

Abstract
Metformin has been considered a potential adjunctive therapy in treating poorly controlled type 1 diabetes with obesity and insulin resistance, owing to its potent effects on improving insulin sensitivity. However, the underlying mechanism of metformin's vascular protective effects remains obscure. Thioredoxin-interacting protein (TXNIP), a key regulator of cellular redox state induced by high-glucose concentration, decreases thioredoxin reductase activity and mediates apoptosis induced by oxidative stress. Here we report that high glucose-induced endothelial dysfunction is associated with induction of TXNIP expression in primary human aortic endothelial cells exposed to high-glucose conditions, whereas the metformin treatment suppresses high-glucose-induced TXNIP expression at mRNA and protein levels. We further show that metformin decreases the high-glucose-stimulated nuclear entry rate of two transcription factors, carbohydrate response element-binding protein (ChREBP) and forkhead box O1 (FOXO1), as well as their recruitment on the TXNIP promoter. An AMP-activated protein kinase inhibitor partially compromised these metformin effects. Our data suggest that endothelial dysfunction resulting from high-glucose concentrations is associated with TXNIP expression. Metformin down-regulates high-glucose-induced TXNIP transcription by inactivating ChREBP and FOXO1 in endothelial cells, partially through AMP-activated protein kinase activation.
Metformin, a first-line oral antidiabetic drug for adult and pediatric type 2 diabetes has long been known to promote its lipid-lowering and insulin sensitivity-improving actions in liver and pancreatic cells (1–5). It inhibits the mitochondrial respiratory chain complex I, which increases the AMP to ATP ratio, thus leading to the activation of AMP-activated protein kinase (AMPK) (6), an important cellular energy sensor that may reflect glucose homeostasis. Activated AMPK is phosphorylated at Thr-172 and subsequently phosphorylates multiple downstream effectors to dictate cellular metabolism for the restoration of energy homeostasis (7), thereby conferring improved insulin sensitivity and lower hepatic lipid content on cells. These properties of metformin make it an attractive candidate for a potential adjunctive therapy for treating type 1 diabetes (T1D). Recent clinical trial data have shown that metformin treatment has a positive impact on improving insulin sensitivity in the overweight youth with T1D (8).
Poor glycemic control increases the incidence of cardiovascular complications that are associated with endothelial dysfunction. Although multiple studies suggest that metformin reduces cardiovascular disease risk in patients with type 2 diabetes (9–11), the underlying mechanism by which metformin improves endothelial cell functions remains poorly understood. Understanding the mechanisms of the vascular-protective effects of metformin during hyperglycemia is necessary before clinicians can evaluate whether metformin might be beneficial for patients with T1D by conferring protection against the micro- and macrovascular complications of T1D.
Thioredoxin-interacting protein (TXNIP), a protein acutely induced by high glucose concentrations, has emerged as a key factor in maintaining glucose homeostasis (12). It binds to, and thus inactivates, the antioxidant protein thioredoxin, which is required for reducing cellular oxidative stress. In addition, it inhibits peripheral glucose uptake, which is independent of thioredoxin binding (12, 13). TXNIP transcription has been shown to be controlled by the transcription complexes carbohydrate response element (ChoRe)-binding protein (ChREBP)-max-like factor X (Mlx), which is predominantly expressed in liver, and by MondoA-Mlx, which mainly exists in skeletal muscle (14). ChREBP-Mlx and MondoA-Mlx bind to the ChoRE in the TXNIP promoter to activate its expression in response to increased glucose influx into cells (14). Many factors, including metformin, have already been found to regulate TXNIP mRNA levels by affecting this binding reaction (15–27). Although metformin has been previously found to repress TXNIP expression in pancreatic β-cells (21), metformin's effects on TXNIP in endothelial cells remain unknown. Of note, previous studies indicate that the regulation of TXNIP transcription may be in a tissue-specific pattern (26, 28–30). Transcription factor forkhead box O1 (FOXO1) up-regulates TXNIP expression in neurons (29) and endothelial cells (30) but inhibits it in liver (28) and pancreatic β-cells (26). Therefore, our study was directed to investigating metformin effects on endothelial TXNIP to elucidate the mechanism that underpins this vascular-protective function of metformin. We demonstrated that metformin benefits endothelial cells by ameliorating high-glucose effects partially via AMPK activation-dependent inactivation of ChREBP and FOXO1 in TXNIP transcriptional initiation, suggesting that metformin may be a potential adjunctive therapy for protection against the micro- and macrovascular complications of T1D.
Materials and Methods
Materials
Metformin (1,1-dimethylbiguanide hydrochloride), mannitol, and anti-β-actin were purchased from Sigma. Compound C was purchased from Millipore. Anti-AMPKα, anti-phospho-AMPKα, anti-phospho-acetyl-CoA carboxylase (ACC), anti-ACC, anti-FOXO1, and FOXO1 small interfering RNA (siRNA) were from Cell Signaling Technology. Anti-TXNIP was from Invitrogen. Anti-ChREBP, siRNAs of AMPK, ChREBP, and the negative control were from Santa Cruz Biotechnology. Protein lysis buffer consisted of 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2 EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin; the mouse aortic endothelial cell (MAEC) line growth media consisted of Medium 199 supplemented with 5 ng/mL vascular endothelial growth factor, 10 mM HEPES, 0.12 mU/mL heparin sodium, 5% fetal bovine serum, and 1% penicillin/streptomycin; and the human aortic endothelial cell (HAEC) growth media consisted of endothelial cell basal medium supplemented with EGM SingleQuots from Lonza.
Cell culture and treatments
MAEC was obtained from Dr Ichiro (Tsurumi University, Yokohama, Japan) (31). MAECs were maintained in MAEC growth media at 37°C and 5% CO2. Unless otherwise indicated, the glucose concentration of the normal growth medium for MAEC was 5.5 mM. MAECs were serum starved overnight in normal growth medium prior to any treatments. Primary HAECs were obtained from Lonza and cultured according to the manufacturer's instructions. Unless otherwise indicated, the glucose concentration of the normal growth medium for HAECs was 5.5 mM, and only the cells at passage 5–6 were used. For metformin treatment, cells were exposed to 5.5 mM D-glucose (normal glucose), 30 mM D-glucose (high glucose), 25 mM mannitol (osmotic control), or high-glucose growth medium with 2 mM metformin in the absence or presence of 10 μM compound C for the times indicated.
Rats and treatments
Male Sprague Dawley rats (weight 250–300 g, age 6–8 weeks) were purchased from Harlan Laboratories. The rats were randomly divided into three groups: nondiabetic, diabetic, and diabetic with metformin treatment. Diabetes was induced by giving a one-time ip injection of streptozotocin (Sigma), 70 mg/kg, as we previously reported (32) and defined as random blood glucose levels of greater than 250 mg/dL for 3 consecutive days. One week after diabetes induction, the streptozotocin-injected rats received metformin in their drinking water for 4 weeks, starting at 50 mg/kg·d body weight and increasing the dose by 50 mg/kg every day to the final dose of 150 mg/kg·d. Water was changed daily. At the end of the treatment period, the rats were humanely euthanized. The aortas were collected then saved snap frozen in liquid nitrogen for further studies.
The rats were housed in a 12-hour light, 12-hour dark cycle with free access to water and regular food. This study was carried out in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the University of Missouri-Kansas City Institutional Animal Care Use Committee protocol 1229.
Assessment of intracellular reactive oxygen species generation
Confluent HAECs in six-well plates were exposed to normal glucose (5.5 mM), high glucose (30 mM), or high glucose plus metformin (2 mM) for 24 hours. For measurement of intracellular reactive oxygen species (ROS) generation, treated cells were then stained with 10 μmol/L dihydroethidium (DHE) for 20 minutes and subsequently photographed by fluorescence microscopy (Fluoview-300; Olympus). Single-cell fluorescence intensities were determined for 60 cells from six randomly selected fields (×200) per group per experiment, using ImageJ analysis software [National Institutes of Health (NIH), Bethesda, Maryland]. Three independent experiments were selected for quantification for each group.
Construction of TXNIP promoter reporter plasmids
The DNA fragment that covers 1 kb Txnip promoter and the first exon of Txnip cDNA was amplified from mouse genomic DNA by PCR and then cloned into a pGL3-basic vector (Promega) to generate a functional Txnip promoter-driven reporter construct named as pTXP-1000. The sequences of the Txnip promoter region in all the constructs were verified by DNA sequencing. All the primers used here are listed in Supplemental Table 1.
Transient transfection
MAECs or HAECs were grown in six-well plates and transfected with mouse-ChREBP-specific siRNA oligonucleotide (m-ChREBPsi) (100 nM), mouse FOXO1-specific siRNA oligonucleotide (m-FOXO1si), human-ChREBP-specific siRNA oligonucleotide (h-ChREBPsi) (100 nM), human-FOXO1-specific siRNA oligonucleotide (h-FOXO1si) (100 nM), human-AMPKα1/2-specific siRNA oligonucleotide (h-AMPKsi) (100 nM), scrambled oligonucleotides (100 nM), or FLAG-FOXO1 (Addgene) (0.4 μg/well) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions (33). All the treatments were performed after an overnight starvation at 24 hours after the transfection.
Luciferase reporter assay
MAECs were seeded in 96-well plates and transfected with a variety of DNA constructs or with an Simian virus 40 driven pGL3 control vector (0.16 μg/well), using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfected cells were treated after an overnight starvation at 24 hours after the transfection. The relative firefly luciferase activities were measured in a TriStar LB 941 multimode microplate reader (Berthold Technologies GmbH & Co KG) using the dual luciferase reporter assay kit from Promega, following the manufacturer's instructions (33). Data were normalized with the Renilla luciferase activity.
Quantitative RT-PCR
A quantitative RT-PCR (qRT-PCR) analysis was performed as described previously (33). Total RNA was extracted using an RNeasy Plus minikit (QIAGEN) according to the manufacturer's instructions. Reverse transcription reactions were performed using a SuperScript II first-strand kit (Invitrogen). qRT-PCR was performed on a Mastercycler ep realplex real-time PCR system (Eppendorf) using iQ SYBR Green Supermix (Bio-Rad Laboratories). Primers used are listed in Supplemental Table 1. All the data were normalized with the expression of β-actin run as an internal standard.
Western blot analysis
Western blot analysis was performed as described previously (33). Whole-cell lysates were prepared on ice using lysis buffer (Cell Signaling Technology) with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche) after two ice-cold rinses with PBS. Immunoreactive bands were visualized by enhanced chemiluminescence Western blotting detection reagent (GE Healthcare Life Sciences) and quantified by the NIH ImageJ software.
Coimmunoprecipitation assay
For immunoprecipitations, MAEC cell lysates prepared in protein lysis buffer were incubated with an anti-FLAG antibody for 4 hours at 4°C and then incubated with protein-G Sepharose beads (GE Healthcare Life Sciences) overnight at 4°C. After three washes in protein lysis buffer, the beads were boiled in Laemmli sample buffer.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed as described elsewhere (33). Briefly, the TXNIP-promoter-ChREBP complex or the TXNIP-promoter-FOXO1 complex was immunoprecipitated with antibodies against ChREBP or FOXO1 from the cell lysates of the MAECs and HAECs treated with 5.5 mM D-glucose, 30 mM D-glucose, or 30 mM D-glucose plus 2 mM metformin. A normal rabbit IgG was used as a negative control. The DNA extracts were then subjected to qRT-PCR analysis. The primers used for ChoRE and the FOXO binding site (FXBS) in the TXNIP promoter are listed in Supplemental Table 1.
Immunostaining and confocal microscopy
HAECs were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then stained with a rabbit anti-ChREBP antibody (1:100; Novus Biologicals) or a rabbit anti-FOXO1 antibody (1:100; Cell Signaling), followed by Alexa Fluor 488 goat antirabbit IgG (1:1000; Invitrogen). Cell imaging was performed on a Zeiss LSM 510 Meta confocal microscope fitted with a PlanApo ×63 oil immersion objective.
Statistical analysis
Results represent means ± SEM. P values were calculated by 1-way ANOVA followed by post hoc analysis for data sets from triplicate experiments using GraphPad Prism 6. Statistical significance was set at P < .05.
Results
Metformin relieves increased intracellular ROS production, proinflammatory state, and early apoptosis in high-glucose-treated endothelial cells by blunting TXNIP overexpression
To investigate the possible vascular-protective function of metformin using TXNIP as a reporter, we first investigated the TXNIP expression profile in high glucose- vs normal glucose-treated MAECs and primary HAECs, by RT-qPCR analysis. We discovered that high glucose exposure significantly increased Txnip mRNA levels and that metformin suppressed the Txnip mRNA level in MAECs in a time-dependent manner (Figure 1A), with no change in 25 mM mannitol treatment (data not shown). We further tested the Txnip promoter activities in MAECs by luciferase reporter assay using a luciferase reporter-containing construct driven by the full-length Txnip promoter. As shown in Figure 1B, metformin (2 mM) effectively inhibited the induction of Txnip promoter activities by high glucose, which is also consistent with its effects on Txnip mRNA expression. We also observed this mRNA trend in HAECs (Figure 1C), indicating that metformin suppresses the high-glucose-induced TXNIP overexpression in endothelial cells. Moreover, metformin also expressed this effect in rat aortas (Figure 1D).
Figure 1. Metformin potently suppresses high glucose-induced TXNIP overexpression in endothelial cells.
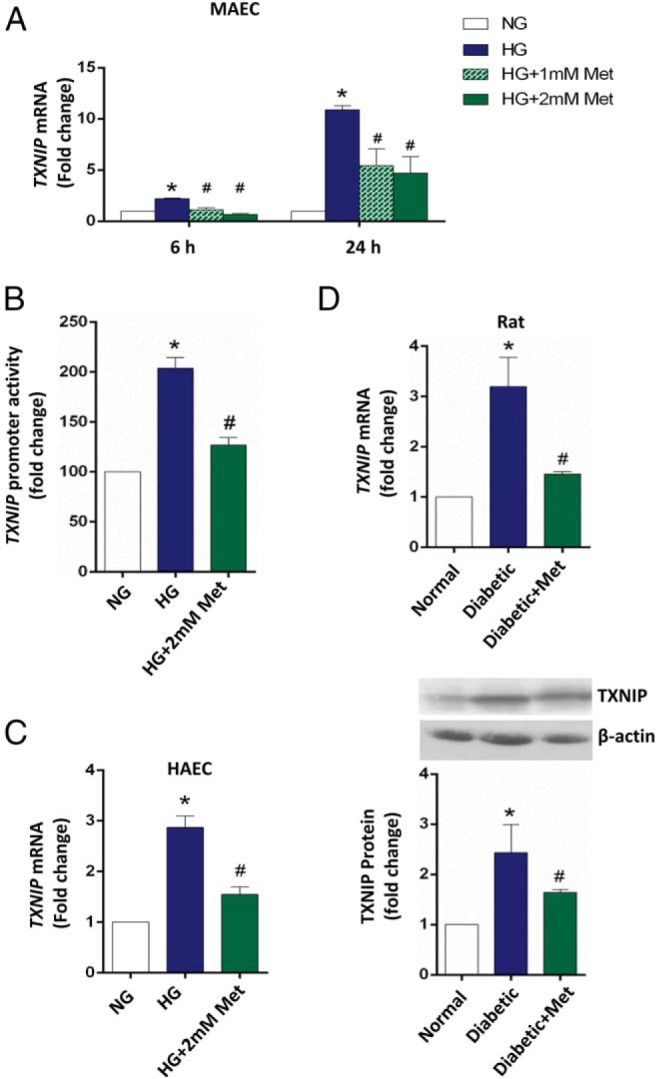
A, Time-dependent effects of metformin on Txnip expression in MAECs. MAECs were harvested after 6 and 24 hours of exposure to 5.5 mM glucose (NG), 30 mM glucose (HG), or metformin treatments indicated in the figure and then subjected to qRT-PCR analysis. B, Metformin suppressed the Txnip promoter activity in MAECs. MAECs were transiently transfected with a full-length Txnip promoter-driven luciferase reporter-containing construct and then at 24 hours after transfection treated for another 24 hours as indicated. C, Metformin suppressed TXNIP mRNA level in primary HAECs. HAECs were harvested and then subjected to qRT-PCR analysis after the 48 hours of treatments as indicated. Data were corrected for β-actin and quantified. D, Metformin suppressed hyperglycemia-induced TXNIP expression in rat aortas. Aortas were collected from saline-injected control (NG; n = 3), streptozotocin-induced diabetic (HG; n = 3), and diabetic plus 150 mg/kg metformin (HG+Met; n = 3) groups of rats after 4-week treatments. TXNIP mRNA (upper panel) and protein (lower panel) levels in rat aortas were analyzed by qRT-PCR and immunoblotting. Data were corrected for β-actin and quantified. In panels A–D, the data are represented as the mean ± SEM (n = 3). *, P < .001 vs normal glucose; #, P < .001 vs high glucose.
Next we determined whether this metformin-suppressed TXNIP overexpression was associated with the possible vascular-protective effects of metformin on endothelial cells. As shown in Figure 2, exposing HAECs to high glucose (30 mM) led to a significant increase in ROS production, intercellular adhesion molecule 1 (ICAM-1; an inflammatory marker) expression, and cleavage of poly(ADP-ribose)polymerase (PARP), an early apoptosis marker, whereas metformin prevented these effects. Transient knockdown of TXNIP mimicked the metformin effects in HAECs (Figure 2, A and B), suggesting that TXNIP mediates at least the high-glucose-induced changes that are consistent with endothelial dysfunction.
Figure 2. Metformin (Met) ameliorates high glucose-induced increase of ROS production, proinflammatory state, and early apoptosis in endothelial cells.
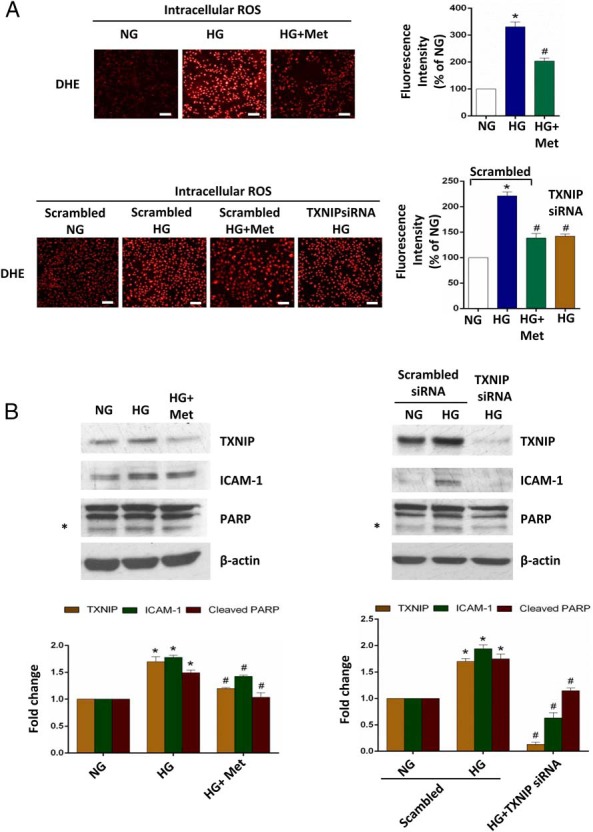
Confluent primary HAECs were treated as indicated for 24 hours. For transient knockdown of TXNIP, 100 nM control siRNA and human-specific TXNIP siRNA were applied to HAECs for 24 hours before the treatments. NG, normal glucose, 5.5 mM glucose; HG, high glucose, 30 mM glucose. A, Measurement of intracellular ROS production using DHE. Scale bar, 100 μm. Fluorescence intensity quantified using NIH ImageJ software is also shown. B, ICAM-1 level and PARP cleavage assessed by Western blot analysis. Transient TXNIP knockdown was also confirmed by Western blot. The protein levels corrected for β-actin and quantified using ImageJ software (NIH) are shown in the right panel. Representative fluorescence images and Western blots from three independent experiments are shown, and the data are represented as the mean ± SEM (n = 3). *, P < .001 vs normal glucose; #, P < .001 vs high glucose.
Metformin impairs high-glucose-induced TXNIP overexpression partially via AMPK pathway
Given that metformin is well known as an AMPK activator (34), we applied an AMPK inhibitor, compound C, along with metformin to endothelial cells under high glucose to inspect the possible involvement of the AMPK pathway in metformin-down-regulated TXNIP expression. We first assessed the phosphorylated AMPK level, which represents the status of AMPK activation. As seen in Figure 3, A and B, without any change in the total amount of AMPK protein, high glucose severely abated AMPK activation, whereas metformin dramatically rescued the cells from this negative effect. Compound C, however, partly reversed the metformin effects on AMPK phosphorylation (Figure 3, A and B). We further observed that compound C abrogated the metformin regulation at both the promoter and protein levels to some extent (Figure 3, A–C). Therefore, AMPK activation at least partially mediates the metformin-dependent down-regulation of TXNIP expression.
Figure 3. Metformin suppresses high glucose-induced TXNIP overexpression at least partially through activation of AMPK.

A and B, Metformin-suppressed TXNIP overexpression is partially reversed by an AMPK inhibitor, compound C, in primary HAECs (A) and MAECs (B). For Western blot analysis of TXNIP, HAECs were harvested after the 48-hour exposure to the treatments indicated; MAECs were harvested after the 24-hour treatments as indicated. For Western blot analysis of phosphorylated AMPK, cells were serum starved overnight and then treated for 1 hour as indicated. C, Metformin-suppressed Txnip promoter activities is partially blocked by compound C. MAECs were transfected with a Simian virus 40 -driven pGL3 control vector (data not shown) or a full-length TXNIP promoter-driven luciferase reporter and then treated 24 hours after transfection for additional 24-hour treatments as indicated. In panels A–C, the data are represented as the mean ± SEM (n = 3). NG, normal glucose, 5.5 mmol/L glucose; HG, high glucose, 30 mmol/L glucose; HG+Met, 30 mmol/L glucose plus 2 mmol/L metformin; HG+Met+C, 30 mmol/L glucose plus 2 mmol/L metformin and 10 μmol/L compound C. *, P < .05 vs normal glucose; #, P < .05 vs high glucose; $, P < .05 vs high glucose plus metformin. In panels A and B, representative Western blots from three independent experiments are shown.
ChREBP and FOXO1 bind to the TXNIP promoter and thus elevate TXNIP transcription in endothelial cells
Based on the in silico analysis of the TXNIP promoter, we hypothesized that the down-regulation of TXNIP transcription by metformin is likely mediated by two highly conserved regulatory elements, FXBS and a ChoRE composed of two E-boxes, within its promoter region (Figure 4A). ChoRE and FXBS are known to be recognized by two transcription factors, ChREBP and FOXO1, respectively, in liver and pancreatic β-cells (26, 28, 35) but have not been well studied in endothelial cells. To establish the correlation between metformin signaling and ChREBP- and FOXO1-involved transcriptional regulation of TXNIP in endothelial cells, we first performed a ChIP assay to determine whether endothelial ChREBP and FOXO1 bind to ChoRE and FXBS, respectively. As expected, a significant enrichment of endogenous ChREBP and FOXO1 at ChoRE and FXBS was observed in both MAECs and HAECs, respectively (Supplemental Figure 1). No enrichment was indicated in the control IgG immunoprecipitates or at the GAPDH internal control (Supplemental Figure 1). Next, to examine the effects of ChREBP and FOXO1 on TXNIP transcription, we measured the Txnip promoter activities by luciferase reporter assay in transient ChREBP and FOXO1 knockdown MAECs, respectively. We found that the induction of Txnip promoter activities by high glucose was blunted by the mouse-specific ChREBP or FOXO1 siRNA but not by scrambled oligonucleotides (Figure 4B). Meanwhile, the Simian virus 40 control promoter activities were unaffected by both the ChREBP and FOXO1 siRNAs as a negative control (data not shown).
Figure 4. Metformin impairs the high-glucose-enhanced binding capacities of ChREBP and FOXO1 to the TXNIP promoter.
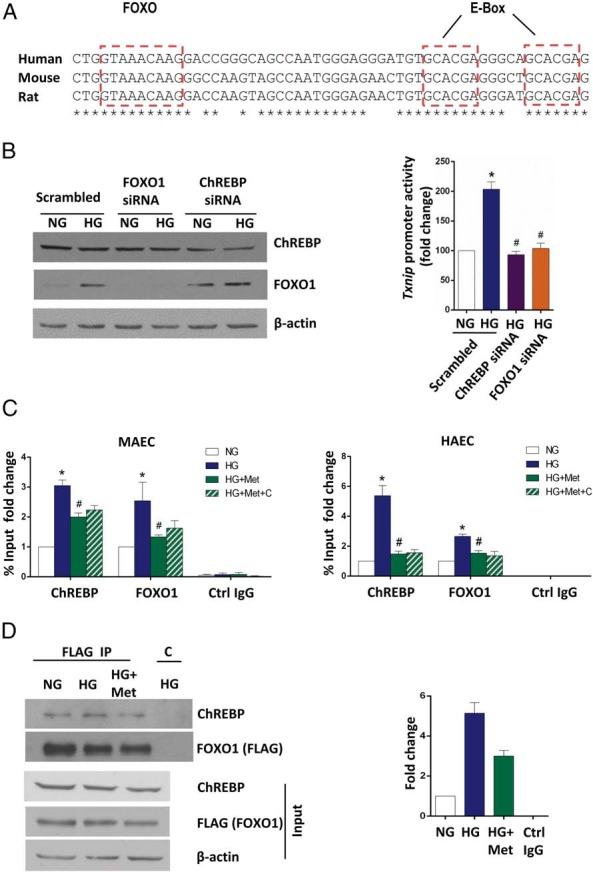
A, Promoter analysis of the TXNIP promoter. The conserved glucose-responsible E-Box and putative FXBS are shown. B, ChREBP and FOXO1 elevate the Txnip promoter activity under high glucose. MAECs were cotransfected with the full Txnip promoter-driven luciferase reporter plasmid and scrambled oligonucleotide (control siRNA), siChREBP or siFOXO1 and 24 hours after transfection subjected to the treatments indicated for an additional 24 hours. Bars indicate relative firefly luciferase activities, and the data are represented as the mean ± SEM (n = 3). *, P < .001 vs normal glucose; #, P < .05 vs high glucose. Successful FOXO1 and ChREBP knockdown was confirmed by immunoblotting (left panel). C, MAECs (left panel) and primary HAECs (right panel) were treated for 24 hours (MAECs) or 48 hours (HAECs) prior to ChIP assays. Ctrl IgG, control IgG. The data are represented as the mean ± SEM (n = 3). *, P < .05 vs normal glucose; #, P < .05 vs high glucose. D, MAEC cells were transiently transfected with a FLAG-tagged FOXO1 and then treated for 24 hours as indicated. FLAG or control IgG (C) immunoprecipitates were immunoblotted with anti-ChREBP and then stripped and reprobed with anti-FOXO1 to control for levels. The whole-cell lysates were immunoblotted with anti-FLAG and anti-ChREBP to detect levels of FLAG-tagged FOXO1 and ChREBP, respectively. Representative Western blots from three independent experiments are shown. B to D, NG, normal glucose, 5.5 mM glucose; HG, high glucose, 30 mM glucose; HG+Met, 30 mM glucose plus 2 mM metformin; HG+Met+C, 30 mmol/L glucose plus 2 mmol/L metformin and 10 μmol/L compound C.
Metformin suppresses the recruitment of ChREBP and FOXO1 on the TXNIP promoter
We next performed ChIP assays to determine whether metformin affects the recruitment of ChREBP and FOXO1 on the TXNIP promoter. In both MAECs and primary HAECs, high glucose potently induced the binding of ChREBP and FOXO1 to ChoRE and FXBS, respectively, whereas metformin significantly aborted this high-glucose effect (Figure 4C). By contrast, no significant change was detected among all the treatments when control IgG was used in the ChIP assays (Figure 4C). Similarly, the GAPDH promoter failed to show any enrichment in the immunoprecipitates as a negative internal control (data not shown), confirming the specificity of the ChIP assays performed. This metformin-disturbed coregulation of TXNIP transcription by ChREBP and FOXO1 prompted us to explore the possible protein-protein interaction between ChREBP and FOXO1 as well as metformin actions on this interaction. By coimmunoprecipitation analysis using MAECs transiently transfected with a FLAG-tagged mouse FOXO1, we revealed that the interaction of ChREBP with FOXO1 existed at a moderate level even under normal glucose conditions. This interaction was significantly increased upon high glucose yet was compromised by the metformin treatment (Figure 4D), conforming to the observed metformin-impaired enrichment of ChREBP and FOXO1 on the promoter.
Metformin inactivates intracellular ChREBP and FOXO1 via the AMPK pathway
Given that ChREBP and FOXO1 require nuclear translocation for their activation, we further investigated metformin's effect on the nuclear entry of endothelial ChREBP and FOXO1 and the role of AMPK activation in this process. Immunofluorescence staining and subcellular fractionation analysis (Figure 5A) indicated that metformin caused a notable decrease in high-glucose-stimulated nuclear accumulation of ChREBP and FOXO1, whereas compound C dramatically reversed this effect. We further created AMPKα (α1 and α2) knockdown HAECs to confirm this observation. As expected, the transient knockdown of AMPKα exactly reproduced the effect of compound C, which was further confirmed by the concomitant decrease in the phosphorylation of ACC, an AMPK downstream target (Figure 5B), supporting the concept that in endothelial cells, metformin represses the activities of ChREBP and FOXO1 by reducing their nuclear entry rate in a manner dependent on AMPK activation.
Figure 5. Metformin hinders nuclear translocations of ChREBP and FOXO1 via the AMPK pathway.

A, HAECs were treated for 48 hours as indicated. Western blot analysis of the nuclear fractions under different conditions is also shown in the bottom panel. The protein levels were corrected for Lamin-A and quantified using ImageJ software (NIH). B, AMPK knockdown diminishes the metformin effect on nuclear translocations of ChREBP and FOXO1 under high-glucose conditions. HAECs were transiently transfected with the human-specific AMPKα (α1 and α2) siRNA or scrambled siRNA and then treated 24 hours after transfection for an additional 24 hours as indicated. Transient AMPK knockdown was confirmed by the Western blots shown in the bottom panel. In panels A and B, representative images from two independent experiments are shown. Immunocytochemistry and confocal imaging were performed as described in Materials and Methods. Scale bars, 5 μm. NG, normal glucose, 5.5 mM glucose; HG, high glucose, 30 mM glucose; HG+Met, 30 mM glucose plus 2 mM metformin; HG+Met+C, 30 mM glucose plus 2 mM metformin and 10 μM compound C. The data are represented as the mean ± SEM (n = 3).
All these findings taken together indicate that the metformin-mediated regulation of the TXNIP promoter activity lies in the recruitment of ChREBP and FOXO1 on the TXNIP promoter. This inhibitory effect of metformin in the nucleus is supported, at least partially, by the metformin-suppressed nuclear translocations of ChREBP and FOXO1.
Metformin mitigates high-glucose-induced TXNIP overexpression by suppressing ChREBP and FOXO1
Given that metformin impairs the transcriptional activities of ChREBP and FOXO1, we examined the possibility that the ablation of endothelial ChREBP and FOXO1 will ameliorate endothelial dysfunction caused by high glucose. Transient knockdown of ChREBP and FOXO1 decreased ROS production and alleviated the proinflammatory state and early apoptosis in primary HAECs under high-glucose conditions, resembling the effects of the metformin treatment (Figure 6, A–D). This finding indicates that metformin imposes its effects on TXNIP transcription via the regulation by ChREBP and FOXO1.
Figure 6. Ablation of ChREBP and FOXO1 ameliorates high-glucose-induced endothelial dysfunction in HAECs.

HAECs were transiently transfected with the human-specific siRNAs of ChREBP and FOXO1 or scrambled siRNA and then treated 24 hours after transfection for an additional 24 hours as indicated. NG, normal glucose, 5.5 mM glucose; HG, high glucose, 30 mM glucose. A, Measurement of intracellular ROS production by DHE staining. Scale bar, 100 μm. Fluorescence intensity quantified using NIH ImageJ software is also shown. B, Protein level analysis of TXNIP and PARP cleavage. Transient ChREBP and FOXO1 knockdown were also confirmed by Western blot. C, Protein level analysis of ICAM-1. The protein levels corrected for β-actin and quantified using NIH ImageJ software are shown in panel D. Representative fluorescence images and Western blots from three independent experiments are shown. The data are represented as the mean ± SEM (n = 3). *, P < .001 vs normal glucose; #, P < .001 vs high glucose.
Altogether, metformin attenuates high-glucose-induced TXNIP transcription by depressing intracellular ChREBP and FOXO1 activities in endothelial cells, partly through AMPK activation.
Discussion
In this study, we discovered an indispensable role of the metformin-AMPK-TXNIP pathway, which contributes to the potential vascular-protective function of metformin by regulating the activities of two transcription factors, ChREBP and FOXO1.
ChREBP (or MondoA) is a central glucose-sensing transcription factor that is responsible for 75% of intracellular glucose-induced transcriptional response (14). TXNIP, which fundamentally mediates glucose homeostasis, is one of its direct targets (36). Recently several publications have reported that another transcription factor, FOXO1, may also regulate TXNIP transcription (26, 28–30). Moreover, FOXO1 has been found to compete with ChREBP to inhibit TXNIP transcription in pancreatic β-cells (26). In the present study, we have identified a cross talk between endothelial ChREBP and FOXO1, which is distinct from the one reported in pancreatic β-cells. Our data show that both ChREBP and FOXO1 are dramatically recruited to the TXNIP promoter to transactivate strong expression of TXNIP upon high glucose, similar to the previously revealed FOXO1-upregulated TXNIP expression in neurons (29). However, in liver and pancreatic β-cells, FOXO1 was observed to inhibit TXNIP expression (26, 28) and was further confirmed in pancreatic β-cells to down-regulate TXNIP transcription by decreasing ChREBP binding to the TXNIP promoter (26). All this evidence, considered alongside our results, indicate that the FOXO1 actions are tissue specific, at least in the context of high-glucose-regulated transcription.
The antidiabetic drug, metformin, has been demonstrated to reduce TXNIP expression in pancreatic β-cells (21) and HeLa cells (22) by preventing some transcription factors, including ChREBP (21), MLX, and nuclear transcription factor Y subunit-α (22), from binding to the TXNIP promoter. In our study, this metformin down-regulated TXNIP expression is also confirmed in endothelial cells at mRNA and protein levels. Furthermore, our ChIP data show that not only ChREBP, but also FOXO1, is inhibited by metformin from binding to the TXNIP promoter in endothelial cells. By coimmunoprecipitation analysis, we further observed the physical interaction that existed between ChREBP and FOXO1. We speculate that this interaction might facilitate more stable binding of ChREBP and FOXO1 to the promoter, owing to the enrichment of ChREBP and FOXO1 on the TXNIP promoter in response to increased glucose influx into cells. Nonetheless, further evidence is needed to verify whether this interaction has any functional consequence in transcriptional regulation.
The AMPK mediation of metformin actions has long been reported. Recently AMPK activation by metformin has been linked to metformin-mediated suppression of TXNIP expression in mouse colon and ileum (37) and in mouse embryonic fibroblast cells (22), consistent with our observations in endothelial cells. Despite these prior observations, the mechanism of the metformin-AMPK-TXNIP pathway remains ill defined. In this study we further delineated another important role of AMPK activation in the regulation of the intracellular ChREBP and FOXO1 activities by metformin. As our data demonstrate, metformin abrogates the high-glucose-stimulated nuclear translocation of ChREBP and FOXO1 via AMPK activation. It has already been found that phosphorylation of ChREBP and FOXO1 facilitates their cytoplasmic retention and that ChREBP and FOXO1 can each be phosphorylated by AMPK (7, 38). These data strongly support our notion that metformin-suppressed nuclear entry of ChREBP and FOXO1 is AMPK dependent. The only existing data supporting FOXO1 phosphorylation by AMPK come from an in vitro kinase assay (38). Our data presented here provide the functional consequence of this phosphorylation. Despite this decisive role, AMPK does not play a central role in terms of metformin-suppressed transcriptional initiation of TXNIP, which was confirmed by our qRT-PCR and ChIP assays in the compound C-treated HAECs and MAECs (Supplemental Figure 2 and Figure 4C). We deduce that an as-yet-unknown pathway within the nucleus mediates the metformin-induced compromised binding of ChREBP and FOXO1 to the TXNIP promoter.
Overall, metformin negatively regulates TXNIP transcription in endothelial cells by impairing the transcriptional activities of ChREBP and FOXO1 on multiple steps that occur both in cytosol and in the nucleus. First, metformin prevents the nuclear entry of ChREBP and FOXO1 from cytosol. Next, for those escaped or already translocated ChREBP and FOXO1, metformin further inhibits their binding capacity to the promoter, thus eventually ensuring potent and effective suppression of TXNIP transcription (Figure 7). The inhibitory effects of metformin on nuclear translocation require AMPK-mediated phosphorylation (Figure 7). Because our data are based on ex vivo cell studies, a more detailed underlying mechanism of metformin actions on endothelial TXNIP still needs to be defined and confirmed in animal models.
Figure 7. Model for metformin action on TXNIP transcription in endothelial cells under high-glucose conditions.
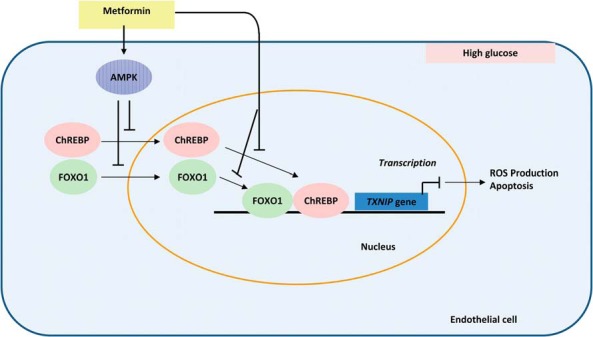
Metformin prevents the nuclear entry of ChREBP and FOXO1 from cytosol and further inhibits their binding capacity to the TXNIP promoter and thus suppresses TXNIP transcription at last. The inhibitory effect of metformin on nuclear translocation is AMPK phosphorylation dependent.
Acknowledgments
We thank Peiying Tong and Chaoying Zhang for their help on some of the animal experiments, We also thank Dr Shui-Qing Ye (Children's Mercy Hospital and University of Missouri-Kansas City, Kansas City, Missouri) for a critical reading of this manuscript. The authors also thank the Medical Writing Center at Children's Mercy Hospital for editing this manuscript.
This work was supported by Children's Mercy Hospital Physician Scientist Award (to Y.Y. and M.A.C.), partially supported by Diabetes Action Research and Education Foundation Grant 399 (to Y.Y. and M.A.C.), and partially supported by The Endocrine Society Helmsley Charitable Trust Abstract Award (to X.L.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by Children's Mercy Hospital Physician Scientist Award (to Y.Y. and M.A.C.), partially supported by Diabetes Action Research and Education Foundation Grant 399 (to Y.Y. and M.A.C.), and partially supported by The Endocrine Society Helmsley Charitable Trust Abstract Award (to X.L.).
Footnotes
- ACC
- acetyl-CoA carboxylase
- AMPK
- AMP-activated protein kinase
- ChIP
- chromatin immunoprecipitation
- ChoRE
- carbohydrate response element
- ChREBP
- ChoRE-binding protein
- DHE
- dihydroethidium
- FOXO1
- forkhead box O1
- FXBS
- FOXO binding site
- HAEC
- human aortic endothelial cell
- ICAM-1
- intercellular adhesion molecule 1
- MAEC
- mouse aortic endothelial cell
- Mlx
- max-like factor X
- PARP
- poly(ADP-ribose)polymerase
- qRT-PCR
- quantitative RT-PCR
- ROS
- reactive oxygen species
- siRNA
- small interfering RNA
- T1D
- type 1 diabetes
- TXNIP
- thioredoxin-interacting protein.
References
- 1. Yu B, Pugazhenthi S, Khandelwal RL. Effects of metformin on glucose and glucagon regulated gluconeogenesis in cultured normal and diabetic hepatocytes. Biochem Pharmacol. 1994;48:949–954. [DOI] [PubMed] [Google Scholar]
- 2. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lupi R, Del Guerra S, Fierabracci V, et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes. 2002;51(suppl 1):S134–S137. [DOI] [PubMed] [Google Scholar]
- 4. Leclerc I, Woltersdorf WW, da Silva Xavier G, et al. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: impact on glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2004;286:E1023–E1031. [DOI] [PubMed] [Google Scholar]
- 5. Zang M, Zuccollo A, Hou X, et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898–47905. [DOI] [PubMed] [Google Scholar]
- 6. Hardie DG. AMPK. a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond). 2008;32(suppl 4):S7–S12. [DOI] [PubMed] [Google Scholar]
- 7. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nadeau KJ, Chow K, Alam S, et al. Effects of low dose metformin in adolescents with type I diabetes mellitus: a randomized, double-blinded placebo-controlled study. Pediatr Diabetes. 2015;16(3):196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garber AJ. Metformin and vascular protection: a diabetologist's view. Diabetes Metab. 2003;29:6S113–6S116. [DOI] [PubMed] [Google Scholar]
- 10. Libby P. Metformin and vascular protection: a cardiologist's view. Diabetes Metab. 2003;29:6S117–6S120. [DOI] [PubMed] [Google Scholar]
- 11. Abbasi F, Chu JW, McLaughlin T, Lamendola C, Leary ET, Reaven GM. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metabolism. 2004;53:159–164. [DOI] [PubMed] [Google Scholar]
- 12. Wu N, Zheng B, Shaywitz A, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49:1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parikh H, Carlsson E, Chutkow WA, et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peterson CW, Stoltzman CA, Sighinolfi MP, Han KS, Ayer DE. Glucose controls nuclear accumulation, promoter binding, and transcriptional activity of the MondoA-Mlx heterodimer. Mol Cell Biol. 2010;30:2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaadige MR, Looper RE, Kamalanaadhan S, Ayer DE. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc Natl Acad Sci USA. 2009;106:14878–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu FX, Goh SR, Dai RP, Luo Y. Adenosine-containing molecules amplify glucose signaling and enhance txnip expression. Mol Endocrinol. 2009;23:932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J, Fontes G, Saxena G, Poitout V, Shalev A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated β-cell death. Diabetes. 2010;59:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu FX, Chai TF, He H, Hagen T, Luo Y. Thioredoxin-interacting protein (Txnip) gene expression: sensing oxidative phosphorylation status and glycolytic rate. J Biol Chem. 2010;285:25822–25830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chai TF, Leck YC, He H, Yu FX, Luo Y, Hagen T. Hypoxia-inducible factor independent down-regulation of thioredoxin-interacting protein in hypoxia. FEBS Lett. 2011;585:492–498. [DOI] [PubMed] [Google Scholar]
- 20. Peterson CW, Ayer DE. An extended Myc network contributes to glucose homeostasis in cancer and diabetes. Front Biosci (Landmark Ed). 2011;16:2206–2223. [DOI] [PubMed] [Google Scholar]
- 21. Shaked M, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic β-cells. PLoS One. 2011;6:e28804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chai TF, Hong SY, He H, Zheng L, Hagen T, Luo Y, Yu FX. A potential mechanism of metformin-mediated regulation of glucose homeostasis: inhibition of thioredoxin-interacting protein (Txnip) gene expression. Cell Signal. 2012;24:1700–1705. [DOI] [PubMed] [Google Scholar]
- 23. He X, Ma Q. Redox regulation by nuclear factor erythroid 2-related factor 2: gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol Pharmacol. 2012;82:887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han KS, Ayer DE. MondoA senses adenine nucleotides: transcriptional induction of thioredoxin-interacting protein. Biochem J. 2013;453:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kanari Y, Sato Y, Aoyama S, Muta T. Thioredoxin-interacting protein gene expression via MondoA is rapidly and transiently suppressed during inflammatory responses. PLoS One. 2013;8:e59026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kibbe C, Chen J, Xu G, Jing G, Shalev A. FOXO1 competes with carbohydrate response element-binding protein (ChREBP) and inhibits thioredoxin-interacting protein (TXNIP) transcription in pancreatic β cells. J Biol Chem. 2013;288:23194–23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okamoto M, Yamaoka M, Takei M, et al. Endogenous hydrogen sulfide protects pancreatic β-cells from a high-fat diet-induced glucotoxicity and prevents the development of type 2 diabetes. Biochem Biophys Res Commun. 2013;442:227–233. [DOI] [PubMed] [Google Scholar]
- 28. de Candia P, Blekhman R, Chabot AE, Oshlack A, Gilad Y. A combination of genomic approaches reveals the role of FOXO1a in regulating an oxidative stress response pathway. PLoS One. 2008;3:e1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Papadia S, Soriano FX, Leveille F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X, Rong Y, Zhang M, et al. Up-regulation of thioredoxin interacting protein (Txnip) by p38 MAPK and FOXO1 contributes to the impaired thioredoxin activity and increased ROS in glucose-treated endothelial cells. Biochem Biophys Res Commun. 2009;381:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishiyama T, Mishima K, Ide F, et al. Functional analysis of an established mouse vascular endothelial cell line. J Vasc Res. 2007;44:138–148. [DOI] [PubMed] [Google Scholar]
- 32. Kover K, Tong P, Pacicca D, et al. Bone marrow cavity: a supportive environment for islet engraftment. Islets. 2011;3:93–101. [DOI] [PubMed] [Google Scholar]
- 33. Yan Y, Li X, Kover K, Clements M, Ye P. CREB participates in the IGF-I-stimulation cyclin D1transcription. Dev Neurobiol. 2013;73:559–570. [DOI] [PubMed] [Google Scholar]
- 34. Pernicova I, Korbonits M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. [DOI] [PubMed] [Google Scholar]
- 35. Cha-Molstad H, Saxena G, Chen J, Shalev A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem. 2009;284:16898–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci USA. 2008;105:6912–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massollo M, Marini C, Brignone M, et al. Metformin temporal and localized effects on gut glucose metabolism assessed using 18F-FDG PET in mice. J Nucl Med. 2013;54:259–266. [DOI] [PubMed] [Google Scholar]
- 38. Greer EL, Banko MR, Brunet A. AMP-activated protein kinase and FoxO transcription factors in dietary restriction-induced longevity. Ann NY Acad Sci. 2009;1170:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]