

Abstract
Glial cell line-derived neurotrophic factor (GDNF) is a TGFβ family member, and GDNF signals through a glycosyl-phosphatidylinositol-linked cell surface receptor (GFRα1) and RET receptor tyrosine kinase. GDNF signaling plays crucial roles in urogenital processes, ranging from cell fate decisions in germline progenitors to ureteric bud outgrowth and renal branching morphogenesis. Gene ablation studies in mice have revealed essential roles for GDNF signaling in urogenital development, although its role in prostate development is unclear. We investigated the functional role of GDNF signaling in the urogenital sinus (UGS) and the developing prostate of mice. GDNF, GFRα1, and RET show time-specific and cell-specific expression during prostate development in vivo. In the UGS, GDNF and GFRα1 are expressed in the urethral mesenchyme (UrM) and epithelium (UrE), whereas RET is restricted to the UrM. In each lobe of the developing prostate, GDNF and GFRα1 expression declines in the epithelium and becomes restricted to the stroma. Using a well-established organ culture system, we determined that exogenous GDNF increases proliferation of UrM and UrE cells, altering UGS morphology. With regard to mechanism, GDNF signaling in the UrM increased RET expression and phosphorylation of ERK1/2. Furthermore, inhibition of RET kinase activity or ERK kinases suppressed GDNF-induced proliferation of UrM cells but not UrE cells. We therefore propose that GDNF signaling in the UGS increases proliferation of UrM and UrE cells by different mechanisms, which are distinguished by the role of RET receptor tyrosine kinase and ERK kinase signaling, thus implicating GDNF signaling in prostate development and growth.
The prostate gland is an endodermal derivative of the hindgut that is formed late in embryonic development from the urogenital sinus (UGS). Morphogenesis of the mouse prostate begins around embryonic day (E) 16.5 in the UGS and is dependent on androgen action. Androgen produced by the testis activates androgen receptors in the mesodermally derived urethral mesenchyme (UrM), which is adjacent to the urethral epithelium (UrE) and composed of discrete spatial regions with different properties (1–4). In rodents, two or more types of mesenchyme are present in the UrM and likely important in prostate development. The periurethral mesenchyme (PUM) is directly adjacent to and surrounds the UrE and newly formed prostatic buds, and the peripheral mesenchyme, termed the ventral mesenchymal pad (VMP), is adjacent to the PUM and is crucial for prostatic induction (1–4). Around the time of prostate induction, mesenchymal cells at the interface of the PUM and VMP undergo smooth muscle differentiation (1–5). Androgen action in the UrM is thought to induce formation of prostatic ductal progenitors/buds in the UrE through paracrine signaling (6–8). Androgen-dependent signaling also stimulates the outgrowth of the fetal prostatic buds from the UrE into the surrounding UrM, perinatal branching morphogenesis, and cytodifferentiation of the prostatic epithelium (PrE) (6–10).
In the developing prostate, epithelial proliferation is enriched at the tips of the elongating prostatic ducts as they invade the UrM and branch (11, 12), and smooth muscle differentiation leads to the formation of thin layers of smooth muscle that eventually encase the PrE (1–5). This culminates in prostatic ducts of epithelial origin that are surrounded by stromal smooth muscle cells and fibroblasts. Furthermore, individual prostatic ducts are connected to surrounding ducts by a loose connective tissue in the multilobed rodent prostate, which comprises pairs of anterior, ventral, and dorsolateral lobes (3, 10).
Androgen receptor (AR) signaling is involved in nearly all aspects of prostate development and growth (2, 6–8), and AR is thought to act in concert with other signaling pathways to regulate these processes. In addition to AR, a small number of conserved signaling molecules have been found to regulate prostate development and growth. Proximal-to-distal outgrowth of the prostate is dictated by time-specific and cell-specific expression of morphoregulatory genes (13, 14), including secreted signaling ligands of the Hedgehog (15–21), Wnt (13, 22–25), Fgf (26–29), and Tgfβ (30–36) gene superfamilies. These positive and negative regulators are thought to communicate autocrine and paracrine signals through their cognate receptors (13, 14). Subsequent epithelial cues induce differentiation of the mesenchyme to stroma, and AR action in the luminal epithelium normally regulates secretory function in the mature prostate gland (37, 38). Although AR and other signaling pathways are necessary for the development and growth of the prostate, surprisingly, little is known of the mediators of these processes.
Several members of the TGFβ superfamily have also been implicated as key regulators of prostatic branching morphogenesis. The founding member of the superfamily, TGFβ1, which is expressed in UrM and prostatic stroma (PrS), inhibits prostatic growth and changes the pattern of branching morphogenesis (33, 35). Furthermore, regulation of WNT paracrine signaling mediates the role of TGFβ in the inductive effects of the UrM on the adjacent epithelial cells (39). Glial cell line-derived neurotrophic factor (GDNF) is a member of the TGFβ protein superfamily. Interestingly, Golden et al (40) reported mRNA localization using in situ hybridization of Gdnf and Gdnf family receptors (Gfrα1 and Gfrα2) in the UGS and developing prostate. However, the functional roles of GDNF family ligands and their cognate receptor signaling complexes in the UGS and developing prostate were unexplored.
GDNF has crucial functions in the nervous system and outside the nervous system. In the central and peripheral nervous system, it serves as a growth factor promoting survival and differentiation of neurons (41–43). Outside the nervous system, GDNF dosage regulates cell fate decisions of germline progenitors in testis and ureteric branching morphogenesis in kidney development. In mammalian seminiferous tubules, Sertoli cells dictate the microenvironment of the spermatogonial stem cell niche through the release of growth factors including GDNF. At low GDNF levels, the spermatogonial stem cells differentiate, whereas at high levels, the stem cells only self-renew and are unable to differentiate (44). Originating from the metanephric mesenchyme adjacent to the caudal end of the nephric duct, GDNF acts as a mesenchyme-derived paracrine signal promoting ureteric bud outgrowth and renal branching morphogenesis of the permanent kidney (45–48).
Cellular responses to GDNF family ligands are mediated by a multicomponent receptor complex consisting of a glycosyl-phosphatidylinositol-linked ligand-binding receptor known as GDNF family receptor-α (GFRα) and RET receptor tyrosine kinase (48–50). In the embryonic kidney, ureteric buds fail to form in mutant embryos, regardless of which component of the GDNF-GFRα1/RET signaling complex has been ablated, causing renal agenesis and neonatal kidney failure (45–47, 51–55). Golden et al (40) also reported Ret mRNA localization using in situ hybridization in the UGS and developing prostate. Additional studies have reported that GDNF family ligands may also signal independently of RET (56–59).
Signaling through GDNF receptors activates several intracellular signaling cascades that modulate cell survival and proliferation, including the MAPK kinase (MEK1/2) and MAPK (ERK1/2) pathway (MEK-ERK) (60–62). During kidney development, MEK1/2 is normally active in branching ureteric buds, and phosphorylation-mediated activation of ERK1/2 in the ureteric bud requires GDNF (63). The conditional loss of Gfrα1 in ureteric bud-tip cells has been shown to decrease cell proliferation and reduce phosphorylation of ERK1/2 (p-ERK1/2) (64). In germline progenitors, MEK-ERK signaling is activated by GDNF and has been suggested to stimulate proliferation and suppress differentiation (65, 66). ERK1/2 is activated in GFRα1-immunoreactive spermatogonial stem cells under the control of GDNF, which is thought to inhibit their differentiation (67). As the prostate develops, fibroblast growth factor receptor-mediated signaling through the MEK-ERK pathway is required for androgen-induced outgrowth of epithelial buds (68). Based on these previous findings, we hypothesized that GDNF, its signaling receptor components, and downstream signaling pathways are expressed in the UGS and may play specific roles in the UGS and developing prostate. We explore this hypothesis in the present study.
We report here that GDNF shows time-specific and cell-specific expression during prostate development in vivo. In embryos, GDNF and GFRα1 are expressed in the epithelium and mesenchyme of the UGS. Although GDNF and GFRα1 expression declines in the prostatic epithelium during branching morphogenesis, their expression becomes restricted to the stroma of each prostate lobe. In addition, exogenous GDNF increases the proliferation of mesenchymal and epithelial cells in UGS organ culture. We therefore propose that GDNF signaling in the UGS increases proliferation of mesenchymal and epithelial cells by different mechanisms, which are distinguished by the role of RET receptor tyrosine kinase and MEK-ERK signaling. UrM proliferation is mediated by RET receptor tyrosine kinase and involves activation of MEK-ERK signaling, whereas UrE proliferation does not.
Materials and Methods
Animal maintenance and tissue collection
C57BL/6J mice were purchased from Jackson Laboratories. Mice were housed under defined conditions, including a 12-hour light, 12-hour dark cycle, a temperature of 22 ± 1°C, an atmospheric humidity of 50% ± 10%. All of the animal experiments were approved by the University of Illinois at Urbana-Champaign Animal Care and Use Committee. Timed-pregnant females were generated by pairing males with females overnight. We define midday on the day of plug identification as E0.5. For the in vivo development experiments, the UGS from E14.5 to E18 male embryos and the prostates from postnatal day (P) 0, P5, P7, P14, and P90 adult males were dissected in ice-cold DMEM/F12 (1:1) medium (catalog number 21041-025; Invitrogen).
Fetal sex was identified by examination of the fetal gonad using a dissecting microscope and confirmed by PCR amplification of the male-specific Sry gene (GeneBank: X67204), which is located on the Y chromosome (69). Genomic DNA was purified using lysis buffer (100 mM Tris-HCl, pH 8; 5 mM EDTA; 200 mM NaCl; 0.2% sodium dodecyl sulfate; 100 μg/mL proteinase K) followed by alcohol precipitation. Real-time quantitative PCR (QPCR) reactions were performed using Sry primers (5′-CTCATCGGAGGGCTAAAGTG-3′ and 5′-CAGTCTTGCCTGTATGTGATGG-3′) and control primers (5′-TGTCCTTGGAAGACGAATGC-3′ and 5′-TGGATCGCAGTAGAATGCAG-3′), which are complementary to chromosome 11. The QPCR was achieved using the following method: a denaturation and polymerase activation step at 94°C for 1 minute and then 40 cycle consisting of 94°C for 10 seconds, 57°C for 10 seconds, and 72°C for 20 seconds. Comparative threshold cycle (Ct) values for Sry were compared with control to determine whether the Y chromosome was present in an embryo.
UGS organ culture
UGS tissue was dissected from E15.5 male embryos and placed on 0.4 μm Millicell CM filters (catalog number PICM01250; EMD Millipore), floating on 1 mL of organ culture medium [DMEM/F12 supplemented with 1% nonessential amino acids, 2% insulin, transferrin and selenium (catalog number 51500-056; Invitrogen), gentamicin and amphotericin B (catalog number R-015-10; Invitrogen), and 1 g/L of D-glucose] in a 5% CO2 atmosphere (70, 71). Additional supplements included 0.1% ethanol control, 10 nM 5α-dihydrotestosterone (DHT; catalog number A8380; Sigma-Aldrich) in ethanol, 0.1% PBS control, 1–100 ng/mL recombinant GDNF (catalog number 212-GD; R&D Systems) in PBS, 100 ng/mL recombinant neurturin (NRTN; catalog number 1297-NE; R&D Systems) in PBS, 0.1% dimethylsulfoxide (DMSO) control, 20 μM U0126 (catalog number 1144; Tocris Bioscience) in DMSO, 20 μM PD98059 (catalog number 1213; Tocris Bioscience) in DMSO, and 100 μM 1,3-dihydro-5,6-dimethoxy-3-[(4-hydrophenyl)methylene]1-H-indol-2-one (RPI-1; catalog number R8907; Sigma-Aldrich) in DMSO. The culture medium was changed every other day.
For cell proliferation experiments, UGSs were cultured in organ culture medium with the previously mentioned supplements for 7 days and then with 10 μM bromodeoxyuridine (BrdU; catalog number B5002; Sigma-Aldrich) for 2 hours prior to fixation in 4% paraformaldehyde solution. For ERK kinase inhibitor experiments, UGS were cultured with 100 ng/ml GDNF and 20 μM MEK inhibitor (PD98059, U0126) or DMSO control for 48 hours prior to tissue processing. For RET kinase inhibitor experiments, UGS were cultured with 100 ng/ml of GDNF and 100 μM RPI-1 or DMSO control for 24 hours prior to tissue processing. UGSs were processed for immunostaining by fixation in 4% paraformaldehyde solution for 2 hours. Morphological changes in UGS whole mounts were imaged using a Leica M205A stereoscope. Using Image J software (National Institutes of Health, Bethesda, Maryland), UrM thickness was quantified from bright-field images using six linear bilateral measurements of the visible UrM in each UGS. The mean UrM thickness was then determined using three or more UGSs for each condition.
Separation of embryonic UGS mesenchyme and epithelium and adult prostate stroma and epithelium
UGSs from E15.5 male embryos were mechanically separated into UrE and UrM components after incubation in Hanks' calcium- and magnesium-free solution (pH 7.3) containing 1% Difco trypsin 1:250 (catalog number 215240; BD Biosciences) for 1 hour on ice (5). Each tissue was washed with PBS, and lysates for immunoblot analysis were made using RIPA buffer (10 mM Tris-HCl, pH 8.0; 1 mM Na2 EDTA; 150 mM NaCl; 5% glycerol; 0.1% sodium deoxycholate; 0.1% sodium dodecyl sulfate; 1% Triton X-100) supplemented with protease and phosphatase inhibitors. Prostates were dissected from P90 male mice, and minced prostate tissue was enzymatically dissociated in 10 mL of RPMI 1640 medium containing 150 U of collagenase for 1.5 hours at 37°C and mechanically dissociated by trituration. The collagenase-digested tissue was then passed through a 100-μM pore cell strainer, and the cellular suspension was washed several times with plating medium (RPMI 1640; 10% fetal bovine serum; 25 mM HEPES, pH7.4). After 24 hours in culture, the vast majority of viable stromal cells had attached to cell culture-treated plates, whereas the viable prostate epithelial cells remained in suspension. The suspension cells were collected and washed with PrEC medium (catalog number CC-3166; Lonza) and plated on collagen-coated plates in epithelial-selective PrEC medium. The prostate stromal cells were cultured in stromal-selective PrSC medium (catalog number CC-3205; Lonza). After 7 days in culture, the cells from the prostatic stroma and epithelium were washed with PBS, and RIPA buffer supplemented with protease and phosphatase inhibitors was used to prepare lysates for immunoblots.
RNA isolation, reverse transcription, and real-time QPCR
UGS from E14.5 to E18 male embryos and prostate from P0, P5, and adult male mice were dissected and rinsed in PBS. UGSs were cultured in organ culture medium containing 0–100 ng/mL GDNF for 2–7 days. Additional UGSs were cultured in medium containing 100 ng/mL GDNF and 0–100 μM RPI-1 for 7days. UGSs and prostate tissues were washed with PBS, minced, and mechanically homogenized in lysis buffer, and total RNA was extracted using an RNeasy minikit (catalog number 74106; QIAGEN) with on-column deoxyribonuclease treatment (catalog number 79254; QIAGEN). Random-primed cDNA was prepared from 1 μg of total RNA using the ProtoScript first-strand cDNA kit (catalog number E6300L; New England Biolabs) and diluted 5-fold in water. One microliter of diluted cDNA was used as template for QPCR using a StepOnePlus real-time PCR system (Applied Biosystems). Primers were designed using Primer3 (http://frodo.wi.mit.edu), and those that efficiently amplified (72) single products of the expected size were used for QPCR.
Primers used to detect mouse transcripts are as follows: Gdnf sense, 5′-GATATTGCAGCGGTTCCTG-3′; Gdnf antisense, 5′-CCTGGCCTACTTTGTCACTTG-3′; Gfrα1 sense, 5′-GGCAGTCCCGTTCATATCG-3′; Gfrα1 antisense, 5′-AGGCGGACCTGTACTTCTTG-3′; Ret sense, 5′-TGACATCAGCAAGGATCTGG-3′; Ret antisense, 5′-AGAGCCCATCGTCATACAGC-3′; Pygo2 sense, 5′-CCCAGTCAACCCTTCAACC-3′; Pygo2 antisense, 5′-GAGATCATGGGACCAAATCC-3′; Gapdh sense, 5′-GTCGGTGTGAACGGATTTG-3′; Gapdh antisense, 5′-CTTGCCGTGGGTAGAGTCAT-3′; Mek1 sense, 5′-GCCTGGTTATGGCTAGAAAGC-3′; Mek1 antisense, 5′-AGAAGCCCACGATGTACGG-3′; Mek2 sense, 5′-ACCGGCACTCACTATCAACC-3′; Mek2 antisense, 5′-AGCTCTTCCAGCTTCTTCTGG-3′; and Grb2 sense, 5′-CCAAGGCAGAAGAAATGCTC-3′; Grb2 antisense, 5′-ACTTGACGGACAGGGAGAAG-3′.
The QPCR was achieved using the following method: a denaturation and polymerase activation step at 94°C for 1 minute and then 40 cycles consisting of 94°C for 10 seconds, 57°C for 10 seconds, and 72°C for 20 seconds. Data were analyzed using the Ct method (73) and Pygo2 control gene, which is not regulated by the treatments used here (Supplemental Figure 1). After the normalization to Pygo2 cDNA levels, which is reflected in the ΔCt values, the relative quantification (RQ) of the fold change for each treatment compared with the reference control was determined using the following equation: RQ = 2(−ΔCt)/2(−ΔCt reference). The RQ mean and SEM are plotted in log2 scale.
Cell proliferation
E15.5 UGSs were cultured in organ culture medium with or without GDNF (1–100 mg/mL) for 7 days, and 10 μM BrdU (catalog number B5002; Sigma-Aldrich) was added to the media for 2 hours prior to fixation in 4% paraformaldehyde solution and tissue processing for immunohistochemistry. Fluorescence microscopy images were collected using an Olympus BX51 epifluorescence microscope with ×10–40 objectives and Olympus DP Controller/Manager software. The BrdU-positive cells were detected using BrdU antibody (catalog number 347580; BD Biosciences), and they were counted in each of the UGS tissues (UrM and UrE). The UrM and UrE were defined by immunostaining using antibodies for mesenchymal vimentin (catalog number 5741S; Cell Signaling Technology) and epithelial keratins (catalog number 4545S; Cell Signaling Technology), respectively. For each UGS tissue, the percentage of proliferating cells was calculated by dividing the number of BrdU-positive cells by the total number of 6-diamidino-2-pheylindole (DAPI)-positive cells. The cells were counted in three different visual fields of each 4-μm UGS section, and a total of five sections through 5 or more UGSs were analyzed for each condition.
Immunoblots
UGS and prostate tissue lysates were made using ice-cold RIPA buffer supplemented with protease and phosphatase inhibitors. UGS lysates were collected, total protein was quantified using the BCA protein assay (catalog number 23227; Pierce Biotechnology), and lysates containing equal amounts of protein were subjected to 4%–20% mini-TGX gel electrophoresis (catalog number 456-1096; Bio-Rad Laboratories) and then transferred to polyvinyl difluoride membranes. Nonspecific binding was blocked by incubation of the membranes in TBST [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% Tween 20] containing 1% BSA for 1 hour at 22°C. Membranes were incubated overnight at 4°C with primary antibodies diluted in TBST + 1% BSA. Primary antibodies were as follows: p-ERK1/2 (1:1000, catalog number 4370S), ERK1/2 (1:1000, catalog number 4695S), vimentin (1:1000, catalog number 5741S), pan-keratin (1:1000, catalog number 4545S), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:1000, catalog number 2118L) were from Cell Signaling Technology; GDNF (1:1000, catalog number sc-328) and GFRα1 (1:1000, catalog number sc-10716) were from Santa Cruz Biotechnology; RET (0.1 μg/mL, catalog number AF482) was from R&D Systems. Membranes were washed in TBST and incubated for 1 hour with 1:2000 dilutions of horseradish peroxidase-linked antirabbit, antimouse, or antirat IgG (Jackson ImmunoResearch Laboratories) in TBST + 1% BSA. The blots were visualized using Pierce ECL Western Blotting Substrate (catalog number 32106) and HyBlot CL autoradiography film (catalog number E3018; Denville Scientific). Films were scanned and Image J software (National Institutes of Health, Bethesda, Maryland) was used to quantify protein expression relative to GAPDH control or p-ERK1/2 expression relative to total ERK1/2 control.
Immunohistochemistry
UGS and prostate tissues were rinsed in PBS, fixed in 4% paraformaldehyde for 2 hours at 22°C, and then dehydrated through an alcohol gradient consisting of 30- to 60-minute incubations (depending on tissue size) in 25%, 50%, 70%, 83%, 90%, and 100% ethanol. The dehydrated tissues were cleared in xylene, infiltrated with melted paraffin for 1 hour at 60°C, and embedded in paraffin blocks. The tissues were sectioned at 4 μm thickness using a rotary microtome (Leica), and the sections were placed on glass slides. Tissue sections were deparaffinized in xylene, rehydrated, and equilibrated in water. Antigen unmasking was performed by boiling sections in 10 mM sodium citrate buffer for 10 minutes. Nonspecific binding was blocked using 1% BSA in PBS for 30 minutes at 22°C. Immunohistochemistry controls included preincubation of primary antibody with antigenic peptide, when available, prior to incubation with secondary antibodies and incubation in the absence of primary antibodies (Supplemental Data). Samples were incubated overnight at 4°C with 1:200 dilutions of primary antibodies. Primary antibodies included the following: vimentin, pan-keratin, p-ERK1/2, ERK1/2, BrdU (catalog number 347580; BD Biosciences), GDNF, GFRα1, and RET. After several washes, tissue sections were incubated with fluorescent-conjugated secondary antibodies diluted 1:300 in 1% BSA in PBS for 1 hour at 22°C. Secondary antibodies from Molecular Probes included the following: Alexafluor-594 antimouse, Alexafluor-594 antirabbit, Alexfluor-594 antigoat, Alexafluor-488 antimouse, and Alexafluor-488 antirabbit. Tissue sections were incubated with 0.6 μM DAPI for 10 minutes and washed with PBS, and coverslips were applied with mounting medium (catalog number 10981; Sigma-Aldrich). Fluorescence microscopy images were collected using epifluorescence microscopes (Zeiss Axiovert 200M inverted microscope with ApoTome optical sectioning device and Olympus BX51), which were equipped with ×10−40 objectives, high-resolution digital cameras, and Olympus DP Controller/Manager software and Zeiss Axiovision software, respectively.
Statistical analysis
The significance differences between two treatment groups were determined using Student's t test, whereas differences among three or more groups or time points were determined by one-way ANOVA, followed by Tukey's honest significant difference test.
Results
Localization of GDNF in the mouse UGS and developing prostate
We examined the temporal expression pattern of GDNF in the murine UGS and developing prostate over a broad developmental period from E14.5 to P5. This period spans the earliest stages of prostate ductal development, including epithelial bud specification, initiation, and elongation as well as ductal branching morphogenesis (10, 74). Using real-time QPCR analysis, we found that Gdnf mRNA levels increased 2-fold during prostate ductal development in males, peaking at E17.5 (Figure 1A and Supplemental Figure 1). Gdnf mRNA levels declined slightly from E17.5 to P5, although they remained significantly higher than at E14.5 (Figure 1A). Notably, QPCR analysis also revealed that Gdnf transcripts were readily detected in the UrE and UrM of E16.5 UGS and were 3-fold enriched in the UrE compared with the UrM (Supplemental Figure 2).
Figure 1. Localization of GDNF in the UGS and developing prostate.

A, Time course of Gdnf mRNA expression in the UGS and developing prostate (E14.5 to P5) of male mice was determined by QPCR analysis. Relative quantification of Gdnf mRNA is shown using the mean and SEM (n = 6) in log2 scale. During prostate development, Gdnf mRNA levels increased (compare E14.5 with E17.5) and remained elevated through P5. B, Immunohistochemistry revealed the localization of GDNF protein (green) in the UGS and during prostate development in vivo (E16.5 to P7). In the early stages of prostate development, the UGS consists of UrM and UrE (cell nuclei stained with DAPI; blue), and immunodetection of epithelial keratins (red) has been shown to identify the UrE and prostatic epithelium. GDNF is expressed in regions of the UrM and UrE. The boundaries between the PUM and the UrE or the ventral, lateral, and dorsal prostatic epithelium (VPE, LPE, DPE) have been traced (dashed white line), and the regions of VMP with GDNF localization have been labeled. In the developing prostate at P7, GDNF localizes in the dorsal condensed mesenchyme (left side of image), and the white arrowheads indicate robust colocalization (yellow) of GDNF and keratin in the PrE of the dorsolateral lobe of the developing prostate. C, Individual prostate lobes (anterior, ventral, and dorsolateral) were collected from P14 males and P90 adult males and processed for immunohistochemistry. Immunodetection of GDNF (green), keratins (red), and DAPI-stained nuclei (blue) shows GDNF localization is enriched in the PrS of each lobe at P14. In the adult prostate, GDNF localization is highly enriched in the PrS of each lobe. The boundaries between the PrS and PrE have been traced (dashed white line). D, In E16.5 UGS, GDNF localization in the UrM and UrE was imaged through structured illumination using an optical sectioning device. GDNF localization is evident in the VMP and the cytoplasm of UrE, suggesting that GDNF is expressed in these epithelial cells. The boxed epithelial cells are magnified 2-fold in the lower right panels. The scale bars represent 100 μm (B and C) and 20 μm (D).
To investigate further the temporal and spatial expression patterns of GDNF, the localization of GDNF protein during prostate development (E16.5 to P14 and P90 adult males) was determined using immunohistochemistry and fluorescence microscopy. During prostate development in vivo, GDNF showed time-specific and cell-specific localization (Figure 1, B and C). In E15.5 to E17.5 male embryos, GDNF was localized in the keratin-positive UrE and PrE, which buds from the UrE (Figure 1B and Supplemental Figure 3). In addition, GDNF was localized in the keratin-negative UrM (Figure 1B and Supplemental Figure 3). Although GDNF protein was below the level of detection in most mesenchymal cells, it was readily detected during embryonic development in discrete regions of the UrM, including the VMP and to a lesser extent the PUM (Figure 1B). During the process of branching morphogenesis in the developing prostate, GDNF localization was robust in the prostatic epithelial buds and PrE as well as the dorsal condensed mesenchyme (Figure 1B, panels E17.5 to P7). Although by P14, GDNF localization was substantially reduced in the PrE, and it was enriched in the stroma (PrS) of each prostatic lobe (Figure 1, B and C, compare P7 with P14). In the adult prostate, GDNF localization was restricted to the PrS of all prostate lobes (Figure 1C). Additional assessment of the localization of GDNF in E16.5 UGS through structured illumination using an optical sectioning device revealed GDNF localization in the VMP and the cytoplasm of the vast majority of UrE cells (Figure 1D), suggesting that GDNF is expressed in these epithelial cells. Taken together, the changes in GDNF mRNA and protein localization in the developing prostate suggest that the GDNF expression is regulated during prostate development.
Localization of GFRα1 and RET in the mouse UGS and developing prostate
Because GFRα1 is the high-affinity receptor for GDNF and RET functions as a critical coreceptor and signal transducer of GDNF signals (58), we examined the temporal and spatial expression patterns of GFRα1 and RET in the developing prostate from stages E15.5 to P5 and in P90 adult prostate. The peak levels of Gfrα1 and Ret mRNAs were detected in the earliest stages of prostate development (E15.5-E16.5), and they declined during postnatal development (Figure 2, A and B).
Figure 2. Localization of GFRα1and RET in the UGS and developing prostate.

Time course of Gfrα1 mRNA (A) and Ret mRNA levels (B) in the UGS and developing prostate (E14.5 to P5) of male mice was determined by QPCR analysis. Relative quantification of Gfrα1 and Ret mRNAs is shown using the mean and SEM (n = 6) in log2 scale. Gfrα1 and Ret mRNA levels were highest in the early stages of prostate development. C, UrM and UrE were separated from E15.5 male UGSs, and immunoblot analysis shows that GFRα1 protein is localized in the UrM and UrE prior to prostate development in vivo, whereas RET expression is localized to the UrM. Adult prostate tissue was dissociated, and immunoblot analysis of PrS and PrE revealed that GFRα1 and RET expression is restricted to adult stromal cells. The clean separation of the mesenchyme/stroma and epithelium is supported by immunodetection of mesenchymal/stromal vimentin and epithelial keratins. D, Immunohistochemistry revealed the localization of RET protein expression (red) in the UGS and during prostate development in vivo (E15.5 to P0 and the ventral lobe of P90 adult prostate). DAPI-stained nuclei (blue) identify all cells in the sections, and immunodetection of keratins (green) has been shown to identify the UrE as well as the ventral and dorsal prostatic epithelium (VPE and DPE). RET is expressed in the VMP and to a lesser extent in the PUM of the UGS and developing prostate. The boundaries between the PUM and UrE and the PrS and PrE have been traced (dashed white line), and the VMP has been labeled. The scale bars represent 100 μm.
To investigate the temporal and spatial expression patterns of GFRα1 and RET, the localization of these proteins was determined during prostate development using immunoblot analysis and immunohistochemistry and fluorescence microscopy. In our hands, the commercially available GFRα1antibodies were unsuitable for immunohistochemistry. Immunoblot analysis of E15.5 UGS using the GFRα1 antibody (Figure 2C) revealed that GFRα1 is expressed in vimentin-positive UrM and keratin-positive UrE prior to prostate ductal development. Furthermore, GFRα1 protein localization in the adult prostate is restricted to the stromal compartment. During prostate development in vivo, RET showed time-specific and cell-specific expression (Figure 2, C and D). At E15.5 and during the earliest stages of prostate development, RET protein localization was detected only in discrete regions of the UrM, including the VMP and to a lesser extent the PUM (Figure 2, C and D). In the developing prostate, RET localization decreased in the stroma, and RET expression was nearly undetectable in adult PrS using immunohistochemistry (Figure 2D), although it was detectable in immunoblots (Figure 2C). The changes in GFRα1 and RET mRNAs and protein localization in the developing prostate suggest that GFRα1 and RET expression are also regulated during prostate development.
GDNF induces proliferation of UrM and UrE cells in UGS organ culture
We investigated the functional role of GDNF signaling in the UGS using a well-established method for UGS explant culture, which recapitulates the earliest stages of prostate development in vivo (68, 70, 71). We dissected UGS from E15.5 male embryos and cultured them in defined organ culture medium (70, 71) for 7 days with 100 ng/mL GDNF or vehicle control. Because androgen signaling is necessary for prostate development in vivo and in culture (70, 75, 76), UGSs were treated with 10 nM DHT, a potent endogenous androgen, 100 ng/mL GDNF, or vehicle control. As expected, DHT induced prostatic epithelial budding in cultured UGS (Figure 3A), which is consistent with previous reports of prostate development ex vivo (70, 75, 76). The UGS exposed to exogenous GDNF showed striking changes in morphology, including a substantial increase in UrM thickness (Figure 3A), indicating that the UGS is indeed responsive to GDNF.
Figure 3. Exogenous GDNF induces proliferation of UrM and UrE cells in UGS organ culture.

A, UGSs from E15.5 male mouse embryos were cultured with 10 nM DHT to induce prostate development, 100 ng/mL exogenous GDNF, or vehicle control for 7 days. In UGS culture, DHT robustly induced prostatic epithelial budding (black arrowheads). Visible thickening of the UrM was observed in GDNF-treated UGSs. UGSs were imaged at the same magnification. Below the images, the boundary of the UrM and PrS has been traced (black lines) and labeled to distinguish it from the UrE and PrE. B, E15.5 UGSs were cultured with 100 ng/mL GDNF, 100 ng/mL NRTN, or vehicle control for 7 days. Treatment with GDNF led to visible thickening of the UrM compared with NRTN and vehicle control. C, Quantification of the average UrM thickness using six linear bilateral measurements shows that GDNF increased UrM thickness 1.8-fold at 48 hours and 5-fold at 168 hours, whereas NRTN increased UrM thickness 1.8-fold at 168 hours, albeit not significantly (n = 3–5). D, E15.5 UGSs were cultured with exogenous GDNF (1, 10, or 100 ng/mL) or vehicle control for 7 days, and 10 μM BrdU was added 2 hours prior to tissue processing for immunohistochemistry. Immunodetection of BrdU-positive cells (green), vimentin-positive mesenchymal cells (red), and DAPI-stained nuclei (blue) revealed an increase in the number of proliferating cells in UGS treated with GDNF. E, Quantification of the percentage of BrdU-positive cells in vimentin-positive UrM and vimentin-negative UrE indicates a dose-dependent increase in the number of proliferating cells in UGS treated with GDNF. Exposure to 10–100 ng/mL GDNF increased proliferation in the UrM and UrE, and 1 ng/mL GDNF increased proliferation, albeit not significantly (n = 5). The scale bars represent 500 μm (A and B) or 100 μm (D).
The specificity of GDNF family ligands, such as GDNF and NRTN, is determined by selective GFRα proteins that have unique binding affinities for each ligand. GDNF binds specifically and reversibly to the GFRα1 receptor with an approximate dissociation constant (Kd) of 3 pM (49, 77), and NRTN binds to the GFRα2 receptor with an approximate Kd of 10 pM (77). We assessed the specificity of GDNF signaling in the growth of the UrM using E15.5 UGSs that were cultured with 100 ng/mL doses of GDNF and NRTN or vehicle control for up to 7 days (Figure 3, B and C). At 24 hours, the morphology of GDNF-treated and NRTN-treated UGS was indistinguishable from control UGS (data not shown). At 48 hours, the UrM of GDNF-treated UGS was modestly thicker compared with vehicle control and NRTN-exposed tissue, and by 168 hours, the UrM was 5-fold thicker compared with vehicle control (Figure 3, B and C). Notably, NRTN-exposed UrM was slightly thicker than control-treated UrM, although the increase was insignificant. These results indicate that the increase in UrM thickness is selectively regulated by the GFRα1 receptor, which mediates GDNF signaling.
We considered several possible explanations for the thickening of the UrM in response to GDNF, including the spreading of the UrM, increasing cell size, and increasing cell proliferation. Because UrM density and cell size are highly variable, we investigated the proliferation of mesenchymal cells and epithelial cells in the UGS by quantifying the incorporation of BrdU into the DNA of dividing cells during the S phase. E15.5 UGSs were cultured with a range of GDNF doses (1–100 ng/mL) or vehicle control for 7 days, and 10 μM BrdU was added 2 hours prior to tissue processing for immunohistochemistry. Immunodetection of BrdU-positive dividing cells, vimentin-positive mesenchymal cells, and DAPI-stained nuclei revealed an increase in the number of proliferating cells in UGSs treated with GDNF (Figure 3D). Quantification of the percentage of BrdU-positive cells in the vimentin-positive UrM and vimentin-negative UrE indeed demonstrates a dose-dependent increase in the number of proliferating cells in UGS treated with 10–100 ng/mL GDNF (Figure 3E and Supplemental Figure 4). Notably, the percentage of proliferating cells in the UrM and UrE was increased with 1 ng/mL GDNF, although these increases were insignificant. The number of vimentin-positive UrM cells in S phase increased 1.7-fold with 100 ng/mL GDNF compared with control, and the number of vimentin-negative UrE cells in S phase increased 2.2-fold with 100 ng/mL GDNF compared with control (Figure 3E). Regional differences were also observed for UrM cell proliferation (Supplemental Figure 4). Although GDNF increased the proliferation of PUM and VMP cells, the proliferation increase was greater in the VMP compared with the PUM (Supplemental Figure 4, 2.3 ± 0.2-fold increase in the VMP compared with 1.4 ± 0.2-fold increase in the PUM, P < .05). Taken together, these findings indicate that the UGS is responsive to GDNF signals and that elevated levels of GDNF can induce morphological changes in the UGS that are driven by enhanced proliferation of UrM and UrE cells.
GDNF signaling up-regulates RET expression in the UGS and RET localization in the UrM
To investigate the molecular mechanism responsible for GDNF-induced proliferation of the mesenchymal cells and epithelial cells in the UGS, E15.5 UGSs were cultured with GDNF or vehicle control. By 7 days in culture, Ret mRNA was robustly up-regulated in a dose-dependent manner in UGS, reaching a peak induction of 10.5-fold with 100 ng/mL GDNF at 168 hours (Figure 4A). In time-course experiments, Ret mRNA was up-regulated 2.3-fold to 6.7-fold in GDNF-treated UGSs compared with control UGS at 24 hours and 48 hours, respectively (Figure 4B), suggesting that downstream targets of GDNF signaling may regulate the transcription of the Ret gene. Immunoblot analysis of UGSs cultured with 100 ng/mL GDNF family ligands or vehicle control revealed that GDNF exposure increased RET expression 3-fold at 24 hours and 8-fold at 48 hours (Figure 4C). In contrast, RET expression was unaffected by NRTN at 24 hours. Notably, NRTN modestly increased RET expression 3-fold at 48 hours, albeit insignificantly (Figure 4C). In additional experiments, the effect of GDNF signaling on RET localization was determined using immunohistochemistry and fluorescence microscopy. RET localization in the VMP increased with GDNF treatment compared with vehicle control (Figure 4D). The enrichment of RET localization suggests that in addition to an increase in RET expression in a subset of VMP cells, there is an increase in the number of RET-expressing cells in the VMP. These results demonstrate that GDNF signaling up-regulates RET expression in the UGS and localization in the VMP.
Figure 4. GDNF signaling up-regulates RET expression in the UGS and RET localization in the UrM.
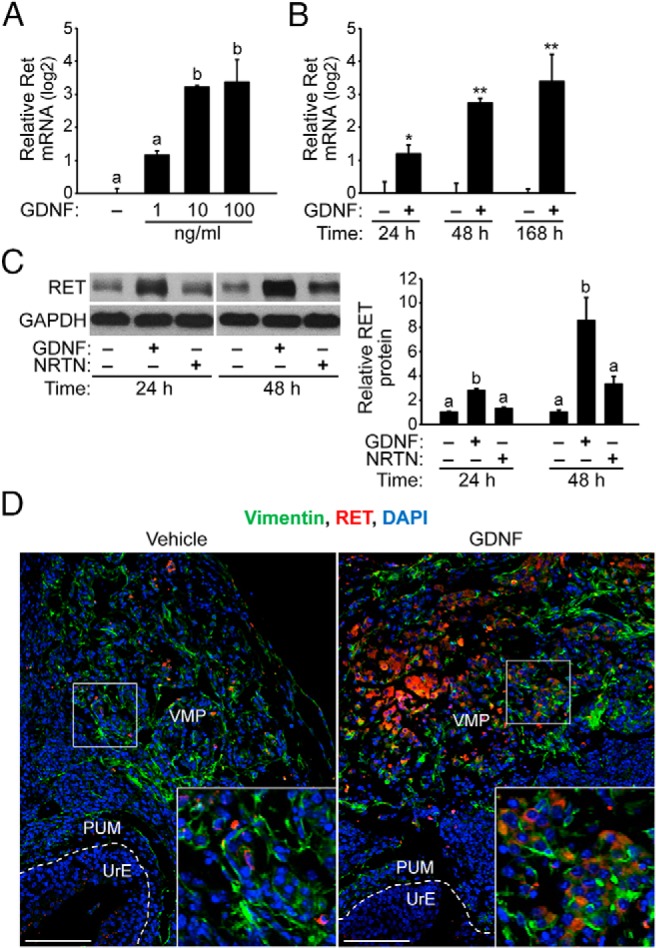
A, E15.5 UGSs were cultured for 7 days with several GDNF doses (1, 10, and 100 ng/mL) or vehicle control. Ret mRNA increased 10.5-fold in UGSs treated with 100 ng/mL GDNF compared with vehicle. Relative quantification of Ret mRNA is shown using the mean and SEM (n = 5) in log2 scale. B, E15.5 UGSs were cultured with 100 ng/mL GDNF or vehicle control for 24–168 hours. Ret mRNA was up-regulated in GDNF-treated UGS compared with control UGS at 24 hours and 48 hours. Relative Ret mRNA is shown using the mean and SEM. *, P < .05; **, P < .01 (n = 5). C, E15.5 UGSs were treated with 100 ng/mL GDNF or vehicle control for 24–48 hours. Immunoblot analysis of UGS lysates reveals that RET expression increased 3-fold in GDNF-treated UGS compared with control UGS and NRTN-treated UGS at 24 hours. By 48 hours, RET protein levels were increased 8-fold in GDNF-treated UGS and 3-fold in NRTN-treated UGS compared with control UGS. Relative RET to GAPDH protein ratios are shown using the mean and SEM (n = 5). D, Immunodetection of RET (red), vimentin (green), and DAPI-stained nuclei (blue) shows enrichment of RET localization in the VMP of GDNF-treated UGS at 48 hours. The boundaries between the PUM and UrE have been traced (dashed white line), and the VMP has been labeled. The boxed mesenchymal cells are magnified 2.5-fold in the inset panels (lower left). The scale bars represent 200 μm.
Inhibition of RET kinase activity suppresses GDNF-induced proliferation of UrM cells
In addition to RET-mediated signaling by GDNF, RET-independent mechanisms of GDNF action have also been reported (56, 57, 59, 78, 79). To assess a functional role for RET in the UGS, we next sought to determine whether the proliferating effects of GDNF in the UGS could be blocked by RET-selective kinase inhibitor RPI-1. E15.5 UGSs were cultured for 7 days with 100 ng/mL GDNF or vehicle control in the absence or presence of 100 μM RPI-1. RPI-1 indeed suppressed the GDNF-induced thickening of the UrM (Figure 5A). In UGSs treated with RPI-1 alone, there were no obvious differences in morphology or UrM proliferation compared with vehicle-treated UGSs (Figure 5, A and B). The UrM exposed to 100 ng/mL GDNF was nearly 4.5-fold thicker compared with vehicle control and 2.5-fold thicker than UGSs cotreated with GDNF and RPI-1 (Figure 5B). Furthermore, cotreated UrM was slightly thicker than control-treated UrM, although the increase was insignificant, implicating RET-mediated signaling in UrM proliferation. The slight increase in UGS size and unusual morphology (Figure 5A) led us to examine further the effects of GDNF and RPI-1 on mesenchymal and epithelial proliferation. Additional BrdU incorporation experiments were performed to quantify changes in GDNF-induced proliferation of UGS exposed to RPI-1 compared with vehicle control. Interestingly, RPI-1 cotreatment suppressed GDNF-induced proliferation in the vimentin-positive UrM (Figure 5, C and D), which is consistent with the morphological changes we observed in the UGS (Figure 5, A and B). However, GDNF-induced proliferation in the vimentin-negative UrE was resistant to RPI-1 (Figure 5, C and D), implicating RET-independent signaling in the regulation of UrE proliferation. These results are consistent with our finding that RET expression is restricted to the UrE and developing prostate stroma (Figure 2, C and D, and Figure 4D).
Figure 5. Inhibition of RET kinase activity suppresses GDNF-induced proliferation of UrM cells.
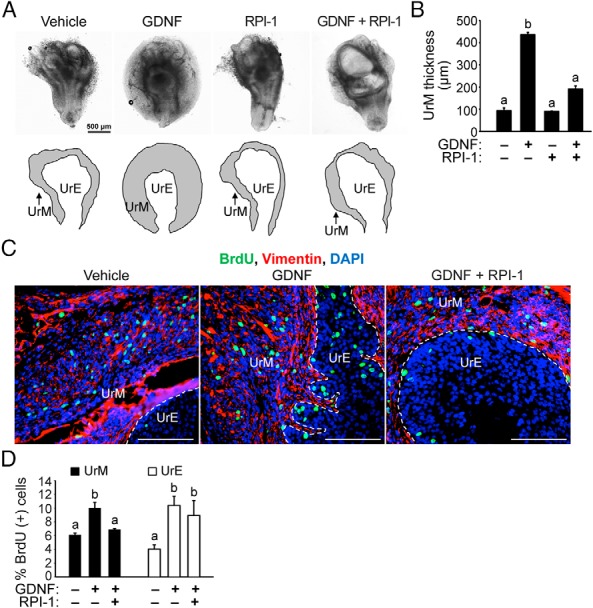
A, E15.5 UGSs were cultured with 100 ng/mL GDNF or vehicle control in the presence or absence of 100 μM RPI-1 for 7 days. UGSs were imaged at the same magnification. Below the images, the boundary of the UrM has been traced (black lines) and labeled to distinguish it from the UrE. B, Quantification of the average UrM thickness using six linear bilateral measurements of the UrM indicates that GDNF induced UrM thickening and RPI-1 inhibited GDNF action in UrM thickening (n = 3–5). C, E15.5 UGSs were cultured with vehicle control or 100 ng/mL GDNF in the presence or absence of 100 μM RPI-1 for 7 days, and 10 μM BrdU was added 2 hours prior to tissue processing for immunohistochemistry. Immunodetection of BrdU-positive cells (green) and vimentin-positive mesenchymal cells (red) and DAPI-stained nuclei (blue) shows an increase in BrdU-positive UrM and UrE cells in UGSs treated with GDNF. However, fewer BrdU-positive UrM cells were observed in UGSs cotreated with GDNF and RPI-1 compared with GDNF alone. The scale bars represent 500 μm (A) or 100 μm (C). D, Quantification of the percentage of BrdU-positive cells in vimentin-positive UrM and vimentin-negative UrE indicates that GDNF-induced proliferation of UrM cells was suppressed by RET inhibitor RPI-1. In addition, the UrE showed similar increases in GDNF-induced proliferation in the presence and absence of RPI-1 (n = 6).
To define further the mechanism of RET up-regulation by GDNF treatment, we tested whether RET kinase inhibitor RPI-1 was able to suppress the up-regulation of Ret mRNA in response to GDNF. Indeed, Ret mRNA levels were suppressed by 68% in UGS cotreated with 100 ng/mL GDNF and RPI-1 compared with GDNF treatment alone (Supplemental Figure 5), suggesting that Ret kinase activity in the UGS may play a central role in the Ret mRNA up-regulation.
GDNF signaling up-regulates MAPK signaling components in the UGS and increases phosphorylation of ERK1/2 in the UrM
To identify additional transcripts that are regulated by GDNF signaling, we assessed the effect of GNDF on the expression of candidate mRNAs that code for signaling components implicated in the MEK-ERK signaling pathway, which is a central pathway controlling cell division and proliferation. By 48 hours, the expression of Mek1, Mek2, and growth factor receptor bound protein 2 (Grb2) mRNAs were significantly up-regulated in UGSs treated with GDNF compared with vehicle control (Figure 6A). Because changes in the expression of these gene products are thought to modulate MAPK signaling through ERK1/2, we hypothesized that GDNF signaling and UGS proliferation may involve activation of the ERK1/2 signaling pathway.
Figure 6. GDNF signaling up-regulates MAPK signaling components in the UGS and increases phosphorylation of ERK1/2 in the UrM.

A, E15.5 UGSs were cultured with 100 ng/mL GDNF or vehicle control for 48 hours, and QPCR analysis of UGS cDNA shows that Mek1, Grb2, and Mek2 mRNAs were significantly increased in GDNF-treated UGS compared to control UGS. The expression of each mRNA relative to Pygo2 mRNA is shown using the mean and SEM. *, P < .05; **, P < .01 (n = 5). B, E15.5 UGSs were cultured with 100 ng/mL of GDNF or vehicle control for 48 hours, and 10 μM BrdU was added for 2 hours prior to tissue processing for immunohistochemistry. Immunodetection of p-ERK1/2, RET, vimentin, and BrdU incorporation in proliferating cells as well as DAPI-stained nuclei (blue) are shown. The boundaries between the PUM and UrE have been traced (dashed white line), and the VMP has been labeled. Elevated levels of p-ERK1/2 and RET proteins are found in UGSs treated with GDNF compared with control. Abundant colocalization of p-ERK1/2 with RET is evident in the VMP of UGS treated with GDNF. BrdU incorporation by S-phase cells indicates that p-ERK1/2-positive cells in the VMP indeed proliferate in response to GDNF. The scale bars represent 200 μm. C, E15.5 UGSs were treated with 100 ng/mL GDNF or vehicle control for 48 hours, and immunoblot analysis of UGS lysates reveals that GDNF exposure increased p-ERK1/2 levels by 3-fold, whereas total ERK1/2 expression levels were unaffected by GDNF treatment. D, E15.5 UGSs were cultured for 48 hours with 100 ng/mL GDNF or vehicle control in the presence or absence of RPI-1. RPI-1 suppressed the increase in p-ERK1/2 levels due to GDNF treatment. Relative p-ERK1/2 to total ERK1/2 protein ratios are shown using the mean and SEM (n = 5).
We next determined whether the ERK1/2 signaling pathway was activated in UGSs treated with GDNF. E15.5 UGSs were cultured for 48 hours with 100 ng/mL GDNF or vehicle control, and the localization of p-ERK1/2 proteins and RET protein was determined using immunohistochemistry and fluorescence microscopy. Immunodetection of p-ERK1/2 and RET revealed increased levels of these proteins with GDNF treatment compared with vehicle control and abundant colocalization of p-ERK1/2 and RET in the VMP (Figure 6B). Additional BrdU incorporation experiments confirmed that the p-ERK1/2-positive cells in the VMP were indeed proliferating in response to GDNF (Figure 6B), which provides evidence for the activation of the ERK signaling pathway in GDNF-induced proliferation of UrM cells located in the VMP. Subsequent immunoblot analysis determined that GDNF exposure markedly increased the p-ERK1/2 levels by 3-fold, whereas total ERK1/2 expression was unchanged by GDNF treatment (Figure 6C). Notably, the p-ERK1/2 levels were not significantly increased by NRTN treatment. We also tested whether the increase in p-ERK1/2 levels due to GDNF exposure could be blocked by RET kinase inhibition using RPI-1. E15.5 UGSs were treated for 48 hours with 100 ng/mL GDNF or vehicle control, and they were cotreated with 100 μM RPI-1 or vehicle control. RPI-1 indeed suppressed the increase in p-ERK1/2 levels due to GDNF treatment (Figure 6D). These findings suggest that RET-mediated GDNF signaling in the VMP region of the UrM increases the activation of the MEK-ERK signaling pathway and, possibly, proliferation of UrM cells.
Inhibition of MEK1/2 kinase activity suppresses GDNF-induced proliferation of UrM cells
The inhibition of ERK1/2 activation with MEK1/2-selective kinase inhibitor UO126 reportedly blocks all morphogenesis, proliferation, and gene expression changes induced by androgens in UGS organ culture, suggesting that MEK-ERK signaling promotes androgen-induced budding morphogenesis, proliferation, and gene expression in the developing prostate (68). We hypothesized that if GDNF-induced proliferation of UrM cells is mediated by activation of the MEK-ERK signaling pathway, then MEK1/2-selective kinase inhibitors should block GDNF-induced proliferation of UrM cells. To assess the effect of combined ERK signaling blockade, E15.5 UGSs were cultured with 100 ng/mL GDNF or vehicle control, and they were cotreated with two different MEK inhibitors (PD98059 and U0126), which are known to inhibit the ERK signaling pathway. From 3 to 7 days in culture, the UGSs that were exposed to 20 μM PD98059 or U0126 and 100 ng/mL GDNF showed striking morphological changes compared with UGSs treated with GDNF alone. The GDNF-induced thickening of the UrM was clearly suppressed by cotreatment with each of the ERK kinase inhibitors (Figure 7, A and B), implicating MEK-ERK-mediated signaling in UrM proliferation. The unusual morphology of the UGSs cotreated with GDNF and the MEK inhibitors prompted us to examine their effects on the proliferation of UrM and UrE cells. Additional BrdU incorporation experiments were performed to quantify changes in GDNF-induced proliferation of UGS exposed to each of the MEK1/2 inhibitors compared with vehicle control. Interestingly, cotreatment with either PD98059 or U0126 suppressed GDNF-induced proliferation in the vimentin-positive UrM (Figure 7, C and D), which is consistent with the morphological changes we observed in the cotreated UGSs (Figure 7, A and B). However, GDNF-induced proliferation in the vimentin-negative UrE was resistant to the MEK inhibitors (Figure 7, C and D), which is remarkably similar to our findings for RPI-1 (Figure 5, C and D).
Figure 7. Inhibition of MEK1/2 kinase activity suppresses GDNF-induced proliferation of UrM cells.

A, E15.5 UGSs were cultured with vehicle control or 100 ng/mL GDNF in the presence or absence of 20 μM MEK inhibitor (PD98059 or U0126) for 7 days. UGSs were imaged at the same magnification. Below the images, the boundary of the UrM has been traced (black lines) and labeled to distinguish it from the UrE. B, Quantification of the average UrM thickness using six linear bilateral measurements of the UrM indicates that GDNF induced UrM thickening and both PD98059 and U0126 inhibited GDNF action in UrM thickening (n = 3–5). C, E15.5 UGSs were cultured with vehicle control or 100 ng/mL GDNF in the presence or absence of 20 μM MEK inhibitor (PD98059 or U0126) for 7 days, and 10 μM BrdU was added 2 hours prior to tissue processing for immunohistochemistry. Immunodetection of BrdU-positive cells (green) and vimentin-positive mesenchymal cells (red) and DAPI-stained nuclei (blue) shows an increase in BrdU-positive UrM and UrE cells in UGSs treated with GDNF. However, fewer BrdU-positive UrM cells were observed in UGSs cotreated with GDNF and PD98059 or U0126 compared with GDNF alone. D, Quantification of the percentage of BrdU-positive cells in vimentin-positive UrM and vimentin-negative UrE indicates that GDNF-induced proliferation of UrM cells was suppressed by MEK inhibitors PD98059 and U0126. In addition, the UrE showed similar increases in GDNF-induced proliferation in the presence and absence of each MEK inhibitor (n = 6). E, E15.5 UGSs were exposed for 48 hours to each MEK inhibitor compared with vehicle control, and the localization of p-ERK1/2 and RET proteins was determined using immunohistochemistry and fluorescence microscopy. Immunodetection of p-ERK1/2 (green) and RET (red) and DAPI-stained nuclei (blue) shows increased levels of these proteins in UGS treated with GDNF compared with vehicle control and abundant colocalization of p-ERK1/2 and RET in the VMP. Cotreatment of UGSs with PD98059 or U0126 suppressed the enrichment of p-ERK1/2 and RET proteins in response to GDNF and the colocalization of these proteins in the VMP. The boundaries between the PUM and UrE have been traced (dashed white line), and the VMP has been labeled. F, E15.5 UGSs were cultured with vehicle control or 100 ng/mL GDNF in the presence of absence of each MEK inhibitor for 48 hours, and immunoblot analysis of UGS lysates show that PD98059 and U0126 indeed suppressed the increase in p-ERK1/2 levels due to GDNF treatment. Relative p-ERK1/2 to total ERK1/2 protein ratios are shown using the mean and SEM (n = 5). G, PD98059 and U0126 also suppressed the increase in RET protein expression due to GDNF treatment. Relative RET to GAPDH protein ratios are shown using the mean and SEM (n = 5). The scale bars represent 500 μm (A), 100 μm (C) or 200 μm (E).
To investigate the functional role of the MEK-ERK signaling pathway in GDNF-stimulated ERK1/2 phosphorylation, RET expression, and their localization in the UrM, E15.5 UGSs were treated with vehicle as a control, GDNF alone, or cotreated with GDNF and each of the MEK1/2 inhibitors for 48 hours, and the localization of p-ERK1/2 and RET proteins was determined using immunohistochemistry and fluorescence microscopy. As expected, immunodetection of p-ERK1/2 and RET showed increased levels of these proteins with GDNF treatment compared with vehicle control and abundant colocalization of p-ERK1/2 and RET in the VMP (Figure 7E). However, cotreatment of UGS with GDNF and either PD98059 or U0126 suppressed the GDNF-stimulated increases in p-ERK1/2 and RET levels in the VMP and the colocalization of these proteins (Figure 7E). Subsequent immunoblot analysis determined that PD98059 or U0126 suppressed increases in p-ERK1/2 and RET expression due to GDNF treatment, whereas total ERK1/2 expression levels were unchanged (Figure 7F). Furthermore, PD98059 or U0126 suppressed the induction of RET expression by GDNF action (Figure 7G). Taken together, these findings reveal substantial clues to the molecular mechanisms responsible for GDNF-induced proliferation of the UrM (RET-MEK-ERK mediated signaling in the VMP) and UrE (RET-MEK-ERK independent signaling) (Figure 8).
Figure 8. Proposed mechanism for GDNF-induced proliferation of mesenchymal and epithelial cells in the UGS.
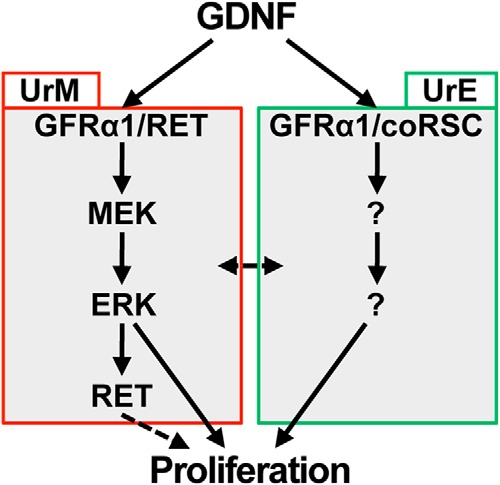
In the UrM, GDNF binds and activates the GFRα1/RET coreceptor complex, and subsequent RET-mediated signaling activates the MEK-ERK pathway, which up-regulates RET expression and stimulates proliferation of mesenchymal cells. In the UrE, GDNF binds and activates another coreceptor complex consisting of GFRα1 and a putative coreceptor signaling component (coRSC), and subsequent RET-independent signaling is responsible for increased proliferation of epithelial cells.
Discussion
GDNF signaling is known to play essential roles in urogenital development, although its role in prostate development had not been explored previously. We have examined the functional role of GDNF signaling in the UGS and the developing prostate of mice. We have defined expression patterns for GDNF-GFRα1/RET signaling components in the UGS and developing prostate and determined that exogenous GDNF stimulates the proliferation of mesenchymal and epithelial cells, altering UGS morphology in organ culture (Figure 3). In support of this finding, we report that UrM thickness is selectively regulated by the GFRα1 receptor, which mediates GDNF signaling (Figure 3). In the VMP region of the UrM, in which GDNF induces proliferation to the greatest extent (Supplemental Figure 4), GDNF also increases RET expression (Figure 4) and phosphorylation of ERK1/2 (Figure 6). Furthermore, inhibition of RET kinase activity or ERK kinases (MEK1/2) suppresses GDNF-induced proliferation of UrM cells but not UrE cells (Figures 5 and 7) and increases in RET expression (Supplemental Figure 5 and Figure 7) and ERK1/2 phosphorylation (Figure 7). Taken together, these findings demonstrate that GDNF signaling influences proliferation in the UGS, and they suggest that different mechanisms are responsible for GDNF signaling in the UrM and UrE. We therefore propose that GDNF signaling stimulates UrM cell proliferation through RET-mediated activation of the MEK-ERK pathway and UrE cell proliferation through an RET-independent mechanism, which is not dependent on MEK-ERK signaling (Figure 8).
We have determined that GDNF, GFRα1, and RET are expressed in the UGS and provide additional evidence for changes in temporal and cell-specific expression patterns of GDNF and its coreceptor components during prostate development in vivo (Figures 1 and 2). Consistent with our findings, Golden et al (40) reported mRNA localization of Gdnf, Nrtn, Ret, and Gdnf family receptors (Gfrα1 and Gfrα2) in the UGS and developing prostate. However, Golden et al did not examine cell-specific mRNA localization or protein expression. We have shown that GDNF and GFRα1 are expressed in the UrE and UrM of the UGS, and RET expression is enriched in a subset of mesenchymal cells located primarily in the VMP (Figures 1 and 2). Gdnf, Gfrα1, and Ret mRNAs decline modestly in the developing prostate during the perinatal period, which is when prostatic epithelial buds elongate and branch. During ductal branching morphogenesis and the subsequent growth period in young adults, GDNF and GFRα1 proteins become localized to the stroma of each prostate lobe. RET expression is also restricted to the mesenchymal/stromal compartment where it decreases in postnatal development, which may serve to limit stromal proliferation in the adult prostate.
In ureteric morphogenesis and kidney development, GDNF is expressed by the metanephric mesenchyme, and RET is expressed by the nephric duct epithelium, from which ureteric budding is promoted by GDNF (47, 80, 81). Although the localization of GDNF and its coreceptor components are different in prostatic and ureteric development, our findings identify intriguing possibilities for autocrine GDNF signaling in the UrM and paracrine GDNF signaling between the UrE and UrM in the UGS and the PrE and PrS in the developing prostate.
In addition to defining expression patterns for GDNF signaling components in the UGS and developing prostate, our experimental approach revealed a functional role for GDNF signaling using a well-established organ culture system that was designed to investigate early signaling events in the UGS and developing prostate (70). Explanted UGS or isolated UGS developing in organ culture provide a more authentic view of the cellular events that occur in vivo. In addition, the UGS organ culture approach is highly amenable to the manipulation of signaling pathways, and it preserves the endogenous tissue architecture of the UGS that is necessary for paracrine signaling and prostate induction (68, 70, 71, 75, 76). Using this organ culture system, we have determined that UrM thickness is selectively regulated by the GFRα1 receptor, which mediates GDNF signaling, and exogenous GDNF stimulates proliferation of both mesenchymal and epithelial cells, altering UGS morphology (Figure 3). Consistent with our findings in the UGS, GDNF has been shown to positively regulate proliferation of several different cell types outside the developing prostate, including glioblastoma cells, ureteric bud tip cells, and spermatogonial stem cells (45, 82, 83). Notably, GDNF-induced proliferation of UrM cells leads to substantial UrM thickening in response to GDNF, whereas GDNF-stimulated cell division in the UrE only subtly altered UrE morphology. Perhaps GDNF signaling may have a more profound effect on epithelial morphology in the developing prostate. Taken together, these findings demonstrate that GDNF signaling regulates cell proliferation in the UGS and implicate GDNF-GFRα1/RET signaling in prostate development and growth.
NRTN is a high-affinity ligand for the GFRα2 receptor. However, its effectiveness in increasing UrM thickness (Figure 3) and up-regulating Ret expression (Figure 4C) and ERK1/2 phosphorylation (Figure 6C) is very low in comparison with GDNF. The slight, albeit insignificant, increase in UrM thickness resulting from exposure to 100 ng/mL (8 nM) NRTN is likely due to cross talk with the GFRα1 receptor. Low-affinity interactions between GDNF and the GFRα2 receptor and between NRTN and the GFRα1 receptor have been reported with an approximate Kd > 1 nM (77). Thus, we speculate that in the developing prostate, GDNF signaling is primarily mediated through GFRα1. Consistent with our findings in NRTN-treated UGS, inactivation of NRTN signaling in Nrtn- or Gfrα2-deficient mice had only a modest effect on prostate growth and an insignificant effect on prostate weight (84). Interestingly, NRTN inactivation is thought to enhance androgen sensitivity in neuroendocrine and reproductive tissues including the prostate (84).
Ligand-bound GFRα receptors including GFRα1 activate a common RET-mediated signaling pathway (85). However, GDNF family ligands are thought have different biological effects upon binding to different GFRα-RET coreceptor complexes (86–88). In addition, the downstream signaling pathways of the selective GFRα receptors are thought to involve phosphorylation activation of distinct RET tyrosines, including Tyr905, Tyr1015, Tyr1062, and Tyr1096 (87, 89). Among these crucial RET tyrosines, phosphorylation of Tyr 1062 is thought to reveal a binding site for several adaptor and effector proteins, including docking protein GRB2 and guanine nucleotide exchange factor son of sevenless homologs. Activation of son of sevenless homologs promotes the exchange of GDP for GTP among members of the RAS GTPase protein family, which can activate RAF-MEK-ERK protein kinase signaling (55, 87, 90, 91). The MEK-ERK pathway is a central signaling pathway controlling cell proliferation (92).
We have proposed cell-specific regulation of proliferation in the UGS in which GDNF signaling stimulates UrM cell proliferation through RET-mediated activation of the MEK-ERK pathway (Figure 8). We also identified regional differences in the proliferation of UrM cells in which GDNF increased proliferation to a greater extent in the VMP compared with the PUM (Supplemental Figure 4). Interestingly, RET expression is limited in PUM cells, which are predominantly RET negative, and enriched in VMP cells (Figure 2). In addition, GDNF treatment of UGS dramatically increased RET expression (Figure 4) and ERK1/2 phosphorylation in the VMP, and inhibition of RET kinase activity or ERK kinases (MEK1/2) suppressed these increases (Figures 6 and 7). Furthermore, inhibition of RET kinase activity using RPI-1 or ERK kinases (MEK1/2) suppressed the GDNF-induced proliferation of UrM cells (Figures 5 and 7) including those located in the VMP and PUM (data not shown). Taken together, these results indicate that GDNF-induced proliferation of VMP cells in the UrM is dependent on the expression and activation of RET and the MEK-ERK pathway in these cells. With regard to the modest stimulation of PUM cell proliferation by GDNF, we offer two possible explanations. Either the PUM cells that proliferate in response to GDNF express RET, albeit at low levels, or they are stimulated by a putative paracrine signal, originating from RET-expressing UrM cells located in the VMP. The latter mechanism is consistent with previous reports of paracrine signaling mediated by secreted ligands of the Wnt (13, 22–25), Fgf (26–29), and Tgfβ (30–36) gene superfamilies. Moreover, smooth muscle differentiation, at the interface of the PUM and VMP in the developing prostate (1–5), may also be susceptible to paracrine signals originating from the VMP.
Activation of the MEK-ERK pathway by GDNF signaling is RET mediated in the UrM (Figure 6D) and leads to increases in RET mRNA and protein expression in the VMP (Figure 4). These findings demonstrate that GDNF signaling up-regulates RET expression in a subset of UrM cells, and they further suggest that the RET-expressing cells proliferate, contributing to increased RET localization in the VMP. Consistent with our report of increased RET expression in GDNF-treated UGSs, microarray analysis of ureteric buds cultured in the presence of GDNF demonstrated Ret mRNA up-regulation (93). In addition to the up-regulation of RET expression by GDNF, Mek1, Mek2, and Grb2 mRNAs were increased in GDNF-treated UGS (Figure 6A). Although it is unclear whether the up-regulation of these mRNAs occurs through direct or indirect mechanisms, GDNF robustly stimulated the MEK-ERK signaling pathway in the VMP region of the UrM (Figures 6 and 7). As the MEK-ERK pathway is a central signaling pathway controlling cell proliferation (92), identifying the transcriptional regulators that are activated by the MEK-ERK pathway and the target genes, such as Ret (Figure 4), that they regulate will inform mechanism and may reveal putative secreted ligands that could be tested for paracrine activation of PUM cell proliferation.
We have also proposed that GDNF signaling stimulates proliferation of UrE cells through a RET-independent mechanism (Figure 8). We determined that the GDNF-induced proliferation of UrE cells, which express GFRα1 and lack detectible RET expression (Figure 2), is resistant to the inhibitory effects of RPI-1 (Figure 5) and ERK kinase inhibitors (Figure 7). Several studies have provided evidence of RET-independent signaling for GDNF and GFRα1 through an association with cell adhesion molecules and activation of nonreceptor tyrosine kinases (56–59). GDNF can bind to neural cell adhesion molecule (NCAM) independently of GFRα1, although both receptors are necessary for GDNF to activate signaling pathways downstream of NCAM, including the activation of the SRC family kinase FYN and focal adhesion kinase (FAK) (57, 58). In cultured rat Schwann cells and mouse neurons lacking RET expression, association of NCAM with GFRα1 promotes high-affinity binding of GDNF, activation of FYN and FAK, Schwann cell migration, and axonal growth (57). This type of RET-independent signaling may be present in the epithelium of the UGS and developing prostate because human prostate tissue has been shown to contain NCAM-like molecules (94), and elevated expression of NCAM2 has been observed in prostate cancer cell lines, whereas normal prostate cell lines express low levels of NCAM2 (95). Another cell adhesion molecule, integrin-β1, has been reported to mediate GDNF signaling in the absence of RET through the activation of FAK and SRC family kinases (56). Integrin-β1 is expressed on the basolateral surfaces of epithelial cells (96), and it is known to regulate many developmental processes (96, 97), including PrE differentiation (98, 99). Furthermore, Integrin-β1 expression is up-regulated and mislocalized during prostate cancer progression (96, 100), demonstrating the potential clinical significance of signaling through integrin-β1 in hyperproliferative disorders of the PrE. Based on these reports and our own findings, GDNF-induced proliferation in the UrE may involve the activation of one or more SRC family kinases via NCAM or integrin-β1cell adhesion molecules.
Our finding that that GDNF signaling influences cell proliferation in the UGS thus implicates GDNF-GFRα1/RET signaling in prostate development and growth. In addition to the proliferative effects of GDNF in the UGS, we expect that GDNF signaling may regulate additional aspects of early prostate development, possibly through communication between epithelial and mesenchymal/stromal cells via a paracrine mechanism. The functional role GDNF signaling in epithelial proliferation is likely restricted to a finite window during prostatic development because GDNF and GFRα1 expression becomes restricted to the prostatic stroma in postnatal development. Perhaps GDNF signaling in the UrE and PrE influences prostatic bud formation and elongation, and upon substantial outgrowth of the prostatic buds, the effect of GDNF on PrE cell proliferation is diminished. In contrast to an endogenous androgen like DHT, GDNF by itself is not sufficient to induce prostatic branching morphogenesis in UGS organ culture (Figure 3). Notably, NRTN inactivation in Nrtn- or Gfrα2-deficient mice was recently shown to enhance androgen sensitivity in neuroendocrine and reproductive tissues including the prostate (84). These findings are indeed consistent with GDNF family ligands playing important roles in the development and function of androgen-dependent tissues. Thus, additional studies are needed to determine whether GDNF signaling functions as a positive or negative regulator of prostate development.
With regard to abnormal GDNF-GFRα1/RET signaling in adult prostatic disease, Dawson et al (101) reported RET overexpression in high-grade prostatic intraepithelial neoplasia and prostate cancer compared with benign PrE. In addition, RET expression was inversely correlated with cellular differentiation, which was assessed by the Gleason pattern. Dawson et al further suggested that RET may play a role in the growth of both benign and malignant neoplastic cells in the prostate (101, 102). In light of the above findings, it is conceivable that constitutive GFRα1 or RET signaling may lead to hyperplasia and enlargement of the prostate. These observations warrant further investigation for possible dysregulation of GDNF signaling in prostate neoplasms.
In conclusion, we have identified a functional role for GDNF signaling in the mouse UGS. Overall, our findings demonstrate that GDNF signaling influences cell proliferation in the UGS, and the mechanism responsible for GDNF-induced proliferation involves different signaling pathways in the UrM and UrE, which are distinguished by the role of RET receptor tyrosine kinase and MEK-ERK signaling. Hence, our findings implicate GDNF-GFRα1/RET signaling in prostate development and growth. Therefore, future studies should explore whether prostate development and growth in vivo are regulated by selective GFRα receptors and their cognate GDNF family ligands and activation of RET tyrosine kinase.
Acknowledgments
We thank Hee Jung Chung, Lori Raetzman, Congcong Chen, and Young-Chae Kim for insightful comments on the manuscript and Congcong Chen, Reiana Mahan, and Jason Dienhart for technical assistance.
This work was supported by University of Illinois at Urbana-Champaign startup funds (to E.C.B.) and Arnold O. Beckman Research Award RB14084 (to E.C.B.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by University of Illinois at Urbana-Champaign startup funds (to E.C.B.) and Arnold O. Beckman Research Award RB14084 (to E.C.B.).
Footnotes
- AR
- androgen receptor
- BrdU
- bromodeoxyuridine
- Ct
- comparative threshold cycle
- DAPI
- 6-diamidino-2-pheylindole
- DHT
- 5α-dihydrotestosterone
- DMSO
- dimethylsulfoxide
- E
- embryonic day
- FAK
- focal adhesion kinase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GDNF
- glial cell line-derived neurotrophic factor
- GFRα
- GDNF family receptor-α
- Grb2
- growth factor receptor bound protein 2
- Kd
- dissociation constant
- MEK
- MAPK kinase
- NCAM
- neural cell adhesion molecule
- NRTN
- neurturin
- P
- postnatal day
- p-ERK1/2
- phosphorylation of ERK1/2
- PrE
- prostatic epithelium
- PrS
- prostatic stroma
- PUM
- periurethral mesenchyme
- QPCR
- quantitative PCR
- RPI-1
- 1,3-dihydro-5,6-dimethoxy-3-[(4-hydrophenyl)methylene]1-H-indol-2-one
- RQ
- relative quantification
- TBST
- Tris-HCl, NaCl, and Tween 20
- UGS
- urogenital sinus
- UrE
- urethral epithelium
- UrM
- urethral mesenchyme
- VMP
- ventral mesenchymal pad.
References
- 1. Thomson AA. Mesenchymal mechanisms in prostate organogenesis. Differentiation. 2008;76(6):587–598. [DOI] [PubMed] [Google Scholar]
- 2. Timms BG, Lee CW, Aumuller G, Seitz J. Instructive induction of prostate growth and differentiation by a defined urogenital sinus mesenchyme. Microsc Res Tech. 1995;30(4):319–332. [DOI] [PubMed] [Google Scholar]
- 3. Timms BG, Mohs TJ, Didio LJ. Ductal budding and branching patterns in the developing prostate. J Urol. 1994;151(5):1427–1432. [DOI] [PubMed] [Google Scholar]
- 4. Thomson AA, Timms BG, Barton L, Cunha GR, Grace OC. The role of smooth muscle in regulating prostatic induction. Development. 2002;129(8):1905–1912. [DOI] [PubMed] [Google Scholar]
- 5. Hayward SW, Haughney PC, Rosen MA, et al. . Interactions between adult human prostatic epithelium and rat urogenital sinus mesenchyme in a tissue recombination model. Differeniation. 1998;63(3):131–140. [DOI] [PubMed] [Google Scholar]
- 6. Cunha GR, Chung LW. Stromal-epithelial interactions—I. Induction of prostatic phenotype in urothelium of testicular feminized (Tfm/y) mice. J Steroid Biochem. 1981;14(12):1317–1324. [DOI] [PubMed] [Google Scholar]
- 7. Cunha GR, Lung B. The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J Exp Zool. 1978;205(2):181–193. [DOI] [PubMed] [Google Scholar]
- 8. Lasnitzki I, Mizuno T. Prostatic induction: interaction of epithelium and mesenchyme from normal wild-type mice and androgen-insensitive mice with testicular feminization. J Endocrinol. 1980;85(3):423–428. [DOI] [PubMed] [Google Scholar]
- 9. Donjacour AA, Cunha GR. The effect of androgen deprivation on branching morphogenesis in the mouse prostate. Dev Biol. 1988;128(1):1–14. [DOI] [PubMed] [Google Scholar]
- 10. Sugimura Y, Cunha GR, Donjacour AA. Morphogenesis of ductal networks in the mouse prostate. Biol Reprod. 1986;34(5):961–971. [DOI] [PubMed] [Google Scholar]
- 11. Prins GS, Cooke PS, Birch L, et al. . Androgen receptor expression and 5α-reductase activity along the proximal-distal axis of the rat prostatic duct. Endocrinology. 1992;130(5):3066–3073. [DOI] [PubMed] [Google Scholar]
- 12. Sugimura Y, Cunha GR, Donjacour AA, Bigsby RM, Brody JR. Whole-mount autoradiography study of DNA synthetic activity during postnatal development and androgen-induced regeneration in the mouse prostate. Biol Reprod. 1986;34(5):985–995. [DOI] [PubMed] [Google Scholar]
- 13. Prins GS, Putz O. Molecular signaling pathways that regulate prostate gland development. Differeniation. 2008;76(6):641–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thomson AA, Marker PC. Branching morphogenesis in the prostate gland and seminal vesicles. Differentiation. 2006;74(7):382–392. [DOI] [PubMed] [Google Scholar]
- 15. Berman DM, Desai N, Wang X, et al. . Roles for Hedgehog signaling in androgen production and prostate ductal morphogenesis. Dev Biol. 2004;267(2):387–398. [DOI] [PubMed] [Google Scholar]
- 16. Doles J, Cook C, Shi X, Valosky J, Lipinski R, Bushman W. Functional compensation in Hedgehog signaling during mouse prostate development. Dev Biol. 2006;295(1):13–25. [DOI] [PubMed] [Google Scholar]
- 17. Freestone SH, Marker P, Grace OC, et al. . Sonic hedgehog regulates prostatic growth and epithelial differentiation. Dev Biol. 2003;264(2):352–362. [DOI] [PubMed] [Google Scholar]
- 18. Lamm ML, Catbagan WS, Laciak RJ, et al. . Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev Biol. 2002;249(2):349–366. [DOI] [PubMed] [Google Scholar]
- 19. Podlasek CA, Barnett DH, Clemens JQ, Bak PM, Bushman W. Prostate development requires Sonic hedgehog expressed by the urogenital sinus epithelium. Dev Biol. 1999;209(1):28–39. [DOI] [PubMed] [Google Scholar]
- 20. Pu Y, Huang L, Prins GS. Sonic hedgehog-patched Gli signaling in the developing rat prostate gland: lobe-specific suppression by neonatal estrogens reduces ductal growth and branching. Dev Biol. 2004;273(2):257–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang BE, Shou J, Ross S, Koeppen H, De Sauvage FJ, Gao WQ. Inhibition of epithelial ductal branching in the prostate by sonic hedgehog is indirectly mediated by stromal cells. J Biol Chem. 2003;278(20):18506–18513. [DOI] [PubMed] [Google Scholar]
- 22. Allgeier SH, Lin TM, Vezina CM, et al. . WNT5A selectively inhibits mouse ventral prostate development. Dev Biol. 2008;324(1):10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang L, Pu Y, Hu WY, et al. . The role of Wnt5a in prostate gland development. Dev Biol. 2009;328(2):188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joesting MS, Cheever TR, Volzing KG, et al. . Secreted frizzled related protein 1 is a paracrine modulator of epithelial branching morphogenesis, proliferation, and secretory gene expression in the prostate. Dev Biol. 2008;317(1):161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keil KP, Mehta V, Branam AM, et al. . Wnt inhibitory factor 1 (Wif1) is regulated by androgens and enhances androgen-dependent prostate development. Endocrinology. 2012;153(12):6091–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alarid ET, Rubin JS, Young P, et al. . Keratinocyte growth factor functions in epithelial induction during seminal vesicle development. Proc Natl Acad Sci USA. 1994;91(3):1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Donjacour AA, Thomson AA, Cunha GR. FGF-10 plays an essential role in the growth of the fetal prostate. Dev Biol. 2003;261(1):39–54. [DOI] [PubMed] [Google Scholar]
- 28. Sugimura Y, Foster BA, Hom YK, et al. . Keratinocyte growth factor (KGF) can replace testosterone in the ductal branching morphogenesis of the rat ventral prostate. Int J Dev Biol. 1996;40(5):941–951. [PubMed] [Google Scholar]
- 29. Thomson AA, Cunha GR. Prostatic growth and development are regulated by FGF10. Development. 1999;126(16):3693–3701. [DOI] [PubMed] [Google Scholar]
- 30. Cancilla B, Jarred RA, Wang H, Mellor SL, Cunha GR, Risbridger GP. Regulation of prostate branching morphogenesis by activin A and follistatin. Dev Biol. 2001;237(1):145–158. [DOI] [PubMed] [Google Scholar]
- 31. Cook C, Vezina CM, Allgeier SH, et al. . Noggin is required for normal lobe patterning and ductal budding in the mouse prostate. Dev Biol. 2007;312(1):217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grishina IB, Kim SY, Ferrara C, Makarenkova HP, Walden PD. BMP7 inhibits branching morphogenesis in the prostate gland and interferes with Notch signaling. Dev Biol. 2005;288(2):334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Itoh N, Patel U, Cupp AS, Skinner MK. Developmental and hormonal regulation of transforming growth factor-β1 (TGFβ1), -2, and -3 gene expression in isolated prostatic epithelial and stromal cells: epidermal growth factor and TGFβ interactions. Endocrinology. 1998;139(3):1378–1388. [DOI] [PubMed] [Google Scholar]
- 34. Lamm ML, Podlasek CA, Barnett DH, et al. . Mesenchymal factor bone morphogenetic protein 4 restricts ductal budding and branching morphogenesis in the developing prostate. Dev Biol. 2001;232(2):301–314. [DOI] [PubMed] [Google Scholar]
- 35. Tomlinson DC, Freestone SH, Grace OC, Thomson AA. Differential effects of transforming growth factor-β1 on cellular proliferation in the developing prostate. Endocrinology. 2004;145(9):4292–4300. [DOI] [PubMed] [Google Scholar]
- 36. Tanji N, Tsuji M, Terada N, Takeuchi M, Cunha GR. Inhibitory effects of transforming growth factor-β1 on androgen-induced development of neonatal mouse seminal vesicles in vitro. Endocrinology. 1994;134(3):1155–1162. [DOI] [PubMed] [Google Scholar]
- 37. Donjacour AA, Cunha GR. Assessment of prostatic protein secretion in tissue recombinants made of urogenital sinus mesenchyme and urothelium from normal or androgen-insensitive mice. Endocrinology. 1993;132(6):2342–2350. [DOI] [PubMed] [Google Scholar]
- 38. Simanainen U, Allan CM, Lim P, et al. . Disruption of prostate epithelial androgen receptor impedes prostate lobe-specific growth and function. Endocrinology. 2007;148(5):2264–2272. [DOI] [PubMed] [Google Scholar]
- 39. Li X, Wang Y, Sharif-Afshar AR, et al. . Urothelial transdifferentiation to prostate epithelia is mediated by paracrine TGF-β signaling. Differentiation. 2009;77(1):95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Golden JP, DeMaro JA, Osborne PA, Milbrandt J, Johnson EM Jr. Expression of neurturin, GDNF, and GDNF family-receptor mRNA in the developing and mature mouse. Exp Neurol. 1999;158(2):504–528. [DOI] [PubMed] [Google Scholar]
- 41. Arenas E, Trupp M, Akerud P, Ibanez CF. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron. 1995;15(6):1465–1473. [DOI] [PubMed] [Google Scholar]
- 42. Henderson CE, Phillips HS, Pollock RA, et al. . GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266(5187):1062–1064. [DOI] [PubMed] [Google Scholar]
- 43. Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260(5111):1130–1132. [DOI] [PubMed] [Google Scholar]
- 44. Meng X, Lindahl M, Hyvonen ME, et al. . Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287(5457):1489–1493. [DOI] [PubMed] [Google Scholar]
- 45. Pepicelli CV, Kispert A, Rowitch DH, McMahon AP. GDNF induces branching and increased cell proliferation in the ureter of the mouse. Dev Biol. 1997;192(1):193–198. [DOI] [PubMed] [Google Scholar]
- 46. Pichel JG, Shen L, Sheng HZ, et al. . Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382(6586):73–76. [DOI] [PubMed] [Google Scholar]
- 47. Sainio K, Suvanto P, Davies J, et al. . Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development. 1997;124(20):4077–4087. [DOI] [PubMed] [Google Scholar]
- 48. Vega QC, Worby CA, Lechner MS, Dixon JE, Dressler GR. Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc Natl Acad Sci USA. 1996;93(20):10657–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jing S, Wen D, Yu Y, et al. . GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-α, a novel receptor for GDNF. Cell. 1996;85(7):1113–1124. [DOI] [PubMed] [Google Scholar]
- 50. Trupp M, Arenas E, Fainzilber M, et al. . Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381(6585):785–789. [DOI] [PubMed] [Google Scholar]
- 51. Cacalano G, Farinas I, Wang LC, et al. . GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21(1):53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Costantini F. GDNF/Ret signaling and renal branching morphogenesis: from mesenchymal signals to epithelial cell behaviors. Organogenesis. 2010;6(4):252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM Jr, Milbrandt J. GFRα1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21(2):317–324. [DOI] [PubMed] [Google Scholar]
- 54. Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367(6461):380–383. [DOI] [PubMed] [Google Scholar]
- 55. Jain S, Encinas M, Johnson EM Jr, Milbrandt J. Critical and distinct roles for key RET tyrosine docking sites in renal development. Genes Dev. 2006;20(3):321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cao JP, Yu JK, Li C, et al. . Integrin β1 is involved in the signaling of glial cell line-derived neurotrophic factor. J Comp Neurol. 2008;509(2):203–210. [DOI] [PubMed] [Google Scholar]
- 57. Paratcha G, Ledda F, Ibanez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113(7):867–879. [DOI] [PubMed] [Google Scholar]
- 58. Sariola H, Saarma M. Novel functions and signalling pathways for GDNF. J Cell Sci. 2003;116(Pt 19):3855–3862. [DOI] [PubMed] [Google Scholar]
- 59. Trupp M, Scott R, Whittemore SR, Ibanez CF. Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J Biol Chem. 1999;274(30):20885–20894. [DOI] [PubMed] [Google Scholar]
- 60. van Weering DH, Medema JP, van Puijenbroek A, Burgering BM, Baas PD, Bos JL. Ret receptor tyrosine kinase activates extracellular signal-regulated kinase 2 in SK-N-MC cells. Oncogene. 1995;11(11):2207–2214. [PubMed] [Google Scholar]
- 61. Worby CA, Vega QC, Zhao Y, Chao HH, Seasholtz AF, Dixon JE. Glial cell line-derived neurotrophic factor signals through the RET receptor and activates mitogen-activated protein kinase. J Biol Chem. 1996;271(39):23619–23622. [DOI] [PubMed] [Google Scholar]
- 62. Chiariello M, Visconti R, Carlomagno F, et al. . Signalling of the Ret receptor tyrosine kinase through the c-Jun NH2-terminal protein kinases (JNKS): evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene. 1998;16(19):2435–2445. [DOI] [PubMed] [Google Scholar]
- 63. Fisher CE, Michael L, Barnett MW, Davies JA. Erk MAP kinase regulates branching morphogenesis in the developing mouse kidney. Development. 2001;128(21):4329–4338. [DOI] [PubMed] [Google Scholar]
- 64. Keefe Davis T, Hoshi M, Jain S. Stage specific requirement of Gfrα1 in the ureteric epithelium during kidney development. Mech Dev. 2013;130(9–10):506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He Z, Jiang J, Kokkinaki M, Golestaneh N, Hofmann MC, Dym M. Gdnf upregulates c-Fos transcription via the Ras/Erk1/2 pathway to promote mouse spermatogonial stem cell proliferation. Stem Cells. 2008;26(1):266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lucas BE, Fields C, Joshi N, Hofmann MC. Mono-(2-ethylhexyl)-phthalate (MEHP) affects ERK-dependent GDNF signalling in mouse stem-progenitor spermatogonia. Toxicology. 2012;299(1):10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hasegawa K, Namekawa SH, Saga Y. MEK/ERK signaling directly and indirectly contributes to the cyclical self-renewal of spermatogonial stem cells. Stem Cells. 2013;31(11):2517–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kuslak SL, Marker PC. Fibroblast growth factor receptor signaling through MEK-ERK is required for prostate bud induction. Differentiation. 2007;75(7):638–651. [DOI] [PubMed] [Google Scholar]
- 69. Daneau I, Pilon N, Boyer A, et al. . The porcine SRY promoter is transactivated within a male genital ridge environment. Genesis. 2002;33(4):170–180. [DOI] [PubMed] [Google Scholar]
- 70. Doles JD, Vezina CM, Lipinski RJ, Peterson RE, Bushman W. Growth, morphogenesis, and differentiation during mouse prostate development in situ, in renal grafts, and in vitro. Prostate. 2005;65(4):390–399. [DOI] [PubMed] [Google Scholar]
- 71. Ghosh S, Lau H, Simons BW, et al. . PI3K/mTOR signaling regulates prostatic branching morphogenesis. Dev Biol. 2011;360(2):329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol. 2002;30(6):503–512. [DOI] [PubMed] [Google Scholar]
- 73. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. [DOI] [PubMed] [Google Scholar]
- 74. Cunha GR, Donjacour AA, Cooke PS, et al. . The endocrinology and developmental biology of the prostate. Endocr Rev. 1987;8(3):338–362. [DOI] [PubMed] [Google Scholar]
- 75. Allgeier SH, Lin TM, Moore RW, Vezina CM, Abler LL, Peterson RE. Androgenic regulation of ventral epithelial bud number and pattern in mouse urogenital sinus. Dev Dyn. 2010;239(2):373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Marker PC, Donjacour AA, Dahiya R, Cunha GR. Hormonal, cellular, and molecular control of prostatic development. Dev Biol. 2003;253(2):165–174. [DOI] [PubMed] [Google Scholar]
- 77. Klein RD, Sherman D, Ho WH, et al. . A GPI-linked protein that interacts with Ret to form a candidate neurturin receptor. Nature. 1997;387(6634):717–721. [DOI] [PubMed] [Google Scholar]
- 78. Panicker AK, Buhusi M, Thelen K, Maness PF. Cellular signalling mechanisms of neural cell adhesion molecules. Front Biosci. 2003;8:d900–911. [DOI] [PubMed] [Google Scholar]
- 79. Poteryaev D, Titievsky A, Sun YF, et al. . GDNF triggers a novel ret-independent Src kinase family-coupled signaling via a GPI-linked GDNF receptor α1. FEBS Lett. 1999;463(1–2):63–66. [DOI] [PubMed] [Google Scholar]
- 80. Hellmich HL, Kos L, Cho ES, Mahon KA, Zimmer A. Embryonic expression of glial cell-line derived neurotrophic factor (GDNF) suggests multiple developmental roles in neural differentiation and epithelial-mesenchymal interactions. Mech Dev. 1996;54(1):95–105. [DOI] [PubMed] [Google Scholar]
- 81. Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119(4):1005–1017. [DOI] [PubMed] [Google Scholar]
- 82. Suter-Crazzolara C, Unsicker K. GDNF mRNA levels are induced by FGF-2 in rat C6 glioblastoma cells. Brain Res Mol Brain Res. 1996;41(1–2):175–182. [DOI] [PubMed] [Google Scholar]
- 83. Wang P, Suo LJ, Wang YF, et al. . Effects of GDNF and LIF on mouse spermatogonial stem cells proliferation in vitro. Cytotechnology. 2014;66(2):309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Simanainen U, Gao YR, Desai R, et al. . Evidence for increased tissue androgen sensitivity in neurturin knockout mice. J Endocrinol. 2013;218(2):151–163. [DOI] [PubMed] [Google Scholar]
- 85. Creedon DJ, Tansey MG, Baloh RH, et al. . Neurturin shares receptors and signal transduction pathways with glial cell line-derived neurotrophic factor in sympathetic neurons. Proc Natl Acad Sci USA. 1997;94(13):7018–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Akerud P, Alberch J, Eketjall S, Wagner J, Arenas E. Differential effects of glial cell line-derived neurotrophic factor and neurturin on developing and adult substantia nigra dopaminergic neurons. J Neurochem. 1999;73(1):70–78. [DOI] [PubMed] [Google Scholar]
- 87. Coulpier M, Anders J, Ibanez CF. Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase: importance of tyrosine 1062 for GDNF mediated neuronal differentiation and survival. J Biol Chem. 2002;277(3):1991–1999. [DOI] [PubMed] [Google Scholar]
- 88. Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130(10):2187–2198. [DOI] [PubMed] [Google Scholar]
- 89. Saarma M. GDNF—a stranger in the TGF-β superfamily? Eur J Biochem. 2000;267(24):6968–6971. [DOI] [PubMed] [Google Scholar]
- 90. Rozen EJ, Schmidt H, Dolcet X, Basson MA, Jain S, Encinas M. Loss of Sprouty1 rescues renal agenesis caused by Ret mutation. J Am Soc Nephrol. 2009;20(2):255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jijiwa M, Fukuda T, Kawai K, et al. . A targeting mutation of tyrosine 1062 in Ret causes a marked decrease of enteric neurons and renal hypoplasia. Mol Cell Biol. 2004;24(18):8026–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rossomando AJ, Payne DM, Weber MJ, Sturgill TW. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc Natl Acad Sci USA. 1989;86(18):6940–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lu BC, Cebrian C, Chi X, et al. . Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat Genet. 2009;41(12):1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lipford GB, Wright GL Jr. Comparative study of monoclonal antibodies TURP-27 and HNK-1: their relationship to neural cell adhesion molecules and prostate tumor-associated antigens. Cancer Res. 1991;51(9):2296–2301. [PubMed] [Google Scholar]
- 95. Takahashi S, Kato K, Nakamura K, Nakano R, Kubota K, Hamada H. Neural cell adhesion molecule 2 as a target molecule for prostate and breast cancer gene therapy. Cancer Sci. 2011;102(4):808–814. [DOI] [PubMed] [Google Scholar]
- 96. Knox JD, Cress AE, Clark V, et al. . Differential expression of extracellular matrix molecules and the α6-integrins in the normal and neoplastic prostate. Am J Pathol. 1994;145(1):167–174. [PMC free article] [PubMed] [Google Scholar]
- 97. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. [DOI] [PubMed] [Google Scholar]
- 98. Heer R, Collins AT, Robson CN, Shenton BK, Leung HY. KGF suppresses α2β1 integrin function and promotes differentiation of the transient amplifying population in human prostatic epithelium. J Cell Sci. 2006;119(Pt 7):1416–1424. [DOI] [PubMed] [Google Scholar]
- 99. Bello-DeOcampo D, Kleinman HK, Deocampo ND, Webber MM. Laminin-1 and α6β1 integrin regulate acinar morphogenesis of normal and malignant human prostate epithelial cells. Prostate. 2001;46(2):142–153. [DOI] [PubMed] [Google Scholar]
- 100. Murant SJ, Handley J, Stower M, Reid N, Cussenot O, Maitland NJ. Co-ordinated changes in expression of cell adhesion molecules in prostate cancer. Eur J Cancer. 1997;33(2):263–271. [DOI] [PubMed] [Google Scholar]
- 101. Dawson DM, Lawrence EG, MacLennan GT, et al. . Altered expression of RET proto-oncogene product in prostatic intraepithelial neoplasia and prostate cancer. J Natl Cancer Inst. 1998;90(7):519–523. [DOI] [PubMed] [Google Scholar]
- 102. Lucia MS, Lambert JR. Growth factors in benign prostatic hyperplasia: basic science implications. Curr Urol Rep. 2008;9(4):272–278. [DOI] [PubMed] [Google Scholar]