antibody and inhibition of ERα expression with siRNA both inhibited the stain of ERα including that at the mitotic structures, supporting the specificity of the antibody (Supplemental Figure 5). The subcellular localization pattern of ERα during prophase and metaphase is concordant with that of CCNB1, which localizes in the cytoplasm in interphase, enters nuclei at prophase and associates with spindles at metaphase after nuclear envelope breakdown (Supplemental Figure 6A) (31, 32). We therefore verified the colocalization of ERα and CCNB1 (Figure 5B). As expected, CCNB1 colocalized with ERα in prophase nuclei, evading chromatin (see also a magnified view in Supplemental Figure 6B). Both proteins were condensed at a position close to the metaphase chromatin. Then, CCNB1 disappeared in anaphase and telophase, whereas ERα remained at the microtubules.
Figure 5. Colocalization of UBE3C, ERα, and CCNB1 during mitosis.
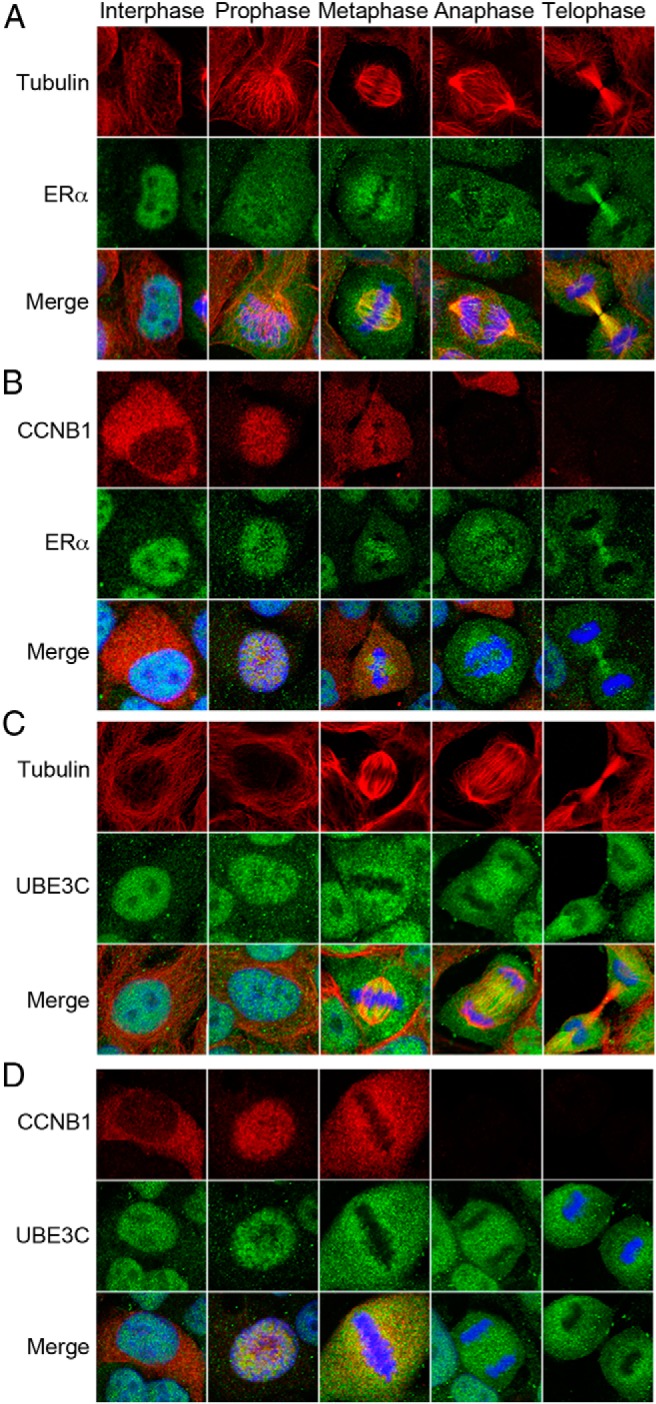
Exponentially growing MCF-7 cells were fixed, permeabilized, and coimmunostained with antibodies to either tubulin and ERα (A), CCNB1 and ERα (B), tubulin and UBE3C (C), or CCNB1 and UBE3C (D). The nucleus was stained with DAPI (blue). Merge indicates an overlay of the images of the 2 proteins and the nucleus. A representative cell in interphase and each phase of mitosis is shown.
We also examined the subcellular localization of UBE3C during mitosis. Significantly, UBE3C also localized in prophase nuclei and at the metaphase spindle in a similar fashion to ERα (Figure 5C and Supplemental Figure 6B) and colocalized with CCNB1 (Figure 5D). The specificity of the anti-UBE3C antibody staining the mitotic structures was verified with a competing peptide and siRNA inhibition of UBE3C (Supplemental Figure 7). Together, the immunofluorescence data indicate that ERα and UBE3C colocalize with CCNB1 during early mitosis before CCNB1 is degraded.
Depletion of UBE3C attenuates estrogen-dependent cell proliferation
The cooperative effect of UBE3C and liganded ERα on the stability of CCNB1 suggests that this cooperative effect affects cell proliferation. To test this possibility, we analyzed the impact of UBE3C on the estrogen-stimulated growth of MCF-7 cells. Cells were transfected with either control or UBE3C-specific siRNA and cultured in the presence or absence of 17β-estradiol. Significantly, inhibition of UBE3C eliminated the effect of 17β-estradiol on day 3 of the culture and reduced it by approximately half on day 5 (Figure 6A). The remaining effect of 17β-estradiol on proliferation in UBE3C-defective cells could be due to genomic actions of liganded ERα. The inhibition of UBE3C did not affect the growth of ERα-negative U2OS and HCT116 cells, supporting the ERα-dependent role of UBE3C on cell proliferation (Supplemental Figure 8). We next evaluated the impact of UBE3C on estrogen-stimulated cell growth in a different way. MCF-7 cells were transfected with either wild-type UBE3C or a catalytically inactive UBE3C (C1051A) mutant (33) and cultured in the presence or absence of 17β-estradiol. Consistent with the siRNA experiment, expression of the mutant UBE3C eliminated the effect of 17β-estradiol on day 3 of culture (Figure 6B). Although these data are consistent with proliferation inhibition due to aberrant regulation of CCNB1 stability, it remained unclear whether altered transcriptional activity of liganded ERα caused by inhibition of UBE3C contributes to the observed phenotype. To exclude this possibility, we first examined the transcriptional activity of ERα using an ERE-TATA luciferase reporter (3xERE-TATA-Luc). MCF-7 cells were transfected with either control, UBE3C- or ERα-specific siRNA. Cells were additionally transfected with 3xERE-TATA-Luc and cultured in the presence or absence of 17β-estradiol for 24 hours before harvest. UBE3C and ERα expressions were both effectively suppressed by the specific siRNAs (Supplemental Figure 9). Inhibition of UBE3C did not have a detectable effect on 17β-estradiol-induced luciferase activity, whereas inhibition of ERα completely abolished this activity (Figure 6C). Similarly, 17β-estradiol-induced luciferase activity was unaffected by the exogenous expression of either wild-type UBE3C or the C1051A mutant (Figure 6D).
Figure 6. UBE3C inhibition suppresses estrogen-dependent cell proliferation without affecting its transcriptional activity.

A and B, Inhibition of UBE3C suppresses estrogen-induced cell proliferation. MCF-7 cells transfected with control or UBE3C-specific siRNA (A), mock transfected, or transfected with wild-type (WT) FLAG-UBE3C or its C1051A (CA) mutant (B) were cultured for the indicated time with or without 17β-estradiol. Live cells were counted, and the mean ± SD from triplicate experiments is shown. C and D, Inhibition of UBE3C does not affect estrogen-induced transactivation by ERα. MCF-7 cells were transfected with control, UBE3C-, or ERα-specific siRNA (C), mock transfected, or transfected with WT FLAG-UBE3C or its C1051A (CA) mutant (D). Cells were additionally transfected with pGL3-3xERE-TATA-Luc and cultured for 24 hours with or without 17β-estradiol. Luciferase-based transcriptional activity values are expressed as the mean ± SD. E, Estrogen-regulated gene expression is not affected by the inhibition of UBE3C. MCF-7 cells transfected with the indicated siRNA were cultured for 6 hours with or without 17β-estradiol, and CCND1 (left panel) or FOS (right panel) mRNA expression levels were analyzed by real-time quantitative PCR. Relative ratios normalized to GAPDH mRNA expression are shown. The mean ± SD is shown.
We further analyzed the effect of UBE3C status on the transcriptional activity of ERα via endogenous gene expression. MCF-7 cells were transfected with either control, ERα- or UBE3C-specific siRNA and cultured with or without 17β-estradiol, and the mRNA expression levels of the estrogen-regulated genes CCND1 and FOS were analyzed by qPCR. Although inhibition of ERα diminished the estrogen-induced expression of CCND1 and FOS, inhibition of UBE3C did not significantly affect their expression (Figure 6E). These results suggest that UBE3C is partly responsible for estrogen-dependent cell proliferation without affecting the transactivation function of ERα.
Finally, we investigated the effect of 17β-estradiol and UBE3C inhibition on mitotic progression. Using a double thymidine block we first analyzed the effect of estrogen on the cell cycle progression during late S to G1 phases (7.5–24 h after release from the double thymidine block) (Figure 7A). Addition of 17β-estradiol did not affect the population in each phase of cell cycle until 13.5 hours after the release. However, it reduced cells in G2/M phase from 31.1% to 16.5%, and increased cells in G1 phase from 60.9% to 76.3% at 15 hours after the release. The difference was gradually adapted but persisted for the next few hours. These findings suggest that the transition from M phase to G1 phase was accelerated by estrogen. We then examined whether the estrogen-induced acceleration of the M/G1 transition would be dependent on UBE3C. For the purpose, MCF-7 cells depleted for UBE3C with siRNA were synchronized with double thymidine block and the cell cycle phases were analyzed at 13.5 and 15 hours after the release (Figure 7B). Either addition of 17β-estradiol or UBE3C inhibition did not affect the cell cycle at 13.5 hours. Importantly, however, addition of 17β-estradiol reduced cells in G2/M phase from 36.9% to 26.1%, and the reduction was inverted to 42.5% by UBE3C depletion at 15 hours. Reciprocally, addition of 17β-estradiol increased cells in G1 phase from 55.3% to 67.7%, and it was inverted to 52.0% by UBE3C depletion at the time point. These findings suggest that the M/G1 transition is accelerated by estrogen in a manner dependent on UBE3C.
Figure 7. UBE3C-dependent acceleration of mitotic progression by 17β-estradiol.

A, MCF-7 cells were treated with double thymidine block, released to the medium with or without 10nM 17β-estradiol for the indicated time length, and subjected to flow cytometry. B, MCF-7 cells transfected with control or UBE3C-specific siRNA were treated with double thymidine block and released with or without 10nM 17β-estradiol for 13.5 or 15 hours. Cell cycle phases were analyzed by flow cytometry.
Discussion
In this study, we demonstrated that ERα acts as a ligand-dependent regulatory subunit of UBE3C E3 ligase; UBE3C ubiquitinates and degrades CCNB1 during mitosis. Our findings provide 2 important new insights into the molecular mechanisms of cellular functions: the ligand-dependent nongenomic action of a NR in mitosis and ligand-dependent enhancement of the activity of a HECT E3 ligase by a NR.
The ligand-dependent nongenomic action of ERα has well been illustrated for its role in PI3K and MAPK pathways (8, 15, 34). Upon estrogen stimuli, plasma membrane-associated ERα colocalizes with eNOS and activates its enzymatic activity in endothelial cells (13). At the membrane ERα interacts with the p85α regulatory subunit of PI3K and activates PI3K as well as its downstream target AKT (15), and phosphorylation of eNOS by AKT stimulates its enzymatic activity (11, 12, 14). The estrogen-induced activation of PI3K requires interaction between c-Src kinase and ERα that is also important for the localization of ERα at the plasma membrane (35). In addition, many other molecules, including protein arginine methyltransferase 1 (36), hematopoietic PBX-interacting protein (37), Src-homology and collagen homology (38), modulator of nongenomic activity of ER (39, 40), and p130Cas (Crk-associated substrate) (41), have been involved in the mechanism. Functional cross talk between the ERα-induced kinase activation and UBE3C activation has not been tested in this study. Although liganded ERα directly interacts with UBE3C and enhances its E3 activity in vitro, the kinases and the associated molecules may affect the ERα-mediated UBE3C function in vivo. The possible connection of these mechanisms remains to be examined.
UBE3C catalyzes Lys29- and Lys48-linked polyubiquitin chains (22) and targets the transcriptional regulator TIP120B/CAND2, a TATA-binding protein-interacting protein (33), and Interferon regulatory factor-3 and Interferon regulatory factor-7, transcription factors that regulate type I interferon (42), for degradation. Importantly, Hul5, a yeast homologue of UBE3C, associates with the proteasome and functions as an E4 that can extend the ubiquitin chains on substrates to facilitate their complete degradation (24, 25). This function is conserved in mammalian UBE3C but not in its homologue UBE3A/E6-AP (26–28). These findings may suggest that UBE3C and the proteasome concertedly function in transcription to degrade transcription factors and cofactors. UBE3A/E6-AP interacts with and degrades estrogen-liganded ERα in collaboration with the proteasome to promote cyclic clearance of transcriptional coactivators, which is required for efficient ERα transactivation (43, 44). However, although UBE3C also interacts with ERα, our data suggest that it does not affect the ligand-dependent transactivation function of ERα. Whether UBE3C plays other roles with ERα during interphase remains to be clarified.
How liganded ERα exerts allosteric effects on the E3 activity of UBE3C is unknown. A recent report demonstrated that ERα noncovalently interacts with ubiquitin chains (45). A ubiquitin-binding surface within the ligand-binding domain in ERα directs interactions with both ubiquitinated proteins and recombinant ubiquitin chains. The noncovalent interaction of ERα with ubiquitin chains may contribute to the enhancement of the E3 ligase activity of UBE3C. UBE3C is a unique leucine-rich protein composed of a number of NR-boxes, 3 LxxLL motifs and 7 LxxLL-like (L/I substitution) motifs. The NR-box interacts with liganded ERα (46). Although SPR analysis demonstrated that UBE3C can interact with ERα in the absence of estrogen, in the presence of estrogen, the NR-boxes in UBE3C may increase its affinity or change its conformation to contribute to the estrogen-dependent augmentation of UBE3C E3 activity in vitro and the estrogen-dependent interaction of ERα with UBE3C in vivo. The LxxLL motif can interact with other members of the nuclear hormone receptor family. UBE3A/E6AP also contains LxxLL motifs and plays important roles as a coactivator of nuclear hormone receptors, including the progesterone receptor (PR), ERα, and the androgen receptor (AR) (18). Notably, UBE3C interacts with the androgen receptor in mitotic cells, and this interaction is increased by androgen (Supplemental Figure 10). This interaction suggests that UBE3C not only plays mitotic roles in ERα-positive cells but may also function in cells with other active NRs.
The cellular abundance of CCNB1 increases toward mitosis, activating CDK1 and promoting a variety of mitotic events, including chromosome condensation, nuclear envelope breakdown, and spindle pole assembly (47, 48). Rapid degradation of CCNB1 during the midphase of mitosis is also important to promote the exit from mitosis, and persistent presence of CCNB1 causes failure of the metaphase-to-anaphase transition (49). The E3 that promotes this degradation is the anaphase-promoting complex/cyclosome (APC/C). However, ERα/UBE3C may have a supportive role in the degradation of CCNB1 during mitosis in estrogen-targeted organs. In C. elegans, the UBE3C ortholog ETC-1 ubiquitinates and degrades CYB-1 and IFY-1, orthologues of human CCNB1 and securin, respectively (23). Depletion of ETC-1 increases the stability of IFY-1 and CYB-1 during meiosis II and causes a delay in anaphase II. Interestingly, inhibition of ETC-1 by siRNA causes synthetic lethality with reduced APC/C activity, suggesting that ETC-1 functions redundantly with APC/C. ETC-1 activity is dispensable for the progression of meiosis I in the presence of APC/C but essential for progression from metaphase I to anaphase I when APC/C activity is compromised (23). Together with the data obtained in this study, these results suggest that UBE3C also functions redundantly with APC/C and degrades CCNB1 during mitosis in mammalian cells in a fashion similar to ETC-1. Although ERα is not conserved in metazoans, other NR(s) may play a supportive role in the ETC-1-mediated degradation of CYB-1 and IFY-1 because the LxxLL motifs are conserved in ETC-1 and steroid hormones regulate many cellular processes in nematodes (50). Similar to ETC-1, UBE3C likely functions independently of APC/C, because depletion of UBE3C stabilizes CCNB1 in cells in which APC/C activity is inhibited by nocodazole.
The physiological role of this redundancy in mitotic progression promoted by either estrogen-dependent activation of UBE3C and subsequent CCNB1 degradation or by APC/C is not clear. However, because depletion of UBE3C reduces estrogen-dependent cell proliferation, this mechanism could be important for estrogen-induced acceleration of cell proliferation in addition to the fundamental role of APC/C in mitotic progression. Estrogen-dependent cell proliferation is also promoted by the transcriptional activation of ERα-targeted genes such as cyclin D1 and E2F1 that drive the G1/S transition (51–53). The gene expression and enhancement of UBE3C activity mediated by ERα may coordinately regulate G1/S and M phases and be a prerequisite for the estrogen-induced acceleration of cell growth. Because the stimulation of UBE3C by liganded ERα is inhibited by the estrogen antagonist tamoxifen, this mechanism could be a viable therapeutic target in clinical applications, including breast cancer treatment. Alternatively, this mechanism could reflect the antiproliferation effects generally observed with antiestrogen agents. In conclusion, we provide the initial evidence for a mitotic role of the ER that facilitates cell proliferation and may represent an additional target in hormone therapy.
Acknowledgments
We thank Yukiko Togashi, Shinko Hanaki, Hikaru Tsuchiya, and Hidehito Yoshihara for technical support of the plasmid preparation, immunoprecipitation, immunoblot experiments, and Aqua analysis.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (T.O. and M.O.) and a Health Labor Sciences Research Grant of Japan (T.O.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (T.O. and M.O.) and a Health Labor Sciences Research Grant of Japan (T.O.).
Footnotes
- AKT
- protein kinase B
- APC/C
- anaphase-promoting complex/cyclosome
- CCNB1
- cyclin B1
- eNOS
- endothelial nitric oxide synthase
- ER
- estrogen receptor
- ERE
- estrogen-response element
- FBS
- fetal bovine serum
- NR
- nuclear receptor
- PI3K
- phosphatidylinositol-3-kinase
- qPCR
- quantitative real-time RT-PCR
- SPR
- surface plasmon resonance.
References
- 1. Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. [DOI] [PubMed] [Google Scholar]
- 2. Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–951. [DOI] [PubMed] [Google Scholar]
- 3. Welboren WJ, Sweep FC, Span PN, Stunnenberg HG. Genomic actions of estrogen receptor α: what are the targets and how are they regulated? Endocr Relat Cancer. 2009;16:1073–1089. [DOI] [PubMed] [Google Scholar]
- 4. Gao H, Dahlman-Wright K. The gene regulatory networks controlled by estrogens. Mol Cell Endocrinol. 2011;334:83–90. [DOI] [PubMed] [Google Scholar]
- 5. Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bennesch MA, Picard D. Minireview: tipping the balance: ligand-independent activation of steroid receptors. Mol Endocrinol. 2015;29:349–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banerjee S, Chambliss KL, Mineo C, Shaul PW. Recent insights into non-nuclear actions of estrogen receptor α. Steroids. 2014;81:64–69. [DOI] [PubMed] [Google Scholar]
- 8. Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor α mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hisamoto K, Ohmichi M, Kurachi H, et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2001;276:3459–3467. [DOI] [PubMed] [Google Scholar]
- 10. Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci USA. 1999;96:4686–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Florian M, Lu Y, Angle M, Magder S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids. 2004;69:637–645. [DOI] [PubMed] [Google Scholar]
- 12. Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors α and β. J Biol Chem. 2005;280:19704–19710. [DOI] [PubMed] [Google Scholar]
- 13. Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–14743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. [DOI] [PubMed] [Google Scholar]
- 15. Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nishikawa H, Ooka S, Sato K, et al. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279:3916–3924. [DOI] [PubMed] [Google Scholar]
- 17. Koike A, Nishikawa H, Wu W, Okada Y, Venkitaraman AR, Ohta T. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res. 2010;70:6746–6756. [DOI] [PubMed] [Google Scholar]
- 18. Nawaz Z, Lonard DM, Smith CL, et al. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol. 1999;19:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramamoorthy S, Nawaz Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal. 2008;6:e006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun J, Zhou W, Kaliappan K, Nawaz Z, Slingerland JM. ERα phosphorylation at Y537 by Src triggers E6-AP-ERα binding, ERα ubiquitylation, promoter occupancy, and target gene expression. Mol Endocrinol. 2012;26:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fang NN, Ng AH, Measday V, Mayor T. Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat Cell Biol. 2011;13:1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. You J, Pickart CM. A HECT domain E3 enzyme assembles novel polyubiquitin chains. J Biol Chem. 2001;276:19871–19878. [DOI] [PubMed] [Google Scholar]
- 23. Wang R, Kaul Z, Ambardekar C, et al. HECT-E3 ligase ETC-1 regulates securin and cyclin B1 cytoplasmic abundance to promote timely anaphase during meiosis in C. elegans. Development. 2013;140:2149–2159. [DOI] [PubMed] [Google Scholar]
- 24. Crosas B, Hanna J, Kirkpatrick DS, et al. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell. 2006;127:1401–1413. [DOI] [PubMed] [Google Scholar]
- 25. Aviram S, Kornitzer D. The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol Cell Biol. 2010;30:985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martínez-Noël G, Galligan JT, Sowa ME, et al. Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol Cell Biol. 2012;32:3095–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chu BW, Kovary KM, Guillaume J, Chen LC, Teruel MN, Wandless TJ. The E3 ubiquitin ligase UBE3C enhances proteasome processivity by ubiquitinating partially proteolyzed substrates. J Biol Chem. 2013;288:34575–34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Besche HC, Sha Z, Kukushkin NV, et al. Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 2014;33:1159–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsumoto ML, Wickliffe KE, Dong KC, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 2010;39:477–484. [DOI] [PubMed] [Google Scholar]
- 30. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. [DOI] [PubMed] [Google Scholar]
- 31. Santos SD, Wollman R, Meyer T, Ferrell JE Jr. Spatial positive feedback at the onset of mitosis. Cell. 2012;149:1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003;5:143–148. [DOI] [PubMed] [Google Scholar]
- 33. You J, Wang M, Aoki T, Tamura TA, Pickart CM. Proteolytic targeting of transcriptional regulator TIP120B by a HECT domain E3 ligase. J Biol Chem. 2003;278:23369–23375. [DOI] [PubMed] [Google Scholar]
- 34. Kousteni S, Bellido T, Plotkin LI, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- 35. Haynes MP, Li L, Sinha D, et al. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. 2003;278:2118–2123. [DOI] [PubMed] [Google Scholar]
- 36. Le Romancer M, Treilleux I, Leconte N, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31:212–221. [DOI] [PubMed] [Google Scholar]
- 37. Manavathi B, Acconcia F, Rayala SK, Kumar R. An inherent role of microtubule network in the action of nuclear receptor. Proc Natl Acad Sci USA. 2006;103:15981–15986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song RX, McPherson RA, Adam L, et al. Linkage of rapid estrogen action to MAPK activation by ERα-Shc association and Shc pathway activation. Mol Endocrinol. 2002;16:116–127. [DOI] [PubMed] [Google Scholar]
- 39. Barletta F, Wong CW, McNally C, Komm BS, Katzenellenbogen B, Cheskis BJ. Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Mol Endocrinol. 2004;18:1096–1108. [DOI] [PubMed] [Google Scholar]
- 40. Cheskis BJ, Greger J, Cooch N, et al. MNAR plays an important role in ERα activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids. 2008;73:901–905. [DOI] [PubMed] [Google Scholar]
- 41. Cabodi S, Moro L, Baj G, et al. p130Cas interacts with estrogen receptor α and modulates non-genomic estrogen signaling in breast cancer cells. J Cell Sci. 2004;117:1603–1611. [DOI] [PubMed] [Google Scholar]
- 42. Yu Y, Hayward GS. The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity. 2010;33:863–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol Cell. 2000;5:939–948. [DOI] [PubMed] [Google Scholar]
- 44. Reid G, Hübner MR, Métivier R, et al. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. [DOI] [PubMed] [Google Scholar]
- 45. Pesiri V, La Rosa P, Stano P, Acconcia F. Identification of an estrogen receptor α non covalent ubiquitin-binding surface: role in 17β-estradiol-induced transcriptional activity. J Cell Sci. 2013;126:2577–2582. [DOI] [PubMed] [Google Scholar]
- 46. Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. [DOI] [PubMed] [Google Scholar]
- 47. Gavet O, Pines J. Progressive activation of cyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol. 2010;189:247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. [DOI] [PubMed] [Google Scholar]
- 50. Maglich JM, Sluder A, Guan X, et al. Comparison of complete nuclear receptor sets from the human, Caenorhabditis elegans and Drosophila genomes. Genome Biol. 2001;2:research0029.1–research0029.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997;272:10882–10894. [DOI] [PubMed] [Google Scholar]
- 52. Planas-Silva MD, Weinberg RA. Estrogen-dependent cyclin E-cdk2 activation through p21 redistribution. Mol Cell Biol. 1997;17:4059–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. [DOI] [PubMed] [Google Scholar]