

Abstract
Sertoli cells (SCs), the only somatic cells within seminiferous tubules, associate intimately with developing germ cells. They not only provide physical and nutritional support but also secrete factors essential to the complex developmental processes of germ cell proliferation and differentiation. The SC transcriptome must therefore adapt rapidly during the different stages of spermatogenesis. We report comprehensive genome-wide expression profiles of pure populations of SCs isolated at 5 distinct stages of the first wave of mouse spermatogenesis, using RNA sequencing technology. We were able to reconstruct about 13 901 high-confidence, nonredundant coding and noncoding transcripts, characterized by complex alternative splicing patterns with more than 45% comprising novel isoforms of known genes. Interestingly, roughly one-fifth (2939) of these genes exhibited a dynamic expression profile reflecting the evolving role of SCs during the progression of spermatogenesis, with stage-specific expression of genes involved in biological processes such as cell cycle regulation, metabolism and energy production, retinoic acid synthesis, and blood-testis barrier biogenesis. Finally, regulatory network analysis identified the transcription factors endothelial PAS domain-containing protein 1 (EPAS1/Hif2α), aryl hydrocarbon receptor nuclear translocator (ARNT/Hif1β), and signal transducer and activator of transcription 1 (STAT1) as potential master regulators driving the SC transcriptional program. Our results highlight the plastic transcriptional landscape of SCs during the progression of spermatogenesis and provide valuable resources to better understand SC function and spermatogenesis and its related disorders, such as male infertility.
In mammals, spermatogenesis is a complex biological process, in which spermatogonial stem cells (SSCs) give rise to mature male gametes, the spermatozoa. Briefly, spermatogenesis is typically divided into 3 stages: the mitotic (spermatogonial self-renewal and proliferation), meiotic, and spermiogenic phases. Sertoli cells (SCs), 1 of the constituent somatic cell populations in the testis, have long been known to play essential roles in spermatogenesis. These cells are in direct physical association with all types of developing germ cells. During embryonic development, SCs play a critical role in the formation of the testis (for review, see Ref. 1), whereas in adult males, they are entirely committed to sustaining spermatogenesis. SCs fulfil several functions, including providing structural support for developing germ cells and controlling spermatogonial self-renewal and survival. They also create an impermeable and immunological barrier, the blood-testis barrier (BTB), and assist in germ cell movement through seminiferous epithelium. Furthermore, they nurse germ cells via their secretory products, produce seminiferous fluid, and are also required for spermiation (for reviews, see Ref. 2). In fact, mature SCs are able to coordinate simultaneously the maintenance of several types of developing germ cells within the spermatogenic cycle and are involved in the complex endocrine and paracrine regulation of spermatogenesis.
Understanding the rapid adaptations in SC gene expression during the differentiation of germ cells is absolutely essential for a better understanding of testicular function. It would also provide insights into the intricate relationship between these supporting cells and spermatogonia stem cells, as well as meiotic and postmeiotic spermatogenic cells. Unfortunately, due to inherent technical limitations, the available studies analyzing SC gene expression do not provide an exhaustive characterization of the SC transcriptome. In fact, these studies relied exclusively on mouse and rat SCs isolated from 18- and 20-day-old animals, respectively (3–10). The use of this single stage cannot reveal the dynamics of gene expression in SCs, because they rapidly adapt to accommodate newly forming meiotic and postmeiotic spermatogenic cells (11). Although interesting, these initial studies possess inherent limitations, such as nonoptimal cell enrichment, or the fact that removing SCs from their in vivo environment and placing them in culture for 2 days alters the expression of various genes (12–14). Finally, the use of microarrays results in incomplete coverage of the murine genome, because it mainly investigates the global expression of protein-coding genes, whereas omitting mRNA splicing variants as well as long noncoding RNAs (lncRNAs) (3–6, 15).
We took advantage of a Sox9-enhanced green fluorescent protein (eGFP) knock-in mouse (16) to label and sort pure fractions of SCs, as close as possible to their in vivo state, by fluorescent-activated cell sorting (FACS) at 5 different time points, corresponding to the key stages of spermatogenesis. Using this tool in combination with high-throughput sequencing technology, we explored how the SC coding and noncoding transcriptome dynamically adapts during the first wave of spermatogenesis. Our work provides an important initial glimpse into the complex SC regulatory network and therefore paves the way for further functional investigations that will lead to a deeper understanding of the mechanisms controlling the dynamic transcriptional landscape of SCs.
Materials and Methods
Animals
Sox9-eGFP knock-in mice were generated by the introduction of an IRES-eGFP-pA cassette into the 3′-untranslated region (UTR) of the endogenous Sox9 gene (16), and genotyped using primers Sox9R1 (5′-GCTTGAGGAGAGCCATTTGA-3′), Sox9R2 (5′-TGATAAAGCTCACCAATGCTC-3′), and Sox9F1 (5′-GGCTTGTCTCCTTCAGAG-3′). Mice were maintained on a C57Bl6/129Sv mixed genetic background and housed and cared for according to the ethical guidelines of the Direction Générale de la Santé of the Canton de Genève (authorization GE/5/13-C).
SC isolation by FACS
Testes from homozygous Sox9-eGFP animals at postnatal day (P)5, P10, P18, P25, and P35 were dissected and decapsulated to release the tubules, which were then incubated in trypsin/EDTA 0.01% medium (Invitrogen), with deoxyribonuclease (DNase) I (0.8 mg/mL final concentration; Sigma) at 37°C for 10 minutes. Trypsin digestion was blocked by adding 10% fetal calf serum (FCS). Tubules were then incubated in collagenase (1.4 mg/mL final; Sigma), hyaluronidase (2.8 mg/mL final; Sigma), and DNase I (0.6 mg/mL final; Sigma) at 37°C for 30 minutes with gentle agitation. At the end of incubation, PBS without calcium and magnesium (Invitrogen) was added, and the suspension was filtered through 40-μm cell stainer (BD Falcon). GFP+ SCs were sorted using a FACS Vantage SE machine (Becton-Dickinson). The purity of FACS-isolated SCs was determined by quantitative real-time reverse transcription-PCR (qRT-PCR), immunofluorescence and FACS (see below), and was above 96% for each stage.
Total RNA isolation and qRT-PCR
Total RNA was extracted using the RNeasy Micro kit from QIAGEN according to the manufacturer's protocol. Genomic DNA was removed by DNase treatment and confirmed by polymerase chain reaction (PCR) assay. A total of 10 ng of total RNAs was reverse transcribed with the TAKARA Reverse Transcription kit (Life Technologies) according to manufacturer's instructions, and 6.25 μL of cDNA were preamplified using the TaqMan preamplification master mix (Life Technology). Finally, 1/20th of the cDNA template was used as template for qRT-PCR, performed as previously described (17). The relevant primers used for qRT-PCR are listed in Supplemental Table 1.
Histology and immunofluorescence
Tissues were fixed overnight either in 4% paraformaldehyde (PFA) and embedded in paraffin. For immunofluorescent (IF) analysis, PFA-fixed sections were incubated overnight at 4°C with primary antibodies: rabbit anti-GATA4 (1:500; Santa Cruz Biotechnology, Inc), rat anti-GCNA1 (1:50; from G. Enders), rabbit anti-GFP (1:100; Invitrogen), and rabbit anti-SOX9 (1:500; from Morohashi). Alexa Fluor-conjugated secondary antibodies (Invitrogen) were then used for signal revelation, and sections were counterstained using DAPI (4′,6-diamidino-2-phenylindole). All images were obtained with a Zeiss Axioscope microscope and processed using the ZEN software.
FACS-isolated-positive cells were spread on Superfrost slides and incubated 10 minutes in ethanol:ether 50/50 before storage at −20°C. Slides were fixed for 10 minutes in PFA 4% at room temperature, washed in PBS, and processed through the usual IF procedure.
RNA sequencing (RNA-seq) library preparation and sequencing
Next generation sequencing of RNA was performed on FACS-isolated populations of mouse SCs at P5, P10, P18, P25, and P35 according to Illumina's protocol, using 500 ng of RNA. For each stage, 3 independent sets of total RNA were isolated and used as a template for RNA-seq library preparation. In addition, for each individual sample, purified SCs originated from a minimum of 3 males. Thus, a minimum of 9 animals per stage was used to minimize/normalize the effect of genetic variability. In short, for each library 500-ng total RNA was selected for poly A+ RNA isolation using Sera-Mag oligo(dT) beads (Thermo Scientific) and fragmented with the NEBNExt DNA library Prep Master Mix Set (New England BioLabs). cDNA synthesis, end-repair, A-base addition, and ligation of the Illumina PCR adaptators were performed according to Illumina's protocol. Libraries were then selected for 300- to 350-bp cDNA fragments on a 3.5% agarose gel and PCR amplified using Phusion DNA polymerase (Finnzymes) for 15–18 cycles. PCR products were then purified on a 2% agarose gel and gel extracted. Each library was quality controlled (product size and concentration) by an Agilent 2100 Bioanalyzer. Single-end libraries were sequenced as 60 mers on a Genome Analyzer II flow cell according to Illumina's protocol at depth of approximately 50–80 million single-end reads per library (for statistics on read counts, see Supplemental Table 2).
Read mapping, transcriptome assembly, and quantification with the Tuxedo suite
Comprehensive database of known transcripts
A comprehensive transcript database was assembled from public databases (Ensembl and National Center for Biotechnology Information [NCBI] [build 37.2], AceView, and mRNAs from University of California Santa Cruz [UCSC] mm9) and merged into a combined set of nonredundant known transcript annotations using Cuffcompare (18, 19).
Mapping reads
RNA-seq-derived reads were aligned independently for each SC sample (3 replicates per time condition) to the mouse genome (mm9) downloaded from the UCSC genome browser website (20) with TopHat (version 1.4.1) (21) using previously published approaches (19, 22). Briefly, to aid these alignments, the database of known nonredundant transcripts and expressed sequence tag (EST) alignments (from UCSC) were used to define an additional junction set (AJS) for each TopHat run. The junction outputs from individual TopHat runs were pooled and added to the AJS to allow TopHat to use junction information from all samples. TopHat was rerun on each sample using the resulting AJS. The output of this second run comprised the final alignment. Finally, individual sample alignments of each SC-isolated stage were pooled.
In order to correctly compare our SC transcriptome with gene expression data from different populations of germ cells, the raw RNA-seq data from Soumillon et al (8) were analyzed anew using the same mapping protocol. These samples included different testicular populations (eg, P20-SCs, spermatogonia, pachytene spermatocytes, round spermatids, and spermatozoa), as well as whole tissues (eg, testis, brain, and liver).
Transcriptome assembly
SC-individual stage transcriptomes were assembled with Cufflinks (version 1.2.0) by finding a parsimonious allocation of reads to the transcripts within a locus (18, 19). Default settings were used. The Cufflinks assembly step yielded a set of approximately 40 500-58 675 transcript fragments (transfrags) for each stage of SCs (Supplemental Table 2).
Merging and classification of transcript fragments
The Cuffcompare program (18, 19) was used to merge the individual transfrags into a combined set (nonredundant “union” of all transcript fragments that share all introns and exons), and to classify the resulting transcripts according to the known transcript annotation database into several classes. SC transcripts corresponding to known (Cuffcompare class code “=,” complete match) or novel (class code “j”) isoformes of known genes as well as novel intronic (class code “i”), or intergenic (class code “u”) transcribed regions were selected for further analysis.
Transcriptome quantification
The isoform-level abundances (expression level) were assessed using Cuffdiff (18, 19) in reads per kilobase of exon model per million reads mapped (RPKM). Transcripts assembled with the SC dataset were also quantified with the Soumillon dataset to estimate their concentration in the different isolated germ cell populations (8).
Expression data preprocessing
We next prepared a matrix of RPKM expression values based on the resulting transcriptome quantification with Cuffdiff. Then, expression data were log2-transformed after adding 0.05 to all RPKM values. Finally, the data were quantile normalized to reduce systematic effects and thus to allow direct comparison of the individual samples. A parallel tissue expression profiling dataset was assembled, including 11 mouse healthy tissues in triplicate, including testis, ovary, brain, embryo, heart, kidney, liver, lung, muscle, spleen, and thymus hybridized on Affymetrix Mouse Exon 1.0 ST GeneChips. This GeneChip data were normalized using the robust multiarray average method (23).
Refinement of transcript fragments
Estimation of background signal
As observed in Ref. 24, manual inspection of the resulting transcript fragments revealed that the vast majority of predicted loci probably corresponded to stochastic transcriptional “noise,” genomic DNA present in the sample, or artifacts due to errors in read mapping and transcript assembly. For removal of low-quality quantifications and to distinguish transcripts from background signal, we defined a background expression cutoff (BEC), approximately 2.7 RPKM, corresponding to the overall median RPKM value of the assembled transcripts that completely match (Cuffcompare class code =) NCBI Reference Sequence (RefSeq) Database curated mRNAs (RefSeq category “NM”) (25).
Long and multiexonic transfrags
We applied 2 additional filtering steps to isolate the most robust transcripts. First, transcript fragments with a total length of less than 200 nt were discarded, because these were most likely sequencing or assembly artifacts. Second, we selected multiexonic transfrags. Together, these refinements produced a set of 15 570 transcript fragments supporting long poly-adenylated RNA molecules in mouse SCs.
Statistical filtration and cluster analysis
Statistical analysis
The statistical filtration of the transcript fragments differentially expressed (DE) among the 5 stages was performed using the Annotation, Mapping, Expression and Network (AMEN) suite of tools (26). We performed every pairwise comparison between cellular time conditions to select 7134 transfrags that exhibited at least 1 fold-change greater or equal to 3.0. Finally, a Linear Models for Microarray Data (LIMMA) statistical test (F value adjusted with the false discovery rate method, P ≤ .01) was employed to identify 4608 significantly DE transfrags in mouse SCs during the progression of spermatogenesis (27).
Cluster analysis
The 4608 DE SC transcripts were grouped into 9 expression patterns (named Clusters 1 to 9, C1–C9) using the k-means algorithm implemented in AMEN. The capacity of the patterns to discriminate transcripts was verified using Silhouette plots. The 9 resulting patterns were ordered according to peak expression levels in the different time conditions. The 10 962 remaining transcripts, for which no significant differential expression was observed, were placed in a 10th group named C0.
Subtractive strategy
In order to refine our analyses of SC-enriched transcripts, the 4608 DE SC transcripts were also quantified using the RNA-seq data from Soumillon et al (8). The quantified SC transcripts were further reclustered in 6 expression patterns (C'1–C'6) using the k-means algorithm and subsequently compared with our 9 SC expression patterns (C1–C9). The transcripts contained in clusters C'5 and C'6 were associated with meiotic and postmeiotic germ cell patterns and thus were subtracted from the SC transcripts, because they were highly enriched in C7 and C8 SC clusters, suggesting contamination with highly expressed germ cell transcripts. The final selection contained 2939 DE transcripts in mouse SCs, clustered in 9 expression patterns (termed SC1–SC9 for Sertoli-associated cluster 1 to 9) and 10 962 transcripts in the SC0 cluster.
Coding potential analysis of novel transcribed regions
Before analyze coding potential of each unannotated intronic or intergenic transcript fragments, we extracted their DNA sequences, corresponding open reading frames and aligned DNA sequences in 4 mammalian species, including human (hg18), rat (rn4), dog (canFam2), and cow (bosTau2), that were generated by MULTIZ and downloaded from the UCSC genome browser (20). Classification of each transfrag as either coding or noncoding was determined using an empirical integrative approach based on the use of 4 distinct predictive tools, including PhyloCSF, HMMER, CPC, and txCdsPredict (20, 27–30). Transcripts that scored higher than 20 using PhyloCSF were retained as potential coding candidates. Any transcript with an E value less than 10−4 in HMMER (vs Pfam-A and Pfam-B) was considered as protein coding. Any transcript classified as “coding” using CPC were retained as potential protein-coding candidates. Transcripts that scored higher than 800 (∼90% predictive of protein-coding genes) in txCdsPredict was considered protein coding. Finally, by comparing results of these 4 tools, we organized the unannotated intergenic and intronic transcripts into 5 classes termed “very high” (transcripts considered protein coding by 4/4 tools), “high” (3/4), “medium” (2/4), “low” (1/4), and “no” (0/4) potential coding regions.
Functional analysis
Gene ontology (GO) analysis was performed using AMEN (26). Functional enrichment analysis was estimated with the Fisher exact probability using the Gaussian hypergeometric test. A term was considered to be significantly enriched in a group of genes when P < .05 (adjusted according to Benjamini and Hochberg's method to control the false discovery rate) and the number of genes bearing this annotation was more than or equal to 5. We also used the gene prioritization system (31) to help interpret the resulting gene expression clusters and to identify potentially important protein-coding genes.
Gene regulatory network analysis
The gene regulatory network representation was drawn using the AMEN software (26). The protein-gene regulation data were from June 17, 2014, from the Transcription Factor encyclopedia database (32). These associations correspond to a consolidation of the human, mouse, and rat data. Briefly, mouse homologs of the human and rat genes were identified through NCBI's HomoloGene database (33). Genes encoding transcription factors (TFs), regulators of cell proliferation and proteins involved in the organic hydroxyl compound metabolic process, were selected on the basis of their association with the “sequence-specific DNA binding TF activity” (GO:0003700), the “regulation of cell proliferation” (GO:0042127), and the “organic hydroxyl compound metabolic process” (GO:1901615) GO terms in the “gene2go” file downloaded from the NCBI website.
Androgen receptor (AR)-regulated gene enrichment analysis
An AR-regulated gene set was assembled by combining the sets of AR target genes published in 6 distinct studies (34–39), among which 5 were focused on mouse SCs. For direct comparisons of these lists of target genes, the entries were sequentially matched with the corresponding gene entries (NCBI Entrez gene IDs) and transcript/loci assembled in our RNA-seq dataset. The standard hypergeometric test was used to identify the gene clusters that were significantly overrepresented in the AR target gene set.
Statistical analysis of biological samples used for the collection of SOX9+ cells
Results are expressed as means ± SEM of n experiments. Parametric unpaired t test was used for statistical analysis. Differences were considered statistically significant if P < .05.
Results
Isolation of highly pure fractions of SCs at key stages during the first wave of spermatogenesis
In adult testis, spermatozoa are produced asynchronously throughout postpubertal life, making the isolation of pure populations of SCs at precise stages of spermatogenesis rather difficult if not impossible unless working at the single cell level. However, during the first wave of spermatogenesis, germ cells multiply and differentiate in a synchronous fashion in the testes. The onset of spermatogenesis occurs at P5. At this stage, seminiferous tubules are exclusively composed of SCs (84%) and primitive type A spermatogonia (16%). New populations of germ cells appear soon afterwards within the seminiferous tubules: spermatocytes at P15, spermatids at P21, and finally spermatozoa at P42. Therefore, by isolating SCs at key stages through the first wave of spermatogenesis, we can assess how the structure and dynamics of the SC transcriptome adapts to and/or controls the development of the meiotic and postmeiotic germ cells.
SCs were isolated at P5, P10, P18, P25, and P35 using a Sox9-eGFP knock-in mouse line that specifically labels SCs (16). SC specificity was confirmed by colocalization of the SOX9 and eGFP proteins in SCs of the developing testis at each relevant stage (Figure 1, A–E). In short, testes from homozygous Sox9-eGFP mice were dissected and dissociated to produce single cell suspensions, which were then sorted using FACS. To confirm cellular specificity and purity, we analyzed the gene expression profiles of eGFP+ fractions at each developmental stage by qRT-PCR, FACS, and immunofluorescence (Figure 1, F–Y). For instance at P5, total RNA isolated from eGFP+ fractions expressed high levels of the SC marker Amh, whereas Leydig (Cyp11A1) and germ cell (Mvh) markers were almost absent (Figure 1F). FACS of isolated eGFP+ cells confirmed the high degree of purity of the fractions (Figure 1K). Furthermore, we found by immunofluorescence that more than or equal to 96% of the cells from eGFP+ fractions were positive for the SC marker GATA4 (Figure 1P), and less than 2.5% were positive for the germ cell marker GNCA1 (Figure 1U). These high levels of purity were also obtained for the P10 (≥96%) (Figure 1, G, L, Q, and V), P18 (≥96%) (Figure 1, H, M, R, and W), P25 (≥98%) (Figure 1, I, N, S, and X), and P35 (≥96%) (Figure 1, J, O, T, and Y) stages. Overall, our purification method allowed us to isolate a highly enriched (≥95%) SC fraction at any stage during the progression of spermatogenesis in less than 2 hours.
Figure 1. SC cell isolation and purification by FACS from a Sox9-eGFP transgenic mouse.

A–E, Coimmunofluorescence for SOX9 and eGFP confirming the colocalization of both proteins in the nucleus of SCs (yellow arrows), whereas eGFP was also expressed in the cytoplasm. Asterisks represent germ cell nuclei. The purities of isolated SCs were determined by qRT-PCR, FACS, and immunofluorescence at P5 (F, K, P, and U), P10 (G, L, Q, and V), P18 (H, M, R, and W), P25 (I, N, S, and X), and P35 (J, O, T, and Y). F–J, RNA quantification by qRT-PCR of purified SC-GFP+ fraction with Sertoli (Amh or Cldn11), Leydig (Cyp11a1), and germ (Mvh) cell-specific markers. Note that the purity of SC populations is more than 97% for each stage. Results are mean ± SEM; ***, P < .0001 vs Sertoli marker (Amh or Cldn11). K–O, The GFP isolated fractions were tested by FACS, and the profiles showed above 95% purity. Quantification by immunofluorescence with Sertoli (GATA4) (P–T) and germ (GCNA1) (U–Y) cell-specific antibodies of GFP+ fractions showed that the purity was up to 95% for all stages. Scale bars are indicated on figure.
The SC transcriptome comprises 15 570 high-confidence isoforms during the progression of spermatogenesis
To assess the SC transcriptome during the progression of spermatogenesis, we isolated polyadenylated RNA from purified fractions at P5, P10, P18, P25, and P35 and performed RNA-seq in biological triplicates with a depth of approximately 65 million reads per library, using Genome Analyzer II sequencer (60 bp, single-end reads).
The resulting reads were then mapped on the mouse reference genome (version mm9), and transcripts were assembled using the Tuxedo suite (see Materials and Methods) (19). We obtained a nonredundant set of 171 704 assembled transcripts corresponding to 49 247 loci (Supplemental Table 2). These transcripts were classified into 4 categories in accordance with the mouse transcriptome annotation, the first 2 being known and novel isoforms of annotated loci, the 3rd and 4th being novel intronic and intergenic transcribed regions (Supplemental Figure 1A and Supplemental Table 3).
Because the potential presence of unspliced pre-mRNA and gDNA can lead to erroneously assembled transcripts (24), we used a highly stringent refinement strategy (see Materials and Methods and Supplemental Figure 1B) resulting in a “high-confidence” set of 15 570 long (>200 bp) and multiexonic transcripts highly detectable (>2.7 RPKM) in at least 1 of the 5 SC stages (Figure 2). This set is almost entirely composed of known (54.4%) and novel (45.4%) isoforms of annotated loci, with very few novel intronic (0.1%) and intergenic (0.1%) transcripts. This refinement strategy drastically decreased the number of novel transcripts that seemingly resulted from transcriptional artifacts. All subsequent analyses are conducted on this final set of high-confidence transcripts.
Figure 2. Isoform classification of the SC transcriptome.
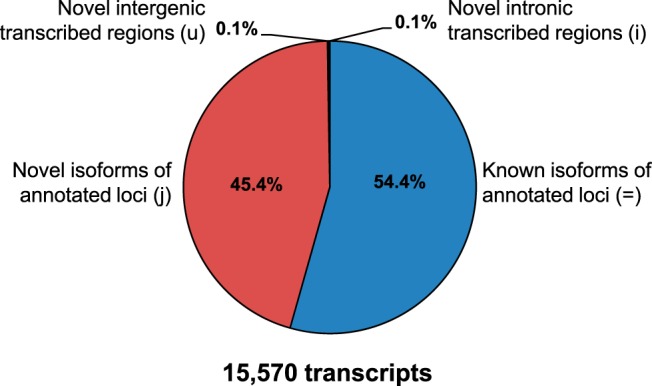
The high-confidence set of 15 570, long, multiexonic, nonredundant transcript isoforms expressed in SCs during the progression of spermatogenesis are classified according to: known annotated protein-coding and noncoding transcripts (=), novel isoforms of known annotated genes (j), and novel intronic (i) or intergenic (u) unannotated transcripts.
These RNA-seq data are available in GEO (accession number GSE59698), and a graphical display of the data is accessible through the ReproGenomics Viewer (http://rgv.genouest.org). In addition, RPKM values and other genomic features of each transcript are also provided as an Excel file with the ability to generate an expression graph for any gene/transcript, as a user friendly resource for the community (Supplemental Dataset 1).
To validate the SC sorting and RNA-seq data, we investigated the expression of a set of genes known to be specifically expressed in SCs. The expression pattern of these genes was consistent with previous reports, confirming the high enrichment of SCs by sorting. The observed variation in abundance of several genes reported as markers of SCs (Gdnf, Gata4, Gata1, Wt1, Inha, Fshr, Cldn11, Cadm1, Cdh2) and as well as several novel ones (Pds5b, Maz, Zfhx2, Smoc1, Gnai1, Os9) was confirmed by qRT-PCR (Supplemental Figure 2). This strongly suggests that the RNA-seq data accurately reflect gene expression patterns in SC cells at different stages of spermatogenesis.
Three thousand DE transcripts reflect the high plasticity of the SC transcriptional landscape during the progression of spermatogenesis
In order to study the dynamics of SC transcriptomes and to obtain a SC signature at specific stages during the progression of spermatogenesis, we performed differential expression analysis and clustering on the high-confidence set of 15 570 transcripts (≥3-fold change, P ≤ .01) (for more details see Materials and Methods and Supplemental Figure 3A). Among them, 4608 were detected as significantly DE and were classified into 9 clusters according to their expression profiles (termed C1–C9) (Supplemental Figure 3B). Each of these clusters reveals transcripts with specific expression profiles related to 1 or more SC maturation stages. To investigate whether these DE transcripts were specific to SCs, and did not result from contamination by transcripts from differentiating germ cells, we took advantage of RNA-seq data from 5 testicular cell type populations, including 1) mitotic spermatogonia, 2) meiotic pachytene spermatocytes, 3) round spermatids undergoing spermiogenesis, 4) spermatozoa, and 5) SCs (8).
Surprisingly, the integration of these germline RNA-seq data allowed us to observe, in our dataset of 4608 DE transcripts, the presence of transcripts expressed in the meiotic and postmeiotic germline, mostly into the expression clusters related to P25 and P35 (clusters C7 and C8) (Supplemental Figure 3C). The presence of germ cell transcripts in our samples is likely due to the phagocytic activity of SCs, required to eliminate both apoptotic germ cells and excess spermatid cytoplasm (40, 41), and to a much lesser extent to germ cell contamination. We therefore performed a subtractive strategy to eliminate the transcripts having obvious meiotic and postmeiotic germline expression profiles from our set of 4608 DE transcripts (C'5 and C'6) (Supplemental Figure 3D) in order to study SC-specific gene expression.
As a result, we finally obtained 13 901 high-confidence transcripts (corresponding to 8459 genes/loci), among which 2939 (2337 loci) displayed a significant SC-related differential expression pattern classified into 9 expression profiles (termed SC1–SC9) (Figure 3 and Supplemental Figure 4). SC1 consists of transcripts highly expressed at P5 only, SC2 contains transcripts expressed at P5 and P10, SC3 those expressed at P10, SC4 at P18, SC5 between P18 and P25, SC6 from P18 onward, SC7 at P25 and P35, SC8 at P35, and finally SC9 at P5 and P35. The expressed transcripts with no significant DE between SC stages were grouped together in cluster SC0. Interestingly, although the number of lncRNAs (173) was relatively low in comparison with coding isoforms (13 688), they were very specific to SCs at different stages of spermatogenesis (Supplemental Figure 4 and Supplemental Table 4). Finally, our RNA-seq analysis also revealed 29 novel unannotated intronic and intergenic transcripts with little (11 candidates) or no (18 candidates) protein-coding potential, with 7 of the 29 being DE (Supplemental Figure 4 and Supplemental Table 4).
Figure 3. The transcriptome of SCs is dynamically regulated during the progression of spermatogenesis.

Among the 13 901 transcripts expressed in SCs, 2939 displayed a dynamic expression profile classified into 9 expression profiles (termed SC1–SC9). Each plot displays the standardized log2 RPKM expression profile for each assembled transcript (gray lines) in a particular expression cluster, overlaid with that cluster's centroid (red line). For each cluster (SC1–SC9), the number of transcripts and the percentage of transcripts within the corresponding cluster as compared with the total number of DE transcripts are in parenthesis.
Dynamic expression profiles reflect changes in SC function during the first wave of spermatogenesis
To assess the functional relevance of the complex transcriptional profiles observed in SCs during the first wave of spermatogenesis, we submitted the list of DE genes to a GO enrichment analysis. We also investigated pathways related to cellular processes in more detail, such as the transition from immature proliferative SCs (P5) to mature quiescent SCs (P18, P25, and P35), BTB formation, SC metabolism, germ cell renewal, and differentiation. We found that SC-related expression profiles (SC1–SC9) were significantly enriched in GO annotations associated with biological process and cellular component terms essential for SC function (P < .05, and the number of genes bearing this annotation was ≥5) (Figure 4 and Supplemental Table 5). Indeed, it is well known that immature SCs proliferate until about P5, before reaching functional maturation and acquiring the ability to sustain spermatogenesis. At P5 (SC1), GO terms “cell cycle,” “DNA replication,” as well as “nucleus” and “chromatin” were significantly enriched, and the expression profiles of these enriched genes effectively confirm the proliferative activity of these immature SCs (eg, Aurora kinase B, cyclin A2, cyclins D2 and D3, cell division cycle 25B, cyclin-dependent kinases 2 and 14, chromatin licensing and DNA replication factor 1/Cdt1, Krüppel-like factor 4/Klf4, Cables1, Fam64a, Dlgap5, Dazl/Tpx2, cytoskeleton-associated protein 2/Cakp2, Anilin, Mcm2-6, Melk, Ki67, Skp2) (Figure 5, A and B). At P10, we found enrichments in genes associated with “negative regulation of cell proliferation” (Cdkn1a/p21, Cdkn1b/p27, Cdkn2a/p19, Nkx3-1, Dab2ip) (Figure 5C). These complementary expression patterns are consistent with previous studies showing that SCs proliferate from E14.5 until P5, when they stop dividing and maturate (42, 43). At later stages, we found enrichments in genes associated with “multiple cellular processes and metabolism” (P18–P35), such as “vesicle-mediated transport” and “cellular membrane organization,” likely regulating cell adhesion and communication as well as lactate and lipid metabolism (44). Expression profiles of transcripts associated with immature SCs include keratin18 (Krt18) (45) and Gata4 (46), whereas expression of Gata1 and AR, the cyclical markers of mature SCs, correlates with previous gene expression studies (Figure 5D) (47, 48). Interestingly, AR expression slightly precedes that of known AR-regulated genes in SCs, such as Drd4, Gpd1, and Claudin11 (Figure 5E) (35, 49). Consistent with SC maturation, we observed an up-regulation of transcripts coding for the cytokine TGF-β3, known to regulate BTB dynamics (50, 51), as well as specific components of the BTB, such as tight junction protein (TJP)1 and TJP2, occludin, junctional adhesion molecule (JAM)-A, JAM-B, and JAM-C, nectin-2, and claudin11 (Figure 5, F and G).
Figure 4. GO enrichment analysis of the SC-related expression patterns.

Each SC-related expression cluster is matched with a selection of enriched GO terms from the ontology “biological process” and “cellular component” that are ordered according to their peak expression at P5 (SC1), P5–P10 (SC2), P10 (SC3), P18–P25 (SC4–SC5), and P25–P35 (SC6–SC8). Numbers of genes associated with a specific GO term and enriched in each cluster are given within rectangles in bold. A color code indicates overrepresentation (red) and underrepresentation (blue) as indicated in the scale bar.
Figure 5. Expression profile of selected genes with known function in SCs during the first wave of spermatogenesis.

Expression profiles in RPKM at the 5 stages for selected genes with known functions as positive (A and B) and negative (C) regulators of cell cycle, markers of immature and mature SCs (D), AR-regulated genes (E), components of the blood-testis-barrier (F and G), genes mediating SSC self-renewal (H), that code for secreted factors (I), mediated RA synthesis or its action (J), and involved in glucose/lactate metabolism (K and L).
Another example of functional relevance is the important role played by SCs in regulating SSC renewal and differentiation through paracrine mechanisms. Secreting factors such as glial cell-line-derived neurotrophic factor (GDNF) and stem cell factor (SCF) were mostly expressed around P5 and P10 in SCs, corresponding to the beginning of SC involvement in SSC renewal (52, 53), whereas the late expression profile of Etv5 is consistent with a role in SSC renewal in adult animals (Figure 5H) (54). Numerous genes encoding factors secreted by SCs that are known to affect SC proliferation or the differentiation of adjacent somatic/germ cells were also identified (Bmp1, Bmp6, Kitl, Ntf5, Pdgfra, Pdgfrb, and Pleiotrophin) (Figure 5I). We also observed very specific expression patterns for genes related to the synthesis and action of retinoic acid (RA), whose synthesis by SCs is essential to induce spermatogonia differentiation during the first spermatogenic cycle (55). These include the retinaldehyde dehydrogenase genes (Aldha1, Aldha2, and Aldha3) and the nuclear receptors Rara, Rxra, and Rxrb (Figure 5J).
It is well known that fully differentiated SCs provide both physical and nutritional support for differentiating germ cells. In fact, the metabolic role of SCs is essential for normal spermatogenesis, because developing germ cells are unable to metabolize glucose and instead consume lactate produced at high rates by SCs (56). Interestingly, we found that the expression profiles of genes coding for proteins involved in glucose/lactate metabolic pathways correlate with the appearance of spermatocytes and spermatids around P18 and P25 (Figure 5, K and L). This includes all the transporter proteins (glucose transporter [GLUT]1/GLUT3 and monocarboxylate transporter MCT1/MCT4) and enzymes responsible for the conversion of glucose into lactate (such as aldolase, lactate deshydrogenase B, C, and D, pyruvate kinase, and glycerol-3-phosphate dehydrogenase 1 (GPD-1)).
Overall, this functional analysis indicates that the dynamic transcriptional profile observed in SCs reflects the evolving roles played by SCs during the first wave of spermatogenesis, from an immature proliferative cell to a mature quiescent supporting cell dedicated to sustaining spermatogenesis and germ cell differentiation.
Genes/loci dynamically expressed in SCs are significantly enriched in AR-regulated genes
Androgens play crucial roles in the initiation and maintenance of spermatogenesis. In SCs, AR displays a dynamic expression profile that varies during postnatal development and between spermatogenic stages in mature testis (57, 58). Consistent with available expression data, we found a similar expression profile with AR transcripts being initially absent in immature SCs and then exhibiting a progressive increase up to P18 followed by a decrease (see Figure 5E). The dynamic/cyclical expression profile of AR in SCs suggests that AR-regulated genes may also exhibit dynamic expression patterns during the first wave of spermatogenesis. Over the past few years, several studies have been focused on the identification of AR-regulated genes in mouse SCs (35–39). By combining all these available datasets and by comparing them to our SC RNA-seq dataset, we were able to identify 1036 AR-regulated genes (NCBI Entrez gene IDs) unambiguously expressed in SCs (SC0–SC9) (see also Supplemental Table 5 for annotated AR-regulated genes). Almost half of these AR-regulated genes/loci (ie, 45%) displayed a SC-related differential expression pattern (SC1–SC9) (463/1036, P = 1.64 × 10−36). This means also that among the 2337 gene/loci exhibiting a dynamic expression profile in SCs during the progression of spermatogenesis, approximately 20% of them (463) are potentially regulated by testosterone. We next investigated the extent to which this enrichment was associated with the transcripts showing an “early” (SC1–SC3), “middle” (SC4–SC5), or “late” (SC6–SC8) transcriptional induction during the first wave of spermatogenesis. We found that this overrepresentation was significantly more pronounced in the pattern containing the AR (“middle” induction, P = .010) (Supplemental Figure 5B) than in the “early” (P = .972) (Supplemental Figure 5A) and “late” (P = .3067) (Supplemental Figure 5C) ones. These results indicate that a significant proportion of gene/loci exhibiting a dynamic expression profile in SCs during the progression of spermatogenesis are regulated by androgens.
Network analysis reveals complex regulatory interactions driving the SC transcriptional landscape
We investigated the extent to which TFs DE in SCs during the first wave of spermatogenesis may control this dynamic expression program. To reveal regulatory interactions of known TFs with their potential target genes, we combined gene annotation and transcript profiling data with regulation data from the Transcription Factor encyclopedia database (32). This analysis revealed a relatively small but densely connected network of gene regulations, composed of 62 genes/proteins in a tight functional relationship (Figure 6). We expect TFs that play a crucial role in regulating the dynamic transcriptional profile of SCs to have a high number of direct interactions and act as key regulatory hubs inside the network. Among the 23 found in this analysis, we detected 3 hub TFs-aryl hydrocarbon receptor nuclear translocator (ARNT) (SC2 expression pattern), EPAS1 (SC5) and signal transducer and activator of transcription 1 (STAT1) (SC3 and SC5), suggesting a central role for these factors in the regulation of SC development and function. Although STAT1 is known to promote inflammation and inhibit proliferation (59), there is very little available data relating to its potential role in mediating SC development and/or function (see Discussion for more details). EPAS1 (or hypoxia-inducible TF [HIF]-2α) and ARNT (or HIF-1β) are 2 HIFs of the basic helix-loop-helix family that regulate the expression of numerous genes involved in SC function, such as glycolysis metabolism or BTB formation (see also Discussion and Ref. 60).
Figure 6. Integration of transcript profiling data and information on regulation data to establish a SC gene regulatory network.

The nodes inside the regulatory network represent genes/proteins color coded according to their SC-related expression pattern. A legend for the color code is shown. Edges correspond to protein-DNA interactions, and the thickness of edges represents the number of publications supporting the interaction. Gene symbol of genes encoding TFs are highlighted in bold. Genes encoding proteins associated with regulation of cell proliferation and organic hydroxyl compound metabolic process terms are displayed with a red and green contour lines, respectively.
Another key issue in this analysis was determining the extent to which those TFs could potentially drive the biological processes significantly associated with the SC-related expression patterns. As expected, we found that those TFs were significantly involved in the regulation of genes associated with regulation of cell proliferation (P < 2 × 10−9) as well as organic hydroxyl compound metabolic processes (P < .04). Overall, our protein network analysis identified the TFs EPAS1/ARNT and STAT1 as potential master regulators of SC development and function.
Discussion
SCs are the only somatic cell type within the seminiferous tubules and support germ cell development. SCs must be flexible enough to create a suitable environment for each germ cell throughout the highly ordered process of spermatogenesis. However, very little data are currently available on the dynamic transcriptional programs that provide this flexibility or on the paracrine factors that act on the germ cell compartment. Here, we report the first high-resolution transcriptional profiling of SCs at various stages of spermatogenesis in the mouse. The RNA-seq analyses revealed about 13 901 high-confidence, nonredundant coding and noncoding transcripts enriched in SCs. Importantly, we found that more than one-fifth of them (2939) exhibit a differential expression profile during the progression of spermatogenesis. In addition, SC transcriptomes were characterized by complex alternative splicing patterns, with more than 45% of the transcripts being novel isoforms of known genes. The number of lncRNAs in SCs was relatively low in comparison with coding isoforms, although they were very specific to different stages of spermatogenesis, suggesting a possible role in gene regulation.
Our analysis demonstrates that the high variability in SC gene expression reflects both its maturation status and the multiple and changing functions of SCs during the progression of spermatogenesis. These changes reflect both the adjustment of SC to the progressive changing of the germ cell complement and the influence that the latter exert on SC activity. These RNA-seq data provide an important, user friendly resource for scientists working in the fields of reproduction and testicular function and are available either through an Excel file containing RPKM normalized values of each of the 13 901 assembled SC transcripts (with the capability to generate an expression graph for any gene/transcript (http://www.unige.ch/medecine/nef/Data.html see Supplemental Dataset 1) or through the ReproGenomics Viewer at http://rgv.genouest.org.
Our results provide several examples emphasizing the constant adaptation of the SC transcriptome throughout testis development and spermatogenesis. In general, functional analysis of each of the 9 expression patterns revealed stage-specific and distinct biological terms and processes essential for SC function. Some were novel, whereas others correlated well with known functions of SCs at different stages during the first wave of spermatogenesis. Immature SCs were mostly associated with proliferation, DNA replication, and cell cycle GO terms. We identified a large set of transcripts coding for activators of cell cycle and cell division, most of them being highly expressed in immature proliferative SCs at P5 (eg, see Figure 5, A and B), whereas transcripts coding for inhibitors of cell proliferation were up-regulated in nondividing SCs mostly from P10 onwards (eg, see Figure 5C). Similarly, the immature SC marker Gata4 and the mature SC marker Gata1 were up-regulated at P5 and from P18, respectively. Importantly, the SC gene expression profile was shown to adapt to spermatogenic stages. SC factors regulating SSC renewal and differentiation were mostly expressed around P5 and P10 (eg, Scf and Gdnf), whereas Etv5, a gene known to be involved SSC self-renewal only in adult animals (54), was expressed from P25 onward. We identified a large set of secreted factors emphasizing the key role played by SCs in regulating different aspects of testis development (Figure 5I). It includes the platelet-derived growth factors α and β that are required for Leydig cell recruitment and differentiation (for a review, see Ref. 61), and Kit ligand, which is important proliferation and survival of spermatogonia (62), as well as factors with undescribed testicular functions such as Bone morphogenetic proteins 1 and 6, neurotrophin4/5, and pleiotrophin.
During the first spermatogenic cycle, SC-derived RA is required to induce spermatogonia differentiation (55) and, during the subsequent spermatogenic cycles, is essential for spermiation (55). RA is synthesized by dedicated enzymes, the retinaldehyde dehydrogenases (encoded by the Aldh1a1 to Aldh1a3 genes) and binds to and activates nuclear RA receptors (RARs) (RARA, RARB, and RARG), which function as heterodimers with their corresponding retinoid X receptors (RXRs) (RXRA, RXRB, and RXRG). Here, we found that all 3 Aldh1a genes and their corresponding nuclear receptors (Rara, Rxra, and Rxrb) are expressed in SCs, although their varying expression profiles suggest that RA synthesis regulation in SCs is complex and that RA possibly acts directly on immature SCs (Figure 5J).
In contrast to immature proliferating SCs, mature quiescent SCs are fully dedicated to sustaining spermatogenesis, which is reflected by significant changes in SC gene expression during the progression of germ cell development, with metabolism of particular interest. Mature SCs ensure the nutritional support of germ cells by secreting nutrients and metabolic intermediates, including amino acids and lipids, as well as carbohydrates such as lactate (63). Although spermatogonia use blood-derived glucose as fuel for ATP production, more developed germ cells (meiotic cells and spermatids) are dependent on lactate supplied by SCs (56). As might be expected, SCs themselves require high energy levels to function correctly and use lipids as well as glycogen and amino acids for energy production (64, 65). However, most of our knowledge concerning SC metabolism derives from in vitro experiments using SC primary cultures, which may differ from the in vivo environment where metabolic substrate availability and metabolic requirements can vary with time. It is therefore interesting to observe that genes involved in different aspects of energy production and metabolism are significantly enriched in SCs between P18 and P35 (see Figure 4 and Supplemental Table 5). For example, we found that transcripts coding for proteins involved in lactate and pyruvate synthesis were up-regulated from P18 onward (eg, the transporters glut1 and glut3 and mct1 and mct4, the enzymes aldolase, pyruvate kinase, Gpd-1, and the lactate dehydrogenases b, c, and d) (see Figure 5, K and L).
Another crucial function of mature SCs is to form the BTB, which physically divides the seminiferous epithelium into basal and apical compartments. The BTB consists of different junction types, including tight junctions, which are regulated by a complex set of autocrine and paracrine factors such as cytokines and steroids (reviewed in Ref. 66). We found that numerous components of the BTB are up-regulated from P18 onwards (eg, TJP1 and TJP2, occludin, JAM-A, JAM-B, and JAM-C, nectin-2, and claudin11) (Figure 5, F and G), which correlates well with the formation of the BTB and the stage-specific roles of maturing SCs.
Androgens are essential for spermatogenesis and male fertility, and their actions are mediated indirectly via the somatic cell types in the testis that express the AR such as the SCs, the Leydig cells, and the peritubular myoid cells (for a review, see Ref. 67). Because of their intimate anatomical and functional interaction with developing germ cells, SCs are the prime candidate for the mediation of androgen action. SC-selective AR knockout models revealed spermatogenic defects due to a block in meiosis but also defects in spermiogenesis, defects in SC maturation, delayed/disturbed SC barrier formation, abnormal basal laminal, and disturbed LC development (68–71). Unfortunately, the understanding of androgen action has been hampered by the fact that few consistent AR target genes have been identified. By combining expression data from 5 recent expression profiling studies using mutant mice lacking AR specifically in SCs (35–39), we identified 1036 AR-regulated genes in mouse SCs. Interestingly, we found that 463 of them display a dynamic expression profile and thus are potentially regulated by testosterone. This represents approximately 20% of the genes dynamically expressed in SC during the progression of spermatogenesis.
In an attempt to identify TFs that orchestrate the highly dynamic transcriptome of SCs during the first wave of spermatogenesis, we used an integrative genomics approach combining gene annotation data, transcript profiling data, and regulomic data. We identified 3 TFs at the center of 2 dense gene networks, namely, ARNT, EPAS1, and STAT1. These TFs may act as master regulators controlling the SC transcriptional landscape.
ARNT and EPAS1 are 2 HIFs important for the regulation of oxygen homeostasis in SCs. The ability to control oxygen homeostasis is essential for spermatogenesis, which requires considerable oxygen consumption due to the high sperm production rate (72). Due to the absence of blood vessels within the seminiferous tubules and diffusion distance, spermatogenesis beyond the stage of leptotene spermatocytes occurs in an hypoxic environment (72). These HIFs are α1β1 heterodimeric TFs of the basic helix-loop-helix family that act as regulators of genes involved in oxygen homeostasis. The α-subunit is composed of HIF-2α or EPAS1 and the β-subunit of HIF-1β, also known as ARNT. After binding to hypoxia-response elements found within the promoters or enhancers of target genes, HIFs up-regulate the expression of numerous genes involved in glycolysis metabolism (phosphoglycerate kinase and lactate dehydrogenase-A, carbonic anhydrase-9, GLUT-1), angiogenesis (vascular endothelial growth factor), adrenomedullin, and BTB formation (TJP1, TJP2, and occludin) (73). As one might expect, the expression levels of all these target genes in SCs mirror the up-regulation of EPAS1 at P18 (see Figures 5 and 6). The crucial importance of EPAS and ARNT as regulators of SC functions has been confirmed in vivo: mice with a deletion of Epas1 during first week after birth were sterile due to a substantial reduction of spermatids and spermatozoa (60). This defect resulted from the incapacity of SCs to form tight junctions, due to decreased expression of multiple genes encoding TJP, including TJP1, TJP2, and occluding, resulting in the failure of BTB formation.
STAT1 is a member of the STAT family of TFs and is known to play essential roles in development, inhibition of proliferation, and immune defense (74). A variety of growth factors and cytokines activate intracellular STAT signaling proteins and stimulate transcription of several target genes. To our knowledge, a role for STAT1 in SCs has not been reported to date. Our protein network analysis reveals that STAT1 is at the center of a large network of TFs (Nr1h2/LXRβ, AR, PRRX2, TEAD1, E2F1, SP1, SP3, TCF3, MYCN, IRF1, GATA1, and NFIL3). Many of these TFs have been implicated in the regulation of important processes in SC development and function, such as cell growth and proliferation (TEAD1, E2F1, MYCN/N-Myc, and IRF1), SC metabolism (NR1H2/LXRβ) (75), BTB formation (SP1 and E2F1) (76, 77), and SC regulation of spermatogenesis (AR, SP1, SP3, and E2F) (78–81).
Although powerful, our results describing the genome-wide expression profile of SCs during the progression of spermatogenesis possess also inherent limitations and potential pitfalls. First, due to the phagocytic activity of SCs, we have developed a subtractive strategy to eliminate the transcripts having obvious meiotic and postmeiotic germline expression profiles. This means that some transcripts expressed both in SCs and GCs may have been subtracted. Second, only long polyA mRNAs have been studied by RNA-seq, no information about small noncoding RNAs as well as long non-polyA noncoding RNAs is provided by this study. This aspect is important, because we have recently shown that small noncoding RNAs are essential for SC survival and the capacity of SC to maturate and properly support meiosis and spermiogenesis (82, 83). Third, the Sox9-eGFP transgene is maintained in a C57Bl6/129Sv mixed genetic background that may also have an influence on SC gene expression. Finally, for technical reasons related with SCs purification procedure, our analysis was performed during the progression of the first wave of spermatogenesis. It remains unclear how similar is the dynamic transcriptional profile of SC that occurs in adult testis during the different stages of spermatogenesis.
In summary, this study provides the first high-throughput sequencing of SCs during the progression of spermatogenesis. The dynamic transcriptional profiles of SCs demonstrate the constant adaptation of the SC transcriptome according to the stage of maturation and the germline associations. In addition, a protein network analysis identified the TFs STAT1 and EPAS/ARNT as potential master regulators controlling the SC transcriptional landscape and functions. This report shall provide crucial resources for the better understanding of SC function and mammalian spermatogenesis and their related disorders such as male infertility.
Acknowledgments
We thank Emmanouil T. Dermitzakis and Antoine D. Rolland for their support and analysis of the data. We also thank Thomas Darde, Olivier Sallou and Olivier Collin (GenOuest Bioinformatics Platform, IRISA) for continued development, data upload, and maintenance of the ReproGenomics Viewer database.
This work was supported by the Swiss SystemsX Interdisciplinary PhD Grant 51PHI0–141994 and by the Département de l'Instruction Publique of the State of Geneva (S.N. and I.S.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the Swiss SystemsX Interdisciplinary PhD Grant 51PHI0–141994 and by the Département de l'Instruction Publique of the State of Geneva (S.N. and I.S.).
Footnotes
- AR
- androgen receptor
- AJS
- additional junction set
- AMEN
- Annotation, Mapping, Expression and Network
- ARNT
- aryl hydrocarbon receptor nuclear translocator
- BTB
- blood-testis barrier
- DE
- differentially expressed
- DNase
- deoxyribonuclease
- eGFP
- enhanced green fluorescent protein
- FACS
- fluorescent-activated cell sorting
- GLUT
- glucose transporter
- GO
- gene ontology
- HIF
- hypoxia-inducible TF
- JAM
- junctional adhesion molecule
- lncRNA
- long noncoding RNA
- NCBI
- National Center for Biotechnology Information
- P
- postnatal day
- PCR
- polymerase chain reaction
- PFA
- paraformaldehyde
- qRT-PCR
- quantitative real-time reverse transcription-PCR
- RA
- retinoic acid
- RAR
- RA receptor
- RNA-seq
- RNA sequencing
- RPKM
- reads per kilobase of exon model per million reads mapped
- RXR
- rexinoid receptor
- SC
- Sertoli cell
- SSC
- spermatogonial stem cell
- STAT1
- signal transducer and activator of transcription 1
- TF
- transcription factor
- TJP
- tight junction protein
- UCSC
- University of California Santa Cruz.
References
- 1. Svingen T, Koopman P. Building the mammalian testis: origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013;27:2409–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petersen C, Soder O. The sertoli cell–a hormonal target and 'super' nurse for germ cells that determines testicular size. Horm Res. 2006;66:153–161. [DOI] [PubMed] [Google Scholar]
- 3. McLean DJ, Friel PJ, Pouchnik D, Griswold MD. Oligonucleotide microarray analysis of gene expression in follicle-stimulating hormone-treated rat Sertoli cells. Mol Endocrinol. 2002;16:2780–2792. [DOI] [PubMed] [Google Scholar]
- 4. Schultz N, Hamra FK, Garbers DL. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci USA. 2003;100:12201–12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shima JE, McLean DJ, McCarrey JR, Griswold MD. The murine testicular transcriptome: characterizing gene expression in the testis during the progression of spermatogenesis. Biol Reprod. 2004;71:319–330. [DOI] [PubMed] [Google Scholar]
- 6. Chalmel F, Rolland AD, Niederhauser-Wiederkehr C, et al. . The conserved transcriptome in human and rodent male gametogenesis. Proc Natl Acad Sci USA. 2007;104:8346–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schlecht U, Demougin P, Koch R, et al. . Expression profiling of mammalian male meiosis and gametogenesis identifies novel candidate genes for roles in the regulation of fertility. Mol Biol Cell. 2004;15:1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soumillon M, Necsulea A, Weier M, et al. . Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 2013;3:2179–2190. [DOI] [PubMed] [Google Scholar]
- 9. Chalmel F, Lardenois A, Evrard B, et al. . High-resolution profiling of novel transcribed regions during rat spermatogenesis. Biol Reprod. 2014;91:5. [DOI] [PubMed] [Google Scholar]
- 10. Gan H, Wen L, Liao S, et al. . Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun. 2013;4:1995. [DOI] [PubMed] [Google Scholar]
- 11. Chalmel F, Lardenois A, Evrard B, et al. . Global human tissue profiling and protein network analysis reveals distinct levels of transcriptional germline-specificity and identifies target genes for male infertility. Hum Reprod. 2012;27:3233–3248. [DOI] [PubMed] [Google Scholar]
- 12. Jégou B. Spermatids are regulators of Sertoli cell function. Ann NY Acad Sci. 1991;637:340–353. [DOI] [PubMed] [Google Scholar]
- 13. Syed V, Hecht NB. Up-regulation and down-regulation of genes expressed in cocultures of rat Sertoli cells and germ cells. Mol Reprod Dev. 1997;47:380–389. [DOI] [PubMed] [Google Scholar]
- 14. Vidal F, Lopez P, López-Fernández LA, et al. . Gene trap analysis of germ cell signaling to Sertoli cells: NGF-TrkA mediated induction of Fra1 and Fos by post-meiotic germ cells. J Cell Sci. 2001;114:435–443. [DOI] [PubMed] [Google Scholar]
- 15. Schlecht U, Erb I, Demougin P, et al. . Genome-wide expression profiling, in vivo DNA binding analysis, and probabilistic motif prediction reveal novel Abf1 target genes during fermentation, respiration, and sporulation in yeast. Mol Biol Cell. 2008;19:2193–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nel-Themaat L, Vadakkan TJ, Wang Y, Dickinson ME, Akiyama H, Behringer RR. Morphometric analysis of testis cord formation in Sox9-EGFP mice. Dev Dyn. 2009;238:1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cederroth CR, Schaad O, Descombes P, Chambon P, Vassalli JD, Nef S. Estrogen receptor α is a major contributor to estrogen-mediated fetal testis dysgenesis and cryptorchidism. Endocrinology. 2007;148:5507–5519. [DOI] [PubMed] [Google Scholar]
- 18. Trapnell C, Williams BA, Pertea G, et al. . Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trapnell C, Roberts A, Goff L, et al. . Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pauli A, Valen E, Lin MF, et al. . Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22:577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Irizarry RA, Hobbs B, Collin F, et al. . Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. [DOI] [PubMed] [Google Scholar]
- 24. Prensner JR, Iyer MK, Balbin OA, et al. . Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI reference sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chalmel F, Primig M. The annotation, mapping, expression and network (AMEN) suite of tools for molecular systems biology. BMC Bioinformatics. 2008;9:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. [DOI] [PubMed] [Google Scholar]
- 28. Kong L, Zhang Y, Ye ZQ, et al. . CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Britto R, Sallou O, Collin O, Michaux G, Primig M, Chalmel F. GPSy: a cross-species gene prioritization system for conserved biological processes–application in male gamete development. Nucleic Acids Res. 2012;40:W458–W465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yusuf D, Butland SL, Swanson MI, et al. . The transcription factor encyclopedia. Genome Biol. 2012;13:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sayers EW, Barrett T, Benson DA, et al. . Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40:D13–D25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ou XM, Chen K, Shih JC. Glucocorticoid and androgen activation of monoamine oxidase A is regulated differently by R1 and Sp1. J Biol Chem. 2006;281:21512–21525. [DOI] [PubMed] [Google Scholar]
- 35. Zhou W, Wang G, Small CL, Liu Z, Weng CC, Yang L, Griswold MD, Meistrich ML. Gene expression alterations by conditional knockout of androgen receptor in adult sertoli cells of Utp14b(jsd/jsd) (jsd) mice. Biol Reprod. 2010;83:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang QX, Zhang XY, Zhang ZM, et al. . Identification of testosterone-/androgen receptor-regulated genes in mouse Sertoli cells. Asian J Androl. 2012;14:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang L, Wang Y, Zhang Q, et al. . Identification of Hsf1 as a novel androgen receptor-regulated gene in mouse Sertoli cells. Mol Reprod Dev. 2014;81:514–523. [DOI] [PubMed] [Google Scholar]
- 38. De Gendt K, Verhoeven G, Amieux PS, Wilkinson MF. Genome-wide identification of AR-regulated genes translated in Sertoli cells in vivo using the RiboTag approach. Mol Endocrinol. 2014;28:575–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mou L, Zhang Q, Wang Y, et al. . Identification of Ube2b as a novel target of androgen receptor in mouse sertoli cells. Biol Reprod. 2013;89:32. [DOI] [PubMed] [Google Scholar]
- 40. Russell LD. Spermatid-Sertoli tubulobulbar complexes as devices for elimination of cytoplasm from the head region late spermatids of the rat. Anat Rec. 1979;194:233–246. [DOI] [PubMed] [Google Scholar]
- 41. Grandjean V, Sage J, Ranc F, Cuzin F, Rassoulzadegan M. Stage-specific signals in germ line differentiation: control of Sertoli cell phagocytic activity by spermatogenic cells. Dev Biol. 1997;184:165–174. [DOI] [PubMed] [Google Scholar]
- 42. Vergouwen RP, Jacobs SG, Huiskamp R, Davids JA, de Rooij DG. Proliferative activity of gonocytes, Sertoli cells and interstitial cells during testicular development in mice. J Reprod Fertil. 1991;93:233–243. [DOI] [PubMed] [Google Scholar]
- 43. Pitetti JL, Calvel P, Zimmermann C, et al. . An essential role for insulin and IGF1 receptors in regulating sertoli cell proliferation, testis size, and FSH action in mice. Mol Endocrinol. 2013;27:814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Su W, Mruk DD, Cheng CY. Regulation of actin dynamics and protein trafficking during spermatogenesis–insights into a complex process. Crit Rev Biochem Mol Biol. 2013;48:153–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tarulli GA, Stanton PG, Lerchl A, Meachem SJ. Adult sertoli cells are not terminally differentiated in the Djungarian hamster: effect of FSH on proliferation and junction protein organization. Biol Reprod. 2006;74:798–806. [DOI] [PubMed] [Google Scholar]
- 46. Viger RS, Mertineit C, Trasler JM, Nemer M. Transcription factor GATA-4 is expressed in a sexually dimorphic pattern during mouse gonadal development and is a potent activator of the Müllerian inhibiting substance promoter. Development. 1998;125:2665–2675. [DOI] [PubMed] [Google Scholar]
- 47. Yomogida K, Ohtani H, Harigae H, et al. . Developmental stage- and spermatogenic cycle-specific expression of transcription factor GATA-1 in mouse Sertoli cells. Development. 1994;120:1759–1766. [DOI] [PubMed] [Google Scholar]
- 48. Bremner WJ, Millar MR, Sharpe RM, Saunders PT. Immunohistochemical localization of androgen receptors in the rat testis: evidence for stage-dependent expression and regulation by androgens. Endocrinology. 1994;135:1227–1234. [DOI] [PubMed] [Google Scholar]
- 49. Willems A, De Gendt K, Allemeersch J, et al. . Early effects of Sertoli cell-selective androgen receptor ablation on testicular gene expression. Int J Androl. 2010;33:507–517. [DOI] [PubMed] [Google Scholar]
- 50. Wong CH, Mruk DD, Lui WY, Cheng CY. Regulation of blood-testis barrier dynamics: an in vivo study. J Cell Sci. 2004;117:783–798. [DOI] [PubMed] [Google Scholar]
- 51. Yan HH, Mruk DD, Lee WM, Cheng CY. Blood-testis barrier dynamics are regulated by testosterone and cytokines via their differential effects on the kinetics of protein endocytosis and recycling in Sertoli cells. FASEB J. 2008;22:1945–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meng X, Lindahl M, Hyvönen ME, et al. . Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287:1489–1493. [DOI] [PubMed] [Google Scholar]
- 53. Yoshinaga K, Nishikawa S, Ogawa M, Hayashi S, Kunisada T, Fujimoto T. Role of c-kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c-kit expression and function. Development. 1991;113:689–699. [DOI] [PubMed] [Google Scholar]
- 54. Chen C, Ouyang W, Grigura V, et al. . ERM is required for transcriptional control of the spermatogonial stem cell niche. Nature. 2005;436:1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Raverdeau M, Gely-Pernot A, Féret B, et al. . Retinoic acid induces Sertoli cell paracrine signals for spermatogonia differentiation but cell autonomously drives spermatocyte meiosis. Proc Natl Acad Sci USA. 2012;109:16582–16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boussouar F, Benahmed M. Lactate and energy metabolism in male germ cells. Trends Endocrinol Metab. 2004;15:345–350. [DOI] [PubMed] [Google Scholar]
- 57. Tan KA, De Gendt K, Atanassova N, et al. . The role of androgens in sertoli cell proliferation and functional maturation: studies in mice with total or Sertoli cell-selective ablation of the androgen receptor. Endocrinology. 2005;146:2674–2683. [DOI] [PubMed] [Google Scholar]
- 58. Zhou X, Kudo A, Kawakami H, Hirano H. Immunohistochemical localization of androgen receptor in mouse testicular germ cells during fetal and postnatal development. Anat Rec. 1996;245:509–518. [DOI] [PubMed] [Google Scholar]
- 59. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. [DOI] [PubMed] [Google Scholar]
- 60. Gruber M, Mathew LK, Runge AC, Garcia JA, Simon MC. EPAS1 is required for spermatogenesis in the postnatal mouse testis. Biol Reprod. 2010;82:1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Basciani S, Mariani S, Spera G, Gnessi L. Role of platelet-derived growth factors in the testis. Endocr Rev. 2010;31:916–939. [DOI] [PubMed] [Google Scholar]
- 62. Besmer P, Manova K, Duttlinger R, et al. . The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Dev Suppl. 1993;125–137. [PubMed] [Google Scholar]
- 63. Rato L, Alves MG, Socorro S, Duarte AI, Cavaco JE, Oliveira PF. Metabolic regulation is important for spermatogenesis. Nat Rev Urol. 2012;9:330–338. [DOI] [PubMed] [Google Scholar]
- 64. Xiong W, Wang H, Wu H, Chen Y, Han D. Apoptotic spermatogenic cells can be energy sources for Sertoli cells. Reproduction. 2009;137:469–479. [DOI] [PubMed] [Google Scholar]
- 65. Kaiser GR, Monteiro SC, Gelain DP, Souza LF, Perry ML, Bernard EA. Metabolism of amino acids by cultured rat Sertoli cells. Metabolism. 2005;54:515–521. [DOI] [PubMed] [Google Scholar]
- 66. Lie PP, Cheng CY, Mruk DD. Signalling pathways regulating the blood-testis barrier. Int J Biochem Cell Biol. 2013;45:621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. De Gendt K, Verhoeven G. Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice. Mol Cell Endocrinol. 2012;352:13–25. [DOI] [PubMed] [Google Scholar]
- 68. De Gendt K, Swinnen JV, Saunders PT, et al. . A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc Natl Acad Sci USA. 2004;101:1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chang C, Chen YT, Yeh SD, et al. . Infertility with defective spermatogenesis and hypotestosteronemia in male mice lacking the androgen receptor in Sertoli cells. Proc Natl Acad Sci USA. 2004;101:6876–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Holdcraft RW, Braun RE. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development. 2004;131:459–467. [DOI] [PubMed] [Google Scholar]
- 71. Lim P, Robson M, Spaliviero J, et al. . Sertoli cell androgen receptor DNA binding domain is essential for the completion of spermatogenesis. Endocrinology. 2009;150:4755–4765. [DOI] [PubMed] [Google Scholar]
- 72. Wenger RH, Katschinski DM. The hypoxic testis and post-meiotic expression of PAS domain proteins. Semin Cell Dev Biol. 2005;16:547–553. [DOI] [PubMed] [Google Scholar]
- 73. Patel SA, Simon MC. Biology of hypoxia-inducible factor-2α in development and disease. Cell Death Differ. 2008;15:628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Reich NC. STA. Ts get their move on. JAKSTAT. 2013;2:e27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Robertson KM, Schuster GU, Steffensen KR, et al. . The liver X receptor-β is essential for maintaining cholesterol homeostasis in the testis. Endocrinology. 2005;146:2519–2530. [DOI] [PubMed] [Google Scholar]
- 76. Li MW, Lee WM, Lui WY. Expression of Itch in Sertoli cells is controlled via the interaction of E2F1/DP1 complex with E2F and GATA motifs. Spermatogenesis. 2011;1:152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sze KL, Lee WM, Lui WY. Expression of CLMP, a novel tight junction protein, is mediated via the interaction of GATA with the Kruppel family proteins, KLF4 and Sp1, in mouse TM4 Sertoli cells. J Cell Physiol. 2008;214:334–344. [DOI] [PubMed] [Google Scholar]
- 78. Wang BD, Yang Q, Ceniccola K, et al. . Androgen receptor-target genes in african american prostate cancer disparities. Prostate Cancer. 2013;2013:763569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Agger K, Santoni-Rugiu E, Holmberg C, Karlström O, Helin K. Conditional E2F1 activation in transgenic mice causes testicular atrophy and dysplasia mimicking human CIS. Oncogene. 2005;24:780–789. [DOI] [PubMed] [Google Scholar]
- 80. Hoja MR, Liu JG, Mohammadieh M, Kvist U, Yuan L. E2F1 deficiency impairs murine spermatogenesis and augments testicular degeneration in SCP3-nullizygous mice. Cell Death Differ. 2004;11:354–356. [DOI] [PubMed] [Google Scholar]
- 81. Wong CC, Lee WM. The proximal cis-acting elements Sp1, Sp3 and E2F regulate mouse mer gene transcription in Sertoli cells. Eur J Biochem. 2002;269:3789–3800. [DOI] [PubMed] [Google Scholar]
- 82. Papaioannou MD, Lagarrigue M, Vejnar CE, et al. . Loss of Dicer in Sertoli cells has a major impact on the testicular proteome of mice. Mol Cell Proteomics. 2011;10:M900587MCP900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Papaioannou MD, Pitetti JL, Ro S, et al. . Sertoli cell Dicer is essential for spermatogenesis in mice. Dev Biol. 2009;326:250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]