

Abstract
Progesterone (P), which signals through the P receptor (PR), is critical in normal development of the breast, but its signaling axis is also a major driver of breast cancer risk. Here we review recent advances in the understanding of P signaling in the normal human breast, with a focus on the importance of the balance between autocrine and paracrine signaling. To date, most data (which derive largely from mouse models or human breast cancer cell line studies) have demonstrated that the vast majority of PR+ cells appear to act as “sensor” cells, which respond to P stimulation by translating these hormonal cues into paracrine signals. However, growing evidence suggests that, dependent on the cellular context, P may also signal in an autocrine manner in a subset of cells in the normal mouse mammary gland and human breast. It has been suggested that it may be dysregulation of this autocrine signaling, resulting in a “switch” from a predominance of paracrine signaling to autocrine signaling in PR+ cells, which is an early event during breast tumorigenesis. This review summarizes current evidence in the literature that demonstrates the mechanisms through which P acts in the normal human breast, as well as highlighting the important questions that remain unanswered.
The ovarian hormone, progesterone (P), plays a pivotal role in normal female reproduction. Although known to be critical in the growth and proliferation of the breast during normal development, signaling through P receptor (PR) has been implicated in breast cancer, and synthetic P analogues have been associated with increased breast cancer risk. Prolonged ovarian activity, either early menarche or late menopause, profoundly influences breast cancer risk, and removal of the ovaries reduces breast cancer risk by more than 50%, implicating the ovarian hormones in breast tumorigenesis (1, 2). A specific role for signaling via PR is demonstrated in animal models, where PR is required for mammary carcinogenesis in mice (3), and this is supported by clinical trials in humans showing that exposure to exogenous hormones, such as progestins in hormone-replacement therapy (HRT) and oral contraceptives (OCs), is associated with increased breast cancer risk and/or mortality (4–9). This highlights the importance of understanding the molecular mechanisms of P signaling, both in the normal breast and in the development and progression of breast cancer. Importantly, due to the limited availability of normal human breast tissue, the vast majority of our knowledge on the mechanisms of these hormones has evolved from animal models and cell line studies, and recapitulation of these mechanisms in the normal human breast largely remains to be confirmed.
The effects of P are mediated by binding to the nuclear PR to regulate hormone-responsive target genes. Newly transcribed cytoplasmic PR is assembled in an inactive multiprotein chaperone complex, which dissociates upon ligand binding and receptor activation. Binding of P to PR induces a conformational change leading to dissociation of chaperones, receptor dimerization, binding of receptor dimers to specific P-response elements in enhancer regions and the promoters of target genes, and recruitment of specific coactivators and general transcription factors (10), resulting in modulation of transcription of those genes. There is evidence that gene targets of P become colocated in the nucleus to form hotspots of transcriptional regulation (11), and these ligand-dependent active transcription units can be visualized as discrete nuclear aggregates, or foci, as opposed to the diffuse, fine granular nuclear distribution of PR in unstimulated cells (12, 13). PR is a member of a large family of ligand-activated nuclear transcription factors, and is expressed as 2 distinct isoforms, PRA and PRB, with molecular masses of approximately 81 and 115 kDa, respectively. These isoforms are transcribed from distinct promoters on a single gene residing on chromosome 11q22-q23 (14), and are identical in sequence except that the shorter form, PRA, lacks 164 amino acids at the N terminus (15). The structure of PR includes a central DNA-binding domain and a C-terminal ligand-binding domain, and a number of activation function (AF) and inhibitory function elements, which enhance and repress transcriptional activation of PR by association of these regions with transcriptional coregulators (16–23). The region of the protein that is unique to PRB contains a transcription AF, AF3, in addition to AF1 and AF2, which are common to PRA (Figure 1) (20).
Figure 1. Schematic diagrams of PRA and PRB structures depicting structural domains of each isoform.
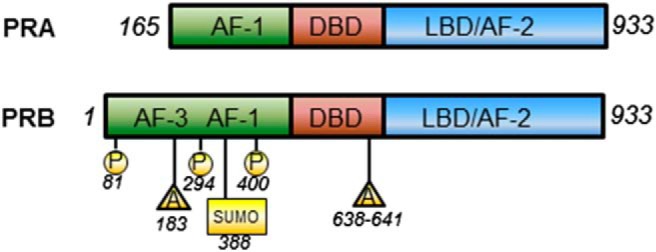
DBD, DNA-binding domain; LBD, ligand-binding domain. Selected posttranslational modifications are also shown. P, phosphorylation; A, acetylation; SUMO, sumoylation.
The activity of PR, and its degradation, are tightly regulated by posttranslational modifications, predominantly in the N-terminal region of each isoform. For example, PR is targeted for down-regulation by the 26S proteasome by phosphorylation at Ser294 by MAPKs, with this turnover being critical for its activity (24), and the Ser400 residue is phosphorylated in response to elevated cyclin-dependent protein kinase 2 activity (25). Ser400 phosphorylation occurs both in the presence and absence of ligand, and indeed, there is evidence that some phosphorylation can occur upon kinase activation in response to growth factors, rendering PR constitutively active in a ligand-independent manner (26). Some modifications can also occur in an isoform-specific manner; the cancer-associated kinase, ck2, has been shown to phosphorylate PRB at Ser81, located in the region unique to PRB (27). In addition to phosphorylation, PR activity can be regulated by other forms of modifications, including acetylation and sumoylation, and together, these different forms of posttranslational modifications are critical in regulating the localization of PR, its stability, and transcriptional activity (reviewed in Ref. 28).
PR proteins are expressed in the nuclei of cells in a variety of human tissues, including the mammary gland, uterus and the ovaries, as well as in nonreproductive tissues, such as the brain, the cardiovascular system, bone and central nervous system. The tissue-specific distribution of PR varies greatly, from positive expression in virtually every cell in the uterus, both epithelial and stromal (29), to being only expressed in a small subset of epithelial cells in the breast (30). Because there are 2 PR isoforms, there is the potential for 3 molecular species (PRB homodimers, PRA homodimers and PRA-PRB heterodimers) to exist concurrently and to contribute to the complexity of PR action. PR homodimers and the heterodimer have the capacity to regulate different suites of genes, and the ratio of PR isoform expression plays an important role in influencing the transcriptional program regulated by PR in target tissues (31–38). In the human breast, most PR+ cells coexpress PRA and PRB at equivalent levels, suggesting that both proteins are required to mediate physiologically relevant P signaling (39, 40). Despite this, data derived from animal models and transient cotransfection studies in various cell lines suggest that these 2 PR isoforms are functionally distinct, and that PRB acts mostly as a transcriptional activator, whereas PRA can act as a transdominant inhibitor of PRB in situations where PRA has little or no transactivational activity (21).
The protein products from PR target genes mediate a diverse array of cellular activities, including cell proliferation, transcription, lipid metabolism and membrane-associated signal transduction, indicating an extensive range of potential P-mediated effects. This wide range of P function is illustrated in PR knockout (PRKO) mouse models, which display pleiotropic reproductive abnormalities, including an inability to ovulate, uterine hyperplasia and inflammation, and severely limited lobuloalveolar development in the mammary gland (41). This review will focus specifically on current understanding of PR expression and the mechanisms of P regulation of proliferation in the different cell types of the normal breast and breast cancer, derived from animal, human, and cell line models.
PR Expression and Distribution in the Breast
The expression and distribution of PR in the breast is likely to be modulated by age and reproductive history. Although there are limited data available during pubertal human breast development, nuclear PR expression is detected in most luminal epithelial cells (42). In contrast, the expression of PR in the adult human breast has been studied in much more detail, with PR detected in up to 20% of luminal cells of the normal premenopausal breast, in either the follicular or luteal phase of the menstrual cycle (42, 43). There is increasing evidence that full-term pregnancy induces long-term gene expression changes in the human breast (44), with data suggesting that parous individuals display reduced expression levels of PR mRNA transcripts, compared with nulliparous subjects (45, 46), a mechanism which may possibly contribute to the long-term protective effect of pregnancy on breast cancer risk. Alterations in the hormonal milieu, as a consequence of exposure to exogenous hormones or endocrine therapy, also play a role in modulating PR expression in the breast. The intensity of PR detection but not the percentage of PR+ cells is increased with use of the OC pill (47), whereas tamoxifen treatment can decrease the proportion of PR+ cells (48). PR expression in the postmenopausal breast can also be regulated by HRT, as evidenced by the increased numbers of PR+ cells in the normal breast of women taking either estrogen (E) only, or combined E+P HRT formulations (49, 50).
The epithelium of the breast is lined by a single layer of luminal cells associated with secretory activity, surrounded by a basal cell layer, consisting mostly of myoepithelial cells with contractile properties, as well as progenitor cells (51). These multiple cell types within the epithelium are thought to descend from a complex hierarchy, arising from multipotent stem cells, which self-renew and give rise to uncommitted bipotent progenitors, and yield the major luminal and basal/myoepithelial lineages (52). To date, the different epithelial cell types have been defined through the use of cell type-specific surface markers to isolate subpopulations enriched for particular cell characteristics. The challenge with this approach is the paucity of reliable markers of stem and bipotent progenitor cells, particularly in the human, and the fact that, although enriched, these are not pure populations. Thus, distinguishing and defining the different cell types is difficult, and although it is known that both myoepithelial cells and progenitor cells express basal markers (52–54), functional assays are the only means by which the identity of a cell can be discerned.
A number of investigators have sought to define the PR+ cell populations in the human breast and mouse mammary gland. PR expression is restricted to a subset of epithelial cells, with receptor positivity observed in approximately 20%–30% of luminal cells in the adult human breast (30, 55). In murine models, and using the type of approach described above, the mammary stem cell (MaSC) compartment has been reported to be PR-negative (56). However, depending on the method and markers used to enrich for these stem cells, there has been the suggestion of the existence of a subpopulation of cells with stem-like characteristics which do express steroid hormone receptors (57). Murine MaSCs have also been proposed to be those which retain their template DNA strands during mitosis (58). These label-retaining epithelial cells (LRECs) comprised approximately 2% of the murine mammary epithelium and were detected in both luminal and basal positions, and interestingly, 30%–40% of these LRECs also expressed PR (57). Similarly, 3H-thymidine labeling of normal human breast epithelium transplanted into mice also suggested that PR was expressed in a significant subset of these LRECs (59). In support of this notion, there is growing evidence that PR can be expressed in human breast progenitor cells. Using cell fractionation strategies to sort primary normal human breast epithelial cells into cell subsets enriched for different lineage characteristics, PR transcripts were detected in the bipotent progenitor-enriched fraction of primary normal human breast epithelial cells (53, 54). The expression of PR protein has been demonstrated in a subset of basal cells expressing smooth muscle actin, cytokeratin-14 and p63 in normal human breast tissue sections (46, 53), and strong PR expression has also been detected in basally located cells enriched for progenitor cells which also expressed cytokeratin-8 (60). PR has also been shown to be expressed in a rare subset of cells expressing CD10 (61, 62), a marker which is enriched in the bipotent progenitor cell compartment (63–65).
Regulation of the expression of the individual PR isoforms in the human breast epithelium is quite distinct from that which occurs in other tissues, as well as in rodent models. For example, in mice, PRA and PRB are frequently expressed in different cells within the reproductive system (66, 67), and PRA or PRB homodimers are critical contributors to P action, consistent with their divergent and tissue-specific roles identified in mouse knockout studies (31, 32). In addition, in the human endometrium during the luteal phase of the menstrual cycle when high circulating levels of P are associated with decreased PR expression, PRA was preferentially decreased, resulting in a distinct predominance of PRB in these cells at this time (39). In the normal breast epithelium, however, PRA and PRB are most commonly expressed at similar levels, whereas an imbalance of PRA and PRB occurs early in breast cancer development and is commonly seen in premalignant lesions (40). This alteration in isoform ratio progressively increases from normal to early lesions, such as atypical hyperplasia, through to ductal carcinoma in situ and invasive cancers, and moreover, there is marked heterogeneity of PRA:PRB expression between neighboring cells in breast cancers (40). Alterations in PRA:PRB expression have also been linked to treatment response, and PRA predominance has been associated with resistance to tamoxifen (68). More recently, PRA predominance in breast tumors was shown to predict a poorer response to treatment with tamoxifen, but was not a discriminator of response to anastrazole (69). In vitro studies in human breast cancer cells have demonstrated that overexpression of the PRA isoform (to mimic the ratios seen in cancers) has distinct effects on P-regulated gene expression and also promotes the acquisition of phenotypes associated with cancer, such as decreased cell adhesion and increased migration into bone marrow stroma (33, 70, 71). Furthermore, transgenic mice which express excess PRA display aberrant mammary development, including ductal hyperplasia, a disorganized basement membrane and decreased cell-cell adhesion, features which are commonly associated with neoplasia (38). These data indicate that correct expression of the 2 PR isoforms is critical for appropriate responsiveness to P signaling, and that disruption of this ratio can have major implications for breast carcinogenesis. Thus, PR isoform expression may be an important indicator of breast cancer risk, prognosis, and/or treatment options.
Mechanisms and Evidence for Paracrine Regulation of Proliferation by P in the Mammary Gland
There are a wealth of historical data demonstrating the well-defined role of P in counteracting the proliferative effects of E in the endometrium (72, 73) and growth inhibitory effects in human breast cancer cells in vitro (74). But it is also clear that context is important in determining the influence of P on proliferation. A recent report showing that PR can physically associate with estrogen receptor (ER) in breast cancer cells to modulate ER activity (75) showed that activation of PR by progestins in breast cancer cell lines resulted in altered E-driven ER genomic interactions, suggesting that PR antagonizes the ER cistrome in this context. At pharmacological levels, the PR agonist R5020 decreased the proliferation marker Ki67 in cultured breast cancer explants, and P reduced the proliferative effect of E in breast cancer cell lines grown as tumor xenografts in mice. This study emphasizes the ability of P to both stimulate and inhibit proliferation depending on context.
Notwithstanding these data in cancer models, P is now recognized as a major proliferative hormone in both the mouse mammary gland and the normal human breast epithelium, and mouse studies have shown P is required to promote the wave of proliferation which occurs during early pregnancy. P stimulates proliferation of a range of mammary cell types. P treatment increases proliferation of luminal cells of mammary glands of ovariectomized mice, as measured by bromo-deoxyuridine (BrdU) incorporation (72) and the expansion of lobular alveolar structures by P requires increased numbers and remodeling of the basal compartment. Data derived from mouse models and using fluorescence-activated cell sorting-based approaches have also shown that P stimulates the MaSC compartment, specifically during pregnancy as well as during the luteal dioestrous phase of the reproductive cycle, a mechanism suggested to account for the increased window of susceptibility to transformation at these times (76, 77).
Because PR is expressed nonuniformly in a subset of cells in the mouse mammary gland, the proliferative action of P is known to predominantly occur indirectly, as ductal side-branching is initiated by PR+ cells but occurs in adjacent PR− cells through stimulation of paracrine mediators that directly regulate proliferation genes in adjacent cells (78, 79). This too has been demonstrated in human breast tissue and human breast epithelial cell 3-dimensional (3D) cell culture models, in which the subset of luminal epithelial cells that proliferate after stimulation by P are predominantly PR−, yet are localized adjacent to PR+ cells that are largely nonproliferating (30, 80).
A number of distinct paracrine targets of P signaling have been characterized and a range of evidence suggests that the proliferation and tissue remodeling driven by P during lobular alveolar development occurs through a coordinated network of paracrine events. Several lines of evidence support this. Many MaSCs do not express ER or PR (56), providing support for the notion that their regulation occurs via paracrine mechanisms. Although proliferating luminal cells are detected in P treated primary breast 3D cultures, most of these do not express PR. P activates a number of the paracrine signaling pathways involved in proliferation and development of the mammary epithelium, including receptor activator of nuclear factor-κB ligand (RANKL), Wnt, Notch, GH/cytokines, amphiregulin (Areg), and calcitonin, as described below.
Receptor activator of nuclear factor-κB ligand
P-mediated proliferation and stimulation of the murine MaSC compartment has been shown to occur via mechanisms involving RANKL in the mouse mammary gland (31, 76, 77, 81). Rapidly induced by exposure to P, RANKL signals through its cognate receptor, RANK, and plays a critical role in parity-induced mammary alveologenesis. The absence of RANKL in the mammary gland results in an almost identical phenocopy of the PRKO mouse model in that RANKL KO virgin females exhibit normal mammary ductal development, but fail to form lobular alveolar structures after pregnancy stimulation (41, 82, 83). Its role as a key paracrine mediator of P signaling was illustrated by the complete rescue of the PRKO phenotype with ectopic expression of RANKL in PR−/− mammary epithelial cell-engrafted mice (81). RANKL, produced in mature luminal cells under P stimulation, is likely to bind to its receptor RANK on basal cells in order to act as a key paracrine effector of P signaling to MaSCs during pregnancy (76, 77). However, recent data using the technique of engrafting of intact pieces of mammary epithelium in order to retain the tissue architecture has suggested that, although RANKL KO grafts generated fewer side branches, RANKL was not essential in the control of MaSCs (84). Thus, the role that RANKL plays in regeneration potential of the mouse mammary gland may be dependent on the physiological context.
Growing evidence has suggested that P may regulate proliferation and tumorigenesis via similar mechanisms in other species. In addition to treatment with the synthetic progestin, medroxyprogesterone acetate (MPA), increasing RANKL transcripts in macaque monkeys, RANKL expression within human breast tissue microstructures was also shown to increase upon stimulation with the P agonist, R5020 (85). Furthermore, RANKL levels fluctuated with P levels during the menstrual cycle in the normal human breast (86), was highly correlated with serum P levels in breast tumor samples (87), and was induced with P or MPA treatment in primary 3D cultures of normal human breast tissue (88). However, not all breast tissue samples in 3D culture displayed an induction of RANKL in response to P (80, 88), pointing to a complex regulatory mechanism that is dependent on the dose and time of P administration in human breast tissue. In animal models, however, proliferative capacity and the occurrence of hyperplasias are significantly higher in monkeys exposed to MPA, and mouse model studies have demonstrated RANKL to be an important mediator of mammary tumorigenesis promoted by HRT progestins, including MPA (89, 90). Thus, the potential of RANKL inhibition as a therapeutic strategy to impede proliferation and increase disease-free survival in breast cancer patients has drawn growing interest (91).
Wnt-4
A member of the Wnt family, Wnt-4, was one of the first paracrine mediators of P action to be identified in the mouse mammary gland (92). Brisken et al (92) demonstrated in 2000 that PR and Wnt-4 are coexpressed in the luminal cell compartment, that Wnt-4 is induced by P treatment, that Wnt-4 expression during pregnancy requires PR, and that Wnt-4 is critical in side-branch formation during early pregnancy. Interestingly, levels of Wnt-4 were not regulated by P in PRKO mouse mammary glands, but were induced normally in PRA- and PRB-specific KO mammary glands, suggesting that the presence of one PR isoform can compensate for the absence of the other in terms of Wnt-4 regulation (31). In contrast to RANKL, which was not critical in MaSC regulation in the tissue engraftment experiments described above, Wnt-4 did play a pivotal role in MaSC activation throughout postnatal mammary gland development (84). This particular study also showed that P induced Wnt-4 specifically in PR+ luminal cells, and secreted Wnt-4 activated canonical Wnt signaling in neighboring basal cells, which in turn acts on MaSCs and lineage-restricted stem cells (84). Although Wnt-4 has been definitively demonstrated to be a crucial paracrine mediator of P action in the mouse, its role has been less clearly defined in the human breast. There is evidence that Wnt-4 transcripts are increased during the luteal phase of the menstrual cycle, when serum P levels are higher (86), as well as within breast tissue microstructures after 24 hours of stimulation with R5020 (85). This regulation was not observed in 3D culture models of normal human breast (80); however, P treatment of primary human breast epithelial cells in nonadherent culture did show increased expression of Wnt-4 (60), again underlining the context-specific actions of P in the human breast.
Notch
The Notch signaling pathway has been associated with regulating breast stem cell self-renewal and proliferation of progenitor cells (93), and members of this pathway are up-regulated in normal breast, as well as in breast cancer cells. Notch signaling is critical for luminal expansion and fate determination in developing murine mammary structures, and regulates the proportion of PR+ luminal cells (94, 95). Moreover, Notch has been shown to be required for restriction of bipotent progenitors to the luminal lineage in primary human breast cell populations (54). Notch ligands are detected in breast stem cells, suggesting a role in progenitor stimulation (96). Progestins induce expression of the Notch ligand, Jagged1, in T47D breast cancer cells (97, 98), and Notch ligands, delta-like 1 and 3, as well as the Notch receptor regulator, presenilin-2, were shown to be up-regulated by P stimulation in the normal human breast (80). This points to a potential role of Notch signaling, via paracrine mechanisms, in mediating the P-stimulated increase in mammary progenitor cells.
GH and chemokines
After the demonstration more than 20 years ago that synthetic progestins induce secretion of GH in the mammary epithelium of dogs (99), this finding was more recently reinforced in normal human breast epithelial cells (100). The study by Lombardi et al (100) showed that GH receptor (GHR)+ cells displayed functional properties of stem/progenitor cells, and that the vast majority of GH+ cells, as well as GHR+ cells, did not express PR but were localized adjacent to PR+ cells, in primary human breast epithelial cells in culture as well as in human breast tissue sections. The demonstration that GH mediated P stimulation of mammosphere formation, supports the hypothesis that PR+ cells stimulate the proliferation of PR−/GHR+ MaSCs via secretion of GH, which acts in a paracrine manner (100). This notion is further supported by the observation that GHR transcripts increase with P levels during the luteal phase of the menstrual cycle in the normal human breast (86). Another recent study has identified the CXCL12-CXCR4 (chemokine receptor) signaling axis as a novel paracrine mechanism essential in P-mediated stimulation of progenitor cells in the mouse mammary gland (101). Shiah et al (101) reported that P stimulated secretion of CXCL12 in luminal cells that bound to CXCR4 in neighboring basal cells, thereby increasing progenitor cell numbers in the mouse mammary gland.
Amphiregulin
Areg, a selective ligand for epidermal growth factor receptor, was first identified as a paracrine mediator of P action in the mouse uterus (102) and has also been shown to be an important PR target in the mammary gland of rodents. Areg transcripts were induced after acute P exposure in the adult murine mammary gland (83), whereas in the developing pubertal gland, P drove terminal end bud formation and proliferation via Areg (103). However, any role of Areg as a mediator of P signaling in the human breast remains to be shown.
Calcitonin
Calcitonin, a peptide hormone involved in calcium regulation and homeostasis, has also been implicated downstream of P signaling in the murine mammary gland. Ismail et al (104) demonstrated in 2004 that, although calcitonin is expressed exclusively in luminal cells, the expression of its receptor is located in myoepithelial cells and is independent of P action, leading to the authors forming the hypothesis that, after induction by P, calcitonin may serve as a paracrine mediator to act on both luminal and myoepithelial cells to contribute to proliferation and tissue remodeling when circulating P levels are high. However, although calcitonin is a P target in the murine mammary gland, as well as in the human endometrium (105), there is little evidence of calcitonin as a paracrine mediator in the human breast.
Thus, although our understanding of paracrine mediators of P action in the mammary gland has grown immensely over recent years, the evidence has for the most part derived from animal models, and the mechanistic details of how P promotes proliferation in the human breast epithelium in vivo remain largely undefined (Figure 2). Whether P-driven signaling networks vary between mouse and human, or whether any discordances identified are merely due to differences between the cell lines or in vitro or in vivo models used, remains to be elucidated. It is clear, however, that depending on the time and duration of administration, formulation and dosage used, P signaling is complex and tightly dependent on the physiological context. Moreover, it is clear that proliferation and development in the mammary gland are controlled by multiple coordinated signaling events and that P signaling pathways converge with many of these.
Figure 2. After P exposure, P enters a PR+ luminal cell (shown in blue) and binds to PR, which dimerises and transcribes target genes (eg, RANKL, WNT-4, NOTCH ligands, GH).

These signaling pathways then stimulate proliferation of neighboring PR− luminal cells (shown in red), potentially also involving cells in the basal cell layer (shown in purple). Not all of these pathways have been shown to be active in both the mouse (M) mammary gland as well as human (H) breast.
Mechanisms and Evidence for Autocrine Regulation of Proliferation by P in the Mammary Gland
Although it is generally agreed that most P signaling occurs via paracrine mechanisms, it is increasingly becoming accepted that, in a subset of cells, there is evidence of P-mediated autocrine signaling. Work performed decades ago showed that N-nitrosomethylurea-induced rat mammary tumors grown in soft agar clonogenic assays were stimulated to grow by both P and the progestin, R5020, and that at least part of this proliferative effect was due to P-induced autocrine growth factor production (106). Similarly in human breast cancer cells, P induced up-regulation of growth-promoting target genes (eg, cyclin D1), resulting in sustained and robust autocrine activation of epidermal growth factor receptor, c-Src and Erk1/2 MAPKs (107). This cyclin D1-dependent autocrine mechanism was later demonstrated to occur in the mouse mammary gland in vivo. Before the large wave of P-mediated proliferation being driven by a paracrine RANKL-dependent mechanism as discussed above, proliferation can also occur via an initial small wave of cell-autonomous signaling, which occurs rapidly, and transiently, within the first 24 hours of P treatment (81). Using BrdU pulse-labeling experiments, Beleut et al (81) demonstrated that at early time points, almost all of the proliferating BrdU+ cells also expressed PR, and that this autocrine signaling was dependent on cyclin D1, a known target of P in T47D breast cancer cells (108), but that cyclin D1 was not required for the second larger wave of proliferation in PR− cells. It remains to be determined whether this mechanism also occurs in the normal human breast; however, proliferation in a small subset of PR+ cells has been observed in primary human breast epithelial cells grown in 3D culture (80). There is some evidence of autocrine signaling in human breast tumors. In human breast cancer cell line xenografts, treatment with P or MPA up-regulated the expression of the stem/progenitor markers cytokeratin-5 (CK5) and CD44 in a subset of cells, by reprogramming a small subset of PR+CK5−CD44− cells into PR−CK5+CD44+ cells through an autocrine mechanism (98, 109).
Mouse studies have also shown that, in addition to being a pivotal paracrine factor, there is a large induction of RANKL in PR+ luminal cells, upon exposure to P and progestins, which stimulates further RANKL expression via an autocrine positive-feedback loop (110). In parallel, secreted RANKL induces up-regulation of RANK expression in adjacent myoepithelial cells, in a paracrine manner, which then regulates proliferation and expansion of the MaSC compartment (89, 90). Interestingly, the study by Shiah et al (101) also showed evidence for P activating the CXCL12-CXCR4 signaling axis within luminal cells via autocrine mechanisms, in addition to paracrine mechanisms.
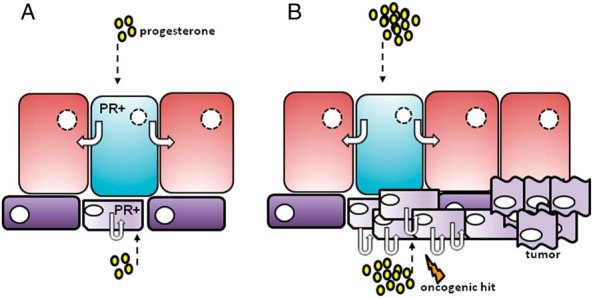
Finally, in the normal mouse mammary gland, P treatment selectively up-regulates specific targets in either basal cells or luminal cells (76, 77), and so P action may potentially be cell-type dependent, and occur via different mechanisms in progenitor cells compared with mature luminal cells. As described above, most evidence supporting P-mediated autocrine signaling has derived from animal studies, and it is now critical to definitively prove whether these mechanisms are conserved in the human. Because P stimulates progenitor cells in normal human breast epithelial cells grown in 3D culture (61, 80), and PR is expressed in a subset of basal cells enriched for progenitor characteristics (53, 60, 61), one could postulate that, although PR acts in a paracrine manner in luminal cells to expand the luminal compartment, there exist a small number of PR+ basal or progenitor cells within the breast lobule that are able to directly respond to the proliferative signals of high circulating P levels via cell-intrinsic mechanisms. This mechanism could serve to expand the epithelial compartment, during both the luteal phase of the menstrual cycle, and during pregnancy in preparation for lactation (Figure 3). However, transient increases in these cell numbers in response to progestins in exogenous formulations, particularly in postmenopausal women with low endogenous levels of E, may also provide a window for increased mutation risk. Thus, it may be this small population of PR+ bipotent progenitor cells that are particularly susceptible to oncogenesis consequent to the increased proliferation provided by exogenous progestin use in HRT and the OC pill.
Figure 3. A, During normal development, P stimulates proliferation of luminal cells by acting on PR+ luminal cells (shown in blue), which signal to neighboring PR− cells (shown in red) in a paracrine manner. In addition, P may further expand the epithelial compartment by direct stimulation of a subset of PR+ basal/progenitor cells (shown in light purple), which proliferate via autocrine mechanisms. B, When P levels are particularly high (during the luteal phase and pregnancy) or after exogenous hormone use, this proliferation may be increased, potentially providing a larger pool of PR+ progenitor cells susceptible to oncogenic hits, which may then increase the risk of tumorigenesis.
Does Paracrine Regulation of Proliferation in Normal Breast Shift to Autocrine Signaling in Breast Cancer?
To summarize, it is established that PR is expressed in a range of mammary cell types and plays proliferative roles in each. In PR+ mature luminal cells P regulates proliferation by stimulation of paracrine signaling intermediates. In contrast, PR expressed in bipotent progenitors may drive their proliferation directly. In the normal breast epithelium there exists a balance of paracrine and autocrine signaling that is cell and context specific. Early in carcinogenesis, current models hold that actively proliferating cells and those with self-renewing capability are the most likely targets of malignancy, so malignant transformation and subsequent expansion of PR+ progenitor cell numbers would expand cells employing an autocrine mode of P action, thereby tipping the balance towards a predominance of autocrine stimulation of proliferation by P in breast cancer. This may result in a critical alteration in the balance of autocrine and paracrine regulation of proliferation by P in breast cancer, and contribute to development and/or progression of breast cancer. This notion is supported by the fact that, although the vast majority of proliferating cells in the normal human breast do not express steroid hormone receptors, an increasing number of proliferating cells express ER and/or PR in breast tumors (30, 55), suggesting that these cells may have the potential to respond directly to P stimulation. However, it is not clear whether PR+ proliferating cells in cancers arise from PR+ cells in the normal breast epithelium in which P signaling occurred through an autocrine mechanism, or whether P in these cells originally signaled via autocrine mechanisms, but “switched” from paracrine to autocrine signaling upon transformation, thereby conferring autocrine control by P of proliferation of these cells.
In human tissue samples, it has been shown that there is increased hormone receptor expression in preinvasive breast samples, compared with normal breast (111–113). Moreover, there is also increased proliferation in preinvasive breast tissue samples (114); however, most of these studies have not looked explicitly at the proportion of proliferating cells within these samples that are also PR+. Although other studies have reported that there is an increased proportion of proliferating hormone receptor-positive cells in premalignant lesions in human breast tissue samples, most of this work has focused on the expression of ER only, or has been extrapolated from studies in rodent models (111, 115–117). So although it may be true that in the human, carcinogenic transformation is accompanied by the acquisition of proliferation by hormone receptor positive cells, it remains to be definitively demonstrated. Although some investigators in the field refer to this change as a “switch” (117–119), an increase in the proportion of proliferating PR+ cells in breast cancer does not prove that this is causative in tumorigenesis. It must be considered equally plausible that there is no “switch” per se. Instead, one could postulate that P stimulates the proliferation of the small subset of PR+ progenitor cells as part of the normal developmental process, but that this process can go awry, for example, when P levels are particularly high during pregnancy, or exogenous hormone use, or after an oncogenic hit. Subsequently, the rate of proliferation of this PR+ progenitor cell subset may increase, gradually expanding the progenitor cell compartment with each round of cell division, leading to an exponential increase in the progenitor pool that may be susceptible to additional oncogenic hits and mutations. This scenario supports the possibility that the increased autocrine signaling observed in breast cancer is contributed to by the process of transformation selecting for PR+ progenitors in which P signaling is predominantly autocrine, thus expanding this pool. Given that paracrine/indirect signaling in the breast is a mechanism for limiting the influence of hormone in that tissue, an increase in the proportion of autocrine/direct signaling may result in the enhancement of hormone action in cancer. Thus, tight regulation of the balance between paracrine and autocrine signaling in the normal breast may be paramount in maintenance of normal homeostasis, and in the prevention of breast cancer. It is important to note that the PRA to PRB ratio also changes from normal breast, through preinvasive lesions and invasive cancer (40), supporting the view that altered P signaling and/or PR expression may be a consequence of, or accompanies, the oncogenic process.
In further support of altered homoeostasis in breast cancer, P signaling in the normal human breast and in breast cancer are completely different. For example, there is minimal overlap of P targets observed in primary normal human breast epithelial cells and breast cancer cells when grown in identical conditions (80), and genome-wide chromatin immunoprecipitation mapping of PR binding has revealed that this is due to nonoverlapping interaction profiles in normal breast epithelial cells and breast cancer cells (120). This is also seen for ER, which has divergent cistromes in breast cancer and osteosarcoma cell lines, driving cell type-specific transcriptional regulation by E (121), and for glucocorticoid receptor, where a very small overlap was seen between glucocorticoid receptor cistromes in mammary and pituitary cell lines (122). However, P action can also be distinct during different phases of development, for example, an increased proportion of colocalization between the PRB isoform and cyclin D1 in proliferating cells is observed during pregnancy in the murine mammary gland, indicating that PR+ cells are capable of proliferation during normal developmental processes (123). Thus, in specific circumstances, aberrant P signaling (which may occur after exogenous hormone use) may activate particular pathways that can trigger autocrine signaling, and consequently, unrestrained proliferation and subsequent tumorigenesis.
In comparing P signaling in normal and cancer, it is also relevant that the nuclear architecture of breast cells influences the transcriptional response to P, suggesting that PR may regulate different targets in the breast depending on the cell type in which it is expressed. Thus, PR may directly regulate proliferation genes in progenitor cells, but these targets may be inaccessible in mature luminal cells where PR acts on genes encoding paracrine signaling intermediates to stimulate proliferation. Moreover, the heightened proliferative response to P in premalignant and malignant breast may reflect the disrupted genomic environment of those cells. The repertoire of genes that are actively transcribed in a cell is tightly controlled by cell type-specific higher order chromatin structure (124). Genomic DNA is compacted by wrapping around a protein octamer made up of 4 core histones into nucleosomes (125), and specific posttranslational modifications of those histones determine whether a particular genomic region is transcribed. Dynamic regulation of those modifications influences accessibility to binding by transcription factors, such as PR, and subsequent transcriptional regulation (124). The relationship between chromatin architecture and the interaction of nuclear receptors with their cognate regulatory elements on genomic DNA is a complex topic that has been well covered recently in excellent reviews (126–128). An emerging consensus is that the cell type-specific pattern of nucleosome-depleted accessible genomic regions influences genomic binding by steroid receptors. That is, receptors are more likely to bind to regulatory elements if they are in accessible DNA regions, although in many instances engagement of the receptor with the target site results in further chromatin remodeling and nucleosome displacement. The genome-wide pattern of accessible regions is dependent on cell type and also on the cell-specific expression of constitutive remodeling factors, such as pioneer factors that bind to condensed chromatin to increase accessibility (122, 129, 130). This provides a mechanism for the cell type-specific determination of nuclear receptor cistromes.
The transition from stem to progenitor through to a lineage-committed mature cell is characterized by specific alterations in chromatin state that permit the expression of transcription factors aimed at maintaining pluripotency in stem cells, but allow stage-specific activation of development genes when progression towards a differentiated state is initiated. This process is managed by progressive remodeling of activating and silencing histone modifications to alter the transcriptional landscape of the cell (131, 132). Histone modification mapping in primary breast epithelial cell populations enriched for progenitor or mature cell types revealed highly specific patterns of modifications, which were linked to cell type-specific transcriptional programs (133). This finding suggests that the targets of P action in mature luminal cells are likely to be different from those in PR+ progenitors and indeed different from P targets in cancer cells. Thus, it is possible to speculate that P may regulate the expression of paracrine signaling molecules in terminally differentiated luminal cells whereas at the same time directly stimulating cell cycle targets in progenitor cells. Moreover, these latter targets could become reactivated in mature cells after a carcinogenic insult.
Unanswered Questions
It has long been known that the hormonal milieu of the breast influences normal tissue development (134), and exposure to exogenous hormone analogues in HRT and OCs contributes adversely to breast cancer risk (5, 7). Although models of mammary development and tumorigenesis have greatly improved, such as the development of patient-derived xenograft models that more accurately recapitulate key aspects of the original tumor (135), the human breast functions in a vastly different manner compared with the mouse mammary gland. There has been a paucity of information on the molecular events underlying the hormonal influence on breast cancer risk in the human, and the precise details of how the normal human breast functions remain largely unknown. This is partly due to the lack of suitable human experimental models; most of our current understanding derives from mouse models or human breast cancer cell line studies. In vivo mouse modeling studies, such as recent lineage tracing experiments where a single cell is marked in order to identify all progeny of that cell (136), have provided a wealth of information regarding mammary development. For the obvious reasons that similar in vivo human models are not possible, it has been necessary to develop primarily in vitro approaches to study human breast development. One could postulate, however, that these in vitro systems lack the various microenvironmental factors, such as immune cells or extracellular matrix components, that are required in order to replicate what occurs in the human breast in vivo.
The recent evidence that P action targets not just luminal cells but can stimulate multiple cell types raises the possibility that these different actions may occur via different mechanisms. Compared with our knowledge regarding the identification of the paracrine mediators of P action in the mouse mammary gland, our understanding of whether these are conserved in the human remains very poor. It is also not clear as to whether these same paracrine mediators are also able to act in an autocrine manner, or whether these different mechanisms employ distinct targets. Furthermore, our understanding of what causes the switch from paracrine to autocrine signaling in cancer is virtually nonexistent. This aspect in particular will be key in uncovering new biomarkers for the early prediction of breast cancer progression, or new pathways that could be directly targeted for interruption, offering potential new therapies for breast cancer patients.
Concluding Remarks
In summary, P action in the breast is clearly context dependent and likely to proceed by multiple coordinated pathways that include both paracrine and autocrine PR signaling events. The avenue through which PR affects transcriptional outcome is likely to be influenced by the cell lineage in which it is expressed, the developmental stage of the tissue (prepubertal, pregnant, postmenopausal) and the position of the cell within the tissue (ductal or lobular). Characterization of P regulated paracrine and autocrine targets, details of which have predominantly been derived from animal studies, paints a complex picture where PR may directly stimulate the initial expansion of bipotent progenitors, immediately followed by a larger wave of paracrine stimulation of the Wnt pathway to initiate new lobuloalveolar expansion, production of RANKL to amplify lobular elaboration and signaling to the Notch pathway to maintain luminal lineage balance. The challenge going forward will be to further characterize the PR+ cell types, particularly in the normal human breast, and to build a network view of P action in this tissue.
Acknowledgments
This work was supported by a Postdoctoral Fellowship cofunded by the Cure Cancer Australia Foundation and the National Breast Cancer Foundation (H.N.H.). C.L.C. is a research fellow of the National Health and Medical Research Council of Australia.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by a Postdoctoral Fellowship cofunded by the Cure Cancer Australia Foundation and the National Breast Cancer Foundation (H.N.H.). C.L.C. is a research fellow of the National Health and Medical Research Council of Australia.
Footnotes
- AF
- activation function
- Areg
- amphiregulin
- BrdU
- bromo-deoxyuridine
- CK5
- cytokeratin-5
- 3D
- 3-dimensional
- E
- estrogen
- ER
- estrogen receptor
- GHR
- GH receptor
- HRT
- hormone-replacement therapy
- LREC
- label-retaining epithelial cell
- MaSC
- mammary stem cell
- MPA
- medroxyprogesterone acetate
- OC
- oral contraceptive
- P
- progesterone
- PR
- P receptor
- PRKO
- PR knockout
- RANKL
- receptor activator of nuclear factor-κB ligand.
References
- 1. Feinleib M. Breast cancer and artificial menopause: a cohort study. J Natl Cancer Inst. 1968;41:315–329. [PubMed] [Google Scholar]
- 2. Trichopoulos D, MacMahon B, Cole P. Menopause and breast cancer risk. J Natl Cancer Inst. 1972;48:605–613. [PubMed] [Google Scholar]
- 3. Lydon JP, Ge G, Kittrell FS, Medina D, O'Malley BW. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Res. 1999;59:4276–4284. [PubMed] [Google Scholar]
- 4. Beral V, Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003;362:419–427. [DOI] [PubMed] [Google Scholar]
- 5. Hunter DJ, Colditz GA, Hankinson SE, et al. Oral contraceptive use and breast cancer: a prospective study of young women. Cancer Epidemiol Biomarkers Prev. 2010;19:2496–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beral V, Reeves G, Bull D, Green J. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J Natl Cancer Inst. 2011;103:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chlebowski RT, Anderson GL. Menopausal hormone therapy and cancer: changing clinical observations of target site specificity. Steroids. 2014;90:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chlebowski RT, Hendrix SL, Langer RD, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the women's health initiative randomized trial. JAMA. 2003;289:3243–3253. [DOI] [PubMed] [Google Scholar]
- 9. Charlton BM, Rich-Edwards JW, Colditz GA, et al. Oral contraceptive use and mortality after 36 years of follow-up in the Nurses' Health Study: prospective cohort study. BMJ. 2014;349:g6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lonard DM, O'Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700. [DOI] [PubMed] [Google Scholar]
- 11. Le Dily F, Baù D, Pohl A, et al. Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 2014;28:2151–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arnett-Mansfield RL, deFazio A, Mote PA, Clarke CL. Subnuclear distribution of progesterone receptors A and B in normal and malignant endometrium. J Clin Endocrinol Metab. 2004;89:1429–1442. [DOI] [PubMed] [Google Scholar]
- 13. Arnett-Mansfield RL, Graham JD, Hanson AR, et al. Focal subnuclear distribution of progesterone receptor is ligand dependent and associated with transcriptional activity. Mol Endocrinol. 2007;21:14–29. [DOI] [PubMed] [Google Scholar]
- 14. Rousseau-Merck MF, Misrahi M, Loosfelt H, Milgrom E, Berger R. Localization of the human progesterone receptor gene to chromosome 11q22–q23. Hum Genet. 1987;77:280–282. [DOI] [PubMed] [Google Scholar]
- 15. Kastner P, Krust A, Turcotte B, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9:1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scarpin KM, Graham JD, Mote PA, Clarke CL. Progesterone action in human tissues: regulation by progesterone receptor (PR) isoform expression, nuclear positioning and coregulator expression. Nucl Recept Signal. 2009;7:e009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huse B, Verca SB, Matthey P, Rusconi S. Definition of a negative modulation domain in the human progesterone receptor. Mol Endocrinol. 1998;12:1334–1342. [DOI] [PubMed] [Google Scholar]
- 18. McEwan IJ. Nuclear receptors: one big family. Methods Mol Biol. 2009;505:3–18. [DOI] [PubMed] [Google Scholar]
- 19. McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474. [DOI] [PubMed] [Google Scholar]
- 20. Sartorius CA, Melville MY, Hovland AR, Tung L, Takimoto GS, Horwitz KB. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol. 1994;8:1347–1360. [DOI] [PubMed] [Google Scholar]
- 21. Vegeto E, Shabbaz MM, Wen DX, Goldman ME, O'Malley BW, McDonnell DP. Human progesterone receptor A form is a cell- and promoter-specific repressor of human progesterone receptor B function. Mol Endocrinol. 1993;7:1244–1255. [DOI] [PubMed] [Google Scholar]
- 22. Hovland AR, Powell RL, Takimoto GS, Tung L, Horwitz KB. An N-terminal inhibitory function, IF, suppresses transcription by the A-isoform but not the B-isoform of human progesterone receptors. J Biol Chem. 1998;273:5455–5460. [DOI] [PubMed] [Google Scholar]
- 23. Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. [DOI] [PubMed] [Google Scholar]
- 24. Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24:10542–10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daniel AR, Knutson TP, Lange CA. Signaling inputs to progesterone receptor gene regulation and promoter selectivity. Mol Cell Endocrinol. 2009;308:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hagan CR, Regan TM, Dressing GE, Lange CA. ck2-dependent phosphorylation of progesterone receptors (PR) on Ser81 regulates PR-B isoform-specific target gene expression in breast cancer cells. Mol Cell Biol. 2011;31:2439–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abdel-Hafiz HA, Horwitz KB. Post-translational modifications of the progesterone receptors. J Steroid Biochem Mol Biol. 2014;140:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Press MF, Udove JA, Greene GL. Progesterone receptor distribution in the human endometrium. Analysis using monoclonal antibodies to the human progesterone receptor. Am J Pathol. 1988;131:112–124. [PMC free article] [PubMed] [Google Scholar]
- 30. Clarke RB, Howell A, Potten CS, Anderson E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 1997;57:4987–4991. [PubMed] [Google Scholar]
- 31. Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA. 2003;100:9744–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000;289:1751–1754. [DOI] [PubMed] [Google Scholar]
- 33. Graham JD, Yager ML, Hill HD, Byth K, O'Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol. 2005;19:2713–2735. [DOI] [PubMed] [Google Scholar]
- 34. Conneely OM, Mulac-Jericevic B, Lydon JP. Progesterone-dependent regulation of female reproductive activity by two distinct progesterone receptor isoforms. Steroids. 2003;68:771–778. [DOI] [PubMed] [Google Scholar]
- 35. Jacobsen BM, Richer JK, Schittone SA, Horwitz KB. New human breast cancer cells to study progesterone receptor isoform ratio effects and ligand-independent gene regulation. J Biol Chem. 2002;277:27793–27800. [DOI] [PubMed] [Google Scholar]
- 36. Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277:5209–5218. [DOI] [PubMed] [Google Scholar]
- 37. Aupperlee MD, Haslam SZ. Differential hormonal regulation and function of progesterone receptor isoforms in normal adult mouse mammary gland. Endocrinology. 2007;148:2290–2300. [DOI] [PubMed] [Google Scholar]
- 38. Shyamala G, Yang X, Silberstein G, Barcellos-Hoff MH, Dale E. Transgenic mice carrying an imbalance in the native ratio of A to B forms of progesterone receptor exhibit developmental abnormalities in mammary glands. Proc Natl Acad Sci USA. 1998;95:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mote PA, Balleine RL, McGowan EM, Clarke CL. Colocalization of progesterone receptors A and B by dual immunofluorescent histochemistry in human endometrium during the menstrual cycle. J Clin Endocrinol Metab. 1999;84:2963–2971. [DOI] [PubMed] [Google Scholar]
- 40. Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72:163–172. [DOI] [PubMed] [Google Scholar]
- 41. Lydon JP, DeMayo FJ, Funk CR, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9:2266–2278. [DOI] [PubMed] [Google Scholar]
- 42. Bartow SA. Use of the autopsy to study ontogeny and expression of the estrogen receptor gene in human breast. J Mammary Gland Biol Neoplasia. 1998;3:37–48. [DOI] [PubMed] [Google Scholar]
- 43. Ricketts D, Turnbull L, Ryall G, et al. Estrogen and progesterone receptors in the normal female breast. Cancer Res. 1991;51:1817–1822. [PubMed] [Google Scholar]
- 44. Belitskaya-Lévy I, Zeleniuch-Jacquotte A, Russo J, et al. Characterization of a genomic signature of pregnancy identified in the breast. Cancer Prev Res (Phila). 2011;4:1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Asztalos S, Gann PH, Hayes MK, et al. Gene expression patterns in the human breast after pregnancy. Cancer Prev Res (Phila). 2010;3:301–311. [DOI] [PubMed] [Google Scholar]
- 46. Taylor D, Pearce CL, Hovanessian-Larsen L, et al. Progesterone and estrogen receptors in pregnant and premenopausal non-pregnant normal human breast. Breast Cancer Res Treat. 2009;118:161–168. [DOI] [PubMed] [Google Scholar]
- 47. Battersby S, Robertson BJ, Anderson TJ, King RJ, McPherson K. Influence of menstrual cycle, parity and oral contraceptive use on steroid hormone receptors in normal breast. Br J Cancer. 1992;65:601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Lima GR, Facina G, Shida JY, et al. Effects of low dose tamoxifen on normal breast tissue from premenopausal women. Eur J Cancer. 2003;39:891–898. [DOI] [PubMed] [Google Scholar]
- 49. Hargreaves DF, Knox F, Swindell R, Potten CS, Bundred NJ. Epithelial proliferation and hormone receptor status in the normal post-menopausal breast and the effects of hormone replacement therapy. Br J Cancer. 1998;78:945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab. 1999;84:4559–4565. [DOI] [PubMed] [Google Scholar]
- 51. Howard BA, Gusterson BA. Human breast development. J Mammary Gland Biol Neoplasia. 2000;5:119–137. [DOI] [PubMed] [Google Scholar]
- 52. Fu N, Lindeman GJ, Visvader JE. The mammary stem cell hierarchy. Curr Top Dev Biol. 2014;197:133–160. [DOI] [PubMed] [Google Scholar]
- 53. Hilton HN, Graham JD, Kantimm S, et al. Progesterone and estrogen receptors segregate into different cell subpopulations in the normal human breast. Mol Cell Endocrinol. 2012;361:191–201. [DOI] [PubMed] [Google Scholar]
- 54. Raouf A, Zhao Y, To K, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. 2008;3:109–118. [DOI] [PubMed] [Google Scholar]
- 55. Anderson E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002;4:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Asselin-Labat ML, Shackleton M, Stingl J, et al. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst. 2006;98:1011–1014. [DOI] [PubMed] [Google Scholar]
- 57. Booth BW, Smith GH. Estrogen receptor-α and progesterone receptor are expressed in label-retaining mammary epithelial cells that divide asymmetrically and retain their template DNA strands. Breast Cancer Res. 2006;8:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Smith GH. Label-retaining epithelial cells in mouse mammary gland divide asymmetrically and retain their template DNA strands. Development. 2005;132:681–687. [DOI] [PubMed] [Google Scholar]
- 59. Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005;277:443–456. [DOI] [PubMed] [Google Scholar]
- 60. Arendt LM, St. Laurent J, Wronski A, et al. Human breast progenitor cell numbers are regulated by WNT and TBX3. PLoS One. 2014;9:e111442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hilton HN, Santucci N, Silvestri A, et al. Progesterone stimulates progenitor cells in normal human breast and breast cancer cells. Breast Cancer Res Treat. 2014;143:423–433. [DOI] [PubMed] [Google Scholar]
- 62. Hilton HN, Doan TB, Graham JD, et al. Acquired convergence of hormone signaling in breast cancer: ER and PR transition from functionally distinct in normal breast to predictors of metastatic disease. Oncotarget. 2014;5:8651–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bachelard-Cascales E, Chapellier M, Delay E, et al. The CD10 enzyme is a key player to identify and regulate human mammary stem cells. Stem Cells. 2010;28:1081–1088. [DOI] [PubMed] [Google Scholar]
- 64. Garbe JC, Pepin F, Pelissier FA, et al. Accumulation of multipotent progenitors with a basal differentiation bias during aging of human mammary epithelia. Cancer Res. 2012;72:3687–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keller PJ, Arendt LM, Skibinski A, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci USA. 2012;109:2772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gava N, Clarke CL, Byth K, Arnett-Mansfield RL, deFazio A. Expression of progesterone receptors A and B in the mouse ovary during the estrous cycle. Endocrinology. 2004;145:3487–3494. [DOI] [PubMed] [Google Scholar]
- 67. Mote PA, Arnett-Mansfield RL, Gava N, et al. Overlapping and distinct expression of progesterone receptors A and B in mouse uterus and mammary gland during the estrous cycle. Endocrinology. 2006;147:5503–5512. [DOI] [PubMed] [Google Scholar]
- 68. Hopp TA, Weiss HL, Hilsenbeck SG, et al. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res. 2004;10:2751–2760. [DOI] [PubMed] [Google Scholar]
- 69. Mote PA, Gompel A, Howe C, et al. Progesterone receptor A predominance is a discriminator of benefit from endocrine therapy in the ATAC trial. Breast Cancer Res Treat. 2015;151:309–318. [DOI] [PubMed] [Google Scholar]
- 70. McGowan EM, Clarke CL. Effect of overexpression of progesterone receptor A on endogenous progestin-sensitive endpoints in breast cancer cells. Mol Endocrinol. 1999;13:1657–1671. [DOI] [PubMed] [Google Scholar]
- 71. McGowan EM, Saad S, Bendall LJ, Bradstock KF, Clarke CL. Effect of progesterone receptor a predominance on breast cancer cell migration into bone marrow fibroblasts. Breast Cancer Res Treat. 2004;83:211–220. [DOI] [PubMed] [Google Scholar]
- 72. Fernandez-Valdivia R, Mukherjee A, Mulac-Jericevic B, et al. Revealing progesterone's role in uterine and mammary gland biology: insights from the mouse. Semin Reprod Med. 2005;23:22–37. [DOI] [PubMed] [Google Scholar]
- 73. Pan H, Deng Y, Pollard JW. Progesterone blocks estrogen-induced DNA synthesis through the inhibition of replication licensing. Proc Natl Acad Sci USA. 2006;103:14021–14026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sutherland RL, Hall RE, Pang GY, Musgrove EA, Clarke CL. Effect of medroxyprogesterone acetate on proliferation and cell cycle kinetics of human mammary carcinoma cells. Cancer Res. 1988;48:5084–5091. [PubMed] [Google Scholar]
- 75. Mohammed H, Russell IA, Stark R, et al. Progesterone receptor modulates ERα action in breast cancer. Nature. 2015;523:313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Asselin-Labat ML, Vaillant F, Sheridan JM, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. [DOI] [PubMed] [Google Scholar]
- 77. Joshi PA, Jackson HW, Beristain AG, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465:803–807. [DOI] [PubMed] [Google Scholar]
- 78. Brisken C, Park S, Vass T, Lydon JP, O'Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci USA. 1998;95:5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rajaram RD, Brisken C. Paracrine signaling by progesterone. Mol Cell Endocrinol. 2012;357:80–90. [DOI] [PubMed] [Google Scholar]
- 80. Graham JD, Mote PA, Salagame U, et al. DNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology. 2009;150:3318–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Beleut M, Rajaram RD, Caikovski M, et al. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc Natl Acad Sci USA. 2010;107:2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fata JE, Kong YY, Li J, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50. [DOI] [PubMed] [Google Scholar]
- 83. Fernandez-Valdivia R, Mukherjee A, Creighton CJ, et al. Transcriptional response of the murine mammary gland to acute progesterone exposure. Endocrinology. 2008;149:6236–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rajaram RD, Buric D, Caikovski M, et al. Progesterone and Wnt4 control mammary stem cells via myoepithelial crosstalk. EMBO J. 2015;34:641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tanos T, Sflomos G, Echeverria PC, et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med. 2013;5:182ra55. [DOI] [PubMed] [Google Scholar]
- 86. Pardo I, Lillemoe H, Blosser R, et al. Next-generation transcriptome sequencing of the premenopausal breast epithelium using specimens from a normal human breast tissue bank. Breast Cancer Res. 2014;16:R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hu H, Wang J, Gupta A, et al. RANKL expression in normal and malignant breast tissue responds to progesterone and is up-regulated during the luteal phase. Breast Cancer Res Treat. 2014;146:515–523. [DOI] [PubMed] [Google Scholar]
- 88. Wang J, Gupta A, Hu H, Chatterton RT, Clevenger CV, Khan SA. Comment on “Progesterone/RANKL is a major regulatory axis in the human breast.” Sci Transl Med 2013;5:215le214. [DOI] [PubMed] [Google Scholar]
- 89. Gonzalez-Suarez E, Jacob AP, Jones J, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468:103–107. [DOI] [PubMed] [Google Scholar]
- 90. Schramek D, Leibbrandt A, Sigl V, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dougall WC, Holen I, Gonzalez Suarez E. Targeting RANKL in metastasis. Bonekey Rep. 2014;3:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Brisken C, Heineman A, Chavarria T, et al. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000;14:650–654. [PMC free article] [PubMed] [Google Scholar]
- 93. Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–R615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bouras T, Pal B, Vaillant F, et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell. 2008;3:429–441. [DOI] [PubMed] [Google Scholar]
- 95. Buono KD, Robinson GW, Martin C, et al. The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev Biol. 2006;293:565–580. [DOI] [PubMed] [Google Scholar]
- 96. Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. [DOI] [PubMed] [Google Scholar]
- 97. Ghatge RP, Jacobsen BM, Schittone SA, Horwitz KB. The progestational and androgenic properties of medroxyprogesterone acetate: gene regulatory overlap with dihydrotestosterone in breast cancer cells. Breast Cancer Res. 2005;7:R1036–R1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA. 2008;105:5774–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Selman PJ, Mol JA, Rutteman GR, van Garderen E, Rijnberk A. Progestin-induced growth hormone excess in the dog originates in the mammary gland. Endocrinology. 1994;134:287–292. [DOI] [PubMed] [Google Scholar]
- 100. Lombardi S, Honeth G, Ginestier C, et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem Cell Reports. 2014;2:780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shiah YJ, Tharmapalan P, Casey AE, et al. A progesterone-CXCR4 axis controls mammary progenitor cell fate in the adult gland. Stem Cell Reports. 2015;4:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Das SK, Chakraborty I, Paria BC, Wang XN, Plowman G, Dey SK. Amphiregulin is an implantation-specific and progesterone-regulated gene in the mouse uterus. Mol Endocrinol. 1995;9:691–705. [DOI] [PubMed] [Google Scholar]
- 103. Aupperlee M, Leipprandt J, Bennett J, Schwartz R, Haslam S. Amphiregulin mediates progesterone-induced mammary ductal development during puberty. Breast Cancer Res. 2013;15:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ismail PM, DeMayo FJ, Amato P, Lydon JP. Progesterone induction of calcitonin expression in the murine mammary gland. J Endocrinol. 2004;180:287–295. [DOI] [PubMed] [Google Scholar]
- 105. Kumar S, Zhu LJ, Polihronis M, et al. Progesterone induces calcitonin gene expression in human endometrium within the putative window of implantation. J Clin Endocrinol Metab. 1998;83:4443–4450. [DOI] [PubMed] [Google Scholar]
- 106. Manni A, Wright C, Badger B, Demers L, Bartholomew M. Polyamines and autocrine control of N-nitrosomethylurea-induced rat mammary tumor growth in vitro by progesterone. Cancer Res. 1988;48:3058–3061. [PubMed] [Google Scholar]
- 107. Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 To induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27:466–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Musgrove EA, Hamilton JA, Lee CS, Sweeney KJ, Watts CK, Sutherland RL. Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol Cell Biol. 1993;13:3577–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sartorius CA, Harvell DM, Shen T, Horwitz KB. Progestins initiate a luminal to myoepithelial switch in estrogen-dependent human breast tumors without altering growth. Cancer Res. 2005;65:9779–9788. [DOI] [PubMed] [Google Scholar]
- 110. Schramek D, Sigl V, Penninger JM. RANKL and RANK in sex hormone-induced breast cancer and breast cancer metastasis. Trends Endocrinol Metab. 2011;22:188–194. [DOI] [PubMed] [Google Scholar]
- 111. Khan SA, Rogers MA, Obando JA, Tamsen A. Estrogen receptor expression of benign breast epithelium and its association with breast cancer. Cancer Res. 1994;54:993–997. [PubMed] [Google Scholar]
- 112. Allred DC, Mohsin SK, Fuqua SA. Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001;8:47–61. [DOI] [PubMed] [Google Scholar]
- 113. Shoker BS, Jarvis C, Clarke RB, et al. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am J Pathol. 1999;1555:1811–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hilton HN, Kantimm S, Graham JD, Clarke CL. Changed lineage composition is an early event in breast carcinogenesis. Histol Histopathol. 2013;28:1197–1204. [DOI] [PubMed] [Google Scholar]
- 115. Lawson JS, Field AS, Champion S, Tran D, Ishikura H, Trichopoulos D. Low oestrogen receptor α expression in normal breast tissue underlies low breast cancer incidence in Japan. Lancet. 1999;354:1787–1788. [DOI] [PubMed] [Google Scholar]
- 116. Shoker BS, Jarvis C, Clarke RB, et al. Abnormal regulation of the oestrogen receptor in benign breast lesions. J Clin Pathol. 2000;53:778–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Obr AE, Edwards DP. The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol Cell Endocrinol. 2012;357:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kim JJ, Kurita T, Bulun SE. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr Rev. 2013;34:130–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Rosen JM. Hormone receptor patterning plays a critical role in normal lobuloalveolar development and breast cancer progression. Breast Dis. 2003;18:3–9. [DOI] [PubMed] [Google Scholar]
- 120. Clarke CL, Graham JD. Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PLoS One. 2012;7:e35859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Krum SA, Miranda-Carboni GA, et al. Unique ERα cistromes control cell type-specific gene regulation. Mol Endocrinol. 2008;22:2393–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. John S, Sabo PJ, Thurman RE, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Aupperlee MD, Smith KT, Kariagina A, Haslam SZ. Progesterone receptor isoforms A and B: temporal and spatial differences in expression during murine mammary gland development. Endocrinology. 2005;146:3577–3588. [DOI] [PubMed] [Google Scholar]
- 124. Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–564. [DOI] [PubMed] [Google Scholar]
- 126. Gadaleta RM, Magnani L. Nuclear receptors and chromatin: an inducible couple. J Mol Endocrinol. 2014;52:R137–R149. [DOI] [PubMed] [Google Scholar]
- 127. Everett LJ, Lazar MA. Cell-specific integration of nuclear receptor function at the genome. Wiley Interdiscip Rev Syst Biol Med. 2013;5:615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Beato M, Vicent GP. Impact of chromatin structure and dynamics on PR signaling. The initial steps in hormonal gene regulation. Mol Cell Endocrinol. 2012;357:37–42. [DOI] [PubMed] [Google Scholar]
- 129. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tewari AK, Yardimci GG, Shibata Y, et al. Chromatin accessibility reveals insights into androgen receptor activation and transcriptional specificity. Genome Biol. 2012;13:R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Christophersen NS, Helin K. Epigenetic control of embryonic stem cell fate. J Exp Med. 2010;207:2287–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Maruyama R, Choudhury S, Kowalczyk A, et al. Epigenetic regulation of cell type–specific expression patterns in the human mammary epithelium. PLoS Genet. 2011;7:e1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Anderson E, Clarke RB. Steroid receptors and cell cycle in normal mammary epithelium. J Mammary Gland Biol Neoplasia. 2004;9:3–13. [DOI] [PubMed] [Google Scholar]
- 135. Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sale S, Pavelic K. Mammary lineage tracing: the coming of age. Cell Mol Life Sci. 2015:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]