

Abstract
Monocarboxylate transporter 8 (MCT8) transports thyroid hormone (TH) across the plasma membrane. Mutations in MCT8 result in the Allan-Herndon-Dudley syndrome, comprising severe psychomotor retardation and elevated serum T3 levels. Because the neurological symptoms are most likely caused by a lack of TH transport into the central nervous system, the administration of a TH analog that does not require MCT8 for cellular uptake may represent a therapeutic strategy. Here, we investigated the therapeutic potential of the biologically active T3 metabolite Triac (TA3) by studying TA3 transport, metabolism, and action both in vitro and in vivo. Incubation of SH-SY5Y neuroblastoma cells and MO3.13 oligodendrocytes with labeled substrates showed a time-dependent uptake of T3 and TA3. In intact SH-SY5Y cells, both T3 and TA3 were degraded by endogenous type 3 deiodinase, and they influenced gene expression to a similar extent. Fibroblasts from MCT8 patients showed an impaired T3 uptake compared with controls, whereas TA3 uptake was similar in patient and control fibroblasts. In transfected cells, TA3 did not show significant transport by MCT8. Most importantly, treatment of athyroid Pax8-knockout mice and Mct8/Oatp1c1-double knockout mice between postnatal days 1 and 12 with TA3 restored T3-dependent neural differentiation in the cerebral and cerebellar cortex, indicating that TA3 can replace T3 in promoting brain development. In conclusion, we demonstrated uptake of TA3 in neuronal cells and in fibroblasts of MCT8 patients and similar gene responses to T3 and TA3. This indicates that TA3 bypasses MCT8 and may be used to improve the neural status of MCT8 patients.
The widely expressed monocarboxylate transporter 8 (MCT8; SLC16A2) has been characterized as an important thyroid hormone (TH) transporter facilitating cellular uptake and export of T3 and T4 (1, 2). The MCT8 gene is localized on the X-chromosome, and mutations in MCT8 are associated with the Allan-Herndon-Dudley syndrome (AHDS) (3, 4). This syndrome is characterized by severe psychomotor retardation in combination with disturbed thyroid function tests, ie, high T3, low-normal T4, and normal to increased TSH. Clinical features include spastic quadriplegia, axial hypotonia, and severe mental retardation (5–9). These patients also suffer from peripheral thyrotoxicosis, producing tachycardia and muscle wasting. Since the genetic cause of AHDS has been elucidated, many patients with MCT8 mutations have been identified worldwide (10). With this increasing prevalence also the need for therapy rises.
Based on the findings of mildly increased serum TSH levels and mildly decreased serum free T4 levels, several AHDS patients have been treated with l-T4 (11, 12). This did not improve mental function, whereas it even further increased the already elevated serum T3 levels and worsened the thyrotoxic state of peripheral tissues. Another therapeutic approach has been the normalization of serum TH levels by block-and-replace therapy using PTU and l-T4, which has been applied to older AHDS patients (13, 14). This treatment improved the peripheral thyrotoxicosis, leading to a normalization of the heart rate as well as a gain in body weight (BW). However, it did not improve psychomotor function.
The pathogenic mechanism of MCT8 mutations is not completely resolved. It is well-known that TH is crucial for brain development (15). Studies in Mct8-knockout (KO) mice (16–18) and in vitro studies using patient-derived fibroblast and transfected cells (19–21) have revealed impaired cellular TH transport, in particular into the brain. These findings support the hypothesis that the key factor in the pathogenic mechanism is the lack of T3 transport via the blood-brain barrier and/or into neurons, causing poor regulation of TH-dependent genes. The lack of transcriptional regulation by T3 could in theory be bypassed by administration of a TH analog. For the effective treatment of patients with MCT8 mutations, such an analog should meet several well-defined criteria. It should have 1) high affinity for T3 receptors, 2) uptake into the brain and into neurons via a transporter other than MCT8, 3) a similar pattern of deiodination by D3 as T3, and 4) T3-like effects on the expression of TH responsive genes in brain cells and should 5) restore neural differentiation in the hypothyroid brain.
The first TH analog that has been studied for possible therapy of AHDS patients is 3,5-diiodothyropropionic acid (DITPA). Its administration to wild-type (WT) and Mct8-KO mice resulted in decreased TSH and T4 levels (22). Although Mct8-KO mice do not replicate the neurological phenotype of patients with MCT8 mutations, they are a good model to evaluate the effects of therapeutic strategies on the hypothalamus-pituitary-thyroid axis and peripheral tissues. Furthermore, DITPA treatment was found to produce TH-like effects in brains of WT and Mct8-KO mice, indicating brain uptake in the absence of functional MCT8. The clinical efficacy of DITPA was studied in 4 younger MCT8 patients (23). Although DITPA treatment alleviated the thyrotoxic state in all 4 and resulted in a weight gain in 1 patient, no improvement was seen in psychomotor development.
The natural TH metabolite 3,3′,5-triiodothyroacetic acid (Triac, TA3) has some advantages compared with DITPA, such as the extensive clinical experience with its use to suppress TSH in patients with thyroid cancer (24) and in those with TH resistance due to TH receptor-β (TRβ) mutations (25, 26). Furthermore, the preserved 3,3′,5-iodination pattern makes that TA3 has retained full affinity for the TRα1 receptor, whereas it even has a somewhat higher affinity than T3 for TRβ (criterion 1) (27). The aim of our study was to explore whether TA3 could be a valuable drug for treatment of AHDS patients by studying whether TA3 also fulfills the other 4 criteria required to be effective in the treatment of patients with MCT8 mutations.
Materials and Methods
Materials
Human SH-SY5Y neuroblastoma cells were obtained from ECACC; FuGENE 6 and XtremeGENE 9 transfection reagents, and the High Pure RNA isolation kit from Roche Diagnostics; fetal bovine serum (FBS), penicillin-streptomycin, DMEM/F12+glutamax, RPMI 1640+l-glutamine, and Dulbecco's PBS from Invitrogen; culture dishes from Corning; TaqMan reagents from Applied Biosystems; SYBR Green from Eurogentec; phorbol 12-myristate 13-acetate (PMA), iodothyronine derivatives, BSA, Na2SeO3, d-glucose and dithiothreitol from Sigma-Aldrich; and Na125I from IDB Holland BV. [125I]T3 and [125I]TA3 were produced as previously described (28).
Cell culture and differentiation
The different cell lines were cultured in DMEM/F12+glutamax medium, supplemented with 9% FBS, 100nM Na2SO4, and 1% penicillin/streptomycin in 75-cm2 flasks at 37°C and 5% CO2. Fibroblasts were cultured in RPMI 1640+l-glutamine with the same supplements as described above.
COS1 cells used for deiodinase activity assays were seeded in 6-well culture dishes in culture medium without penicillin/streptomycin and transfected with 1 μg hD3-pClneo (2) using FuGENE 6 as previously described. COS1 cells intended for uptake studies were cultured in 24-well culture dishes in culture medium without penicillin/streptomycin and transfected with 200 ng pcDNA3 (empty vector), hMCT8-pcDNA3 (2), or hMCT10-pcDNA3.1 (29) using X-tremeGENE 9 following the manufacturer's protocol.
Fibroblasts from MCT8 patients and healthy controls were obtained as explained in Supplemental Table 1. Informed consent was obtained from the patients' parents. Patients and controls were age and gender matched. Cells were subcultured at 90% confluence and seeded into 6-well culture dishes for uptake experiments. Medium was refreshed every 3 to 4 days. Experiments were carried out at confluence.
The human SH-SY5Y neuroblastoma cell line was used as a model for neurons. Cells were passaged at 90% confluence and subcultured in 24-well culture dishes for metabolism studies and in 6-well culture dishes for uptake and gene expression experiments. Medium in the culture dishes was changed to DMEM/F12+glutamax without supplements at 80% confluence. To study gene responses to T3 and TA3, SH-SY5Y cells were incubated for 6 or 48 hours without or with 100nM T3 or TA3.
The human MO3.13 oligodendrocyte cell line was used as a model for oligodendrocyte differentiation (30). At 90% confluence, cells were seeded in 6-well culture dishes for uptake experiments. After 24 hours, cells were initiated to differentiate in DMEM/F12+glutamax using 10 ng PMA/mL. Experiments were carried out in proliferating progenitor cells (24 hours after seeding) and in early differentiated cells (7 days in PMA).
TH transport experiments
TH transport experiments were carried out at confluence for fibroblasts after 2 days of serum deprivation in SH-SY5Y cells and in proliferating and differentiated MO3.13 cells. Cells were rinsed with incubation medium (Dulbecco's PBS, 0.1% d-glucose, and 0.1% BSA) and incubated for 2 to 60 minutes at 37°C and 5% CO2 in incubation medium containing 1nM (105 counts/min) [125I]T3 or [125I]TA3. After incubation, cells were rinsed with incubation medium and lysed in 0.1M NaOH. Radioactivity in the cell lysates was determined using a γ-counter. Uptake was corrected for cellular protein determined with the Bradford protein assay.
Deiodination of T3 and TA3 in intact cells and in cell lysates
Intact cell metabolism experiments using endogenous D3 expressing SH-SY5Y cells were carried out after 2 days of serum deprivation (31). Cells were rinsed with incubation medium followed by incubation for 1 to 24 hours at 37°C and 5% CO2 in incubation medium containing 1nM (5 × 105 cpm) [125I]T3 or [125I]TA3. After incubation, medium was extracted with ethanol and analyzed by HPLC as described previously (2).
Deiodination of T3 and TA3 by recombinant human D3 was determined by incubation of different dilutions of transfected COS1 cell lysates for 60 minutes at 37°C with 1nM [125I]T3 or [125I]TA3 in 0.1M phosphate (pH 7.2), 2mM EDTA and 10mM dithiothreitol. Ethanol extracts of the incubation mixtures were analyzed by HPLC as described previously (2).
RT-PCR and quantitative PCR
Total RNA was extracted from SH-SY5Y and MO3.13 cells with the High Pure RNA isolation kit using the standard protocol. RNA concentrations were determined by Nanodrop 2000. cDNA was produced from 1 μg RNA in a total volume of 50 μL using TaqMan reverse transcription reagents following the manufacturer's protocol.
Quantitative PCR was carried out using 1.25 μL cDNA in a total volume of 13 μL in a 384-well plate. Supplemental Table 2 shows the different primers and probes used for these assays. Expression levels of TH-responsive genes were obtained using 5 pmol/μL primers in combination with SYBR Green following the manufacturer's protocol. Primer-probes and TaqMan Mastermix were used to obtain expression levels of myelin basic protein (MBP) and proteolipid protein (PLP) and the housekeeping gene cyclophilin A (PPIA) following the manufacturer's protocol. TaqMan 7900HT was used for the readout of the plates using SDS version 2.4 software. Data are shown as relative expression to housekeeping gene PPIA and as fold change vs untreated cells.
Mouse studies
The animal studies were approved by the Thüringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz (TLLV Thüringen, Bad Langensalza, Germany). Pax8-KO (32) and WT littermates were generated by mating heterozygous Pax8 mutant mice and genotyped as reported elsewhere (33). Male offspring (4 mice per genotype and treatment) were injected sc once per day between postnatal day 1 (P1) and P12 with either TA3 (200 ng/g BW) or with 0.9% saline as control. The generation and genotyping of Mct8/Oatp1c1-double knockout (dKO) mice) has been described previously (34). To study TA3 effects in this animal model, male Mct8/Oatp1c1-dKO mice were daily injected with either 50 or 400 ng/g BW TA3 between P1 and P12. In addition, WT and Mct8/Oatp1c1-dKO mice treated with saline were included as well. At P12 (and 16 hours after the last injection), the animals were deeply anesthetized with isoflurane and subjected to perfusion fixation using 4% paraformaldehyde/PBS. Forebrain and cerebella were dissected and postfixed overnight in 4% paraformaldehyde/PBS.
To assess the state of Purkinje cell (PC) dendritic development, cerebella were cut sagittally on a vibratome into 50-μm-thick sections, and sections of the vermis area were immunostained with rabbit polyclonal antibody against calbindin D28d (Sigma; 1:500) followed by incubation with Alexa Fluor 555-labeled goat antirabbit IgG antibody (Invitrogen; 1:1000). Thickness of the molecular layer at the primary fissure between anterior and posterior lobe was determined by measuring PC dendritic dimension with ImageJ (35) on 3 consecutive section from each animal (n = 4 per genotype).
Frontal forebrain vibratome sections from the same animals were used to visualize a subset of γ-aminobutyric acid (GABA)ergic interneurons that express parvalbumin (PV). For this purpose, sections were incubated with mouse monoclonal anti-PV antibody (Millipore; 1:1000) followed by incubation with Alexa Fluor 555-labeled goat antimouse IgG antibody (Invitrogen; 1:500). PV immunoreactivity was analyzed in the barrel field of the somatosensory cortex as well as in the retrosplenial cortex using 3 consecutive sections between bregma level −0.8 to −1.1 mm. The number of PV-immunopositive neurons was counted and normalized to the respective cortical area using ImageJ.
To assess myelination, frontal forebrain sections were incubated with rat monoclonal anti-MBP antibody (Millipore, 1:200) and Alexa Fluor 555-labeled goat antirat IgG antibody (Invitrogen, 1:1000). The integrated density of MBP in all areas of the cerebral cortex was determined on 3 consecutive sections located around bregma level 0.1 and normalized to the respective cortical area using ImageJ. For each genotype, 4 animals were used, and results are expressed relative to WT values.
Statistical analysis
The results are shown as means and SEM of a minimum of 2 experiments carried out at least in duplicate. GraphPad Prism version 5.01 for Windows (GraphPad Software) was used for statistical analysis. A 1-way ANOVA with Bonferroni posttest with a cutoff level of P < .05 was used to analyze the difference between the background and the different transporters. Statistical analysis of the effects of T3 and TA3 on different genes as well as of TA3 effects during murine brain development was done using a 2-way ANOVA followed by Bonferroni posttest with a cutoff level of P < .05.
Results
TA3 is taken up by target cells via a transporter other than MCT8
Transport of [125I]T3 and [125I]TA3 by MCT8 and its homolog MCT10 was studied in transiently transfected COS1 cells. Cellular T3 uptake was induced 2.5-fold and 4.4-fold by transfection of cells with MCT8 and MCT10, respectively, vs empty vector control (Figure 1A). In contrast, MCT8 or MCT10 expression did not induce cellular TA3 uptake (Figure 1B). These findings indicate that TA3 is not transported by MCT8 or MCT10.
Figure 1. A and B, Cellular uptake of [125I]T3 (A) and [125I]TA3 (B) after 10 minutes in transiently transfected COS1 cells with pcDNA3 (empty vector [EV]) or hMCT8-pcDNA3 or hMCT10-pcDNA3.1.
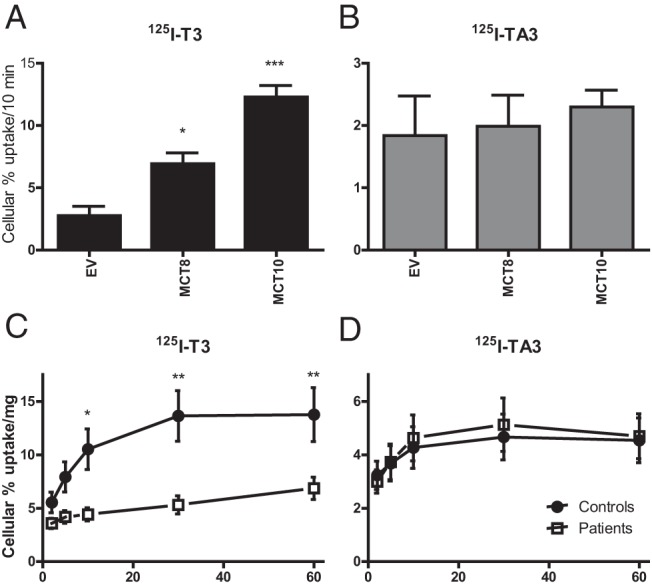
Results are presented as mean ± SEM (n = 3). Significance represents empty vector vs MCT8 or MCT10 using 1-way ANOVA with Bonferroni posttest. C and D, Cellular uptake of [125I]T3 (A) and [125I]TA3 (B) after 2 to 60 minutes of incubation with fibroblasts from MCT8 patients or age- and gender-matched controls. Results are corrected for protein and presented as mean ± SEM (n = 5). Significance represents control fibroblast vs fibroblast of MCT8 patients using 2-way ANOVA with Bonferroni posttest. *, P < .05; **, P < .01; ***, P < .001.
To assure that TA3 is transported into MCT8-deficient cells, [125I]T3 and [125I]TA3 uptake was studied in fibroblasts from AHDS patients and age and gender-matched controls (Figure 1, C and D). As expected, markedly impaired T3 uptake was seen throughout the 2- to 60-minute incubation in fibroblasts from AHDS patients compared with control fibroblasts. In contrast, similar TA3 uptake was observed in fibroblasts from AHDS patients and controls. These findings are in agreement with the results obtained in cells transiently transfected with MCT8, indicating that MCT8 and MCT10 do not facilitate TA3 transport.
Treatment of AHDS patients with TA3 can be beneficial only if TA3 is transported into neural target cells. As a model for neurons, the SH-SY5Y neuroblastoma cell line was used for studies of T3 and TA3 transport. Figure 2A shows the time-dependent cellular uptake of [125I]T3 and [125I]TA3. Uptake of both substrates increased with time between 2 and 60 minutes of incubation, with TA3 uptake (1.8%–5.2%) roughly amounting to half of T3 uptake (∼2.3%–11.7%). However, it should be realized that these uptake studies were done in the presence of 0.1% BSA in the medium, providing a higher free substrate concentration for T3 (17.9%) than for TA3 (9.5%) (unpublished results).
Figure 2. Cellular uptake of [125I]T3 and [125I]TA3 in SH-SY5Y (A), proliferating progenitor MO3.13 (B), and early differentiated MO3.13 cells (C) at increasing incubation times.

Results are corrected for protein and presented as mean ± SEM (n = 2–3).
Other important TH target cells in brain are oligodendrocytes because the differentiation of these cells is also controlled by TH (15). Therefore, these cells would also be an important target cell for TA3 therapy. The MO3.13 oligodendrocyte cell line was used to study transport of [125I]T3 and [125I]TA3 in proliferating cells (Figure 2B) and in early differentiated oligodendrocytes induced by phorbol ester treatment (Figure 2C). Supplemental Figure 1A shows the appearance of immature and mature cells in these cultures. Differentiation was confirmed by the markedly increased expression of mature oligodendrocyte markers (Supplemental Figure 1B). Uptake increased over time for T3 and TA3 in immature as well as mature MO3.13 cells. Together with the data obtained in MCT8-transfected cells and fibroblasts, the uptake results in SH-SY5Y and MO3.13 cells indicate that TA3 is transported into brain target cells by a transporter other than MCT8 (criterion 2).
TA3 shows similar metabolism by D3 as T3
We compared the deiodination of T3 and TA3 by recombinant human D3 expressed in transfected COS1 cells. D3 was found to catalyze the inner ring deiodination of T3 to 3,3′-T2 (Figure 3A) and of TA3 to 3,3′-TA2 (Figure 3B) with equivalent efficacy, although the further inner ring deiodination of 3,3′-TA2 to 3′-TA1 was more rapid than the further deiodination of 3,3′-T2 to 3′-T1.
Figure 3. A and B, Deiodination of T3 and TA3 by recombinant human D3.

Dilutions of lysates of COS1 cells transfected with human D3 were incubated for 60 minutes at 37°C with [125I]T3 (A) or [125I]TA3 (B). C and D, Conversion of T3 and TA3 by endogenous D3 in intact SH-SY5Y cells. Cells were incubated for 4 or 24 hours at 37°C with [125I]T3 (C) or [125I]TA3 (D). Results are presented as mean ± SEM (n = 2–3).
The metabolism of T3 and TA3 was also studied in intact SH-SY5Y cells, which express D3 endogenously (31), providing an integral view of T3 and TA3 transport and metabolism and, consequently, their availability for the T3 receptor. Initial experiments showed a marked TA3 conversion of 87% after 24 hours. We repeated these experiments to compare TA3 and T3 using a different batch of FBS and found somewhat lower conversion of TA3 after 24 hours. Between 4 and 24 hours of incubation, we observed a time-dependent increase in [125I]T3 and [125I]TA3 metabolism, amounting to 41%–86% for T3 (Figure 3C) and to 15%–74% for TA3 (Figure 3D). These results support our uptake data, demonstrating that the T3 and TA3 taken up by SH-SY5Y cells are also available for intracellular deiodination by D3. Therefore, TA3 and T3 are likewise converted by D3 in neuronal cells, allowing the regulation of intracellular TA3 levels by D3 very much like endogenous T3 in physiological conditions (criterion 3).
TA3 has T3-like effects on gene expression in neuronal cells
We also used SH-SY5Y cells as a model for central neurons to study the expression of genes in response to incubation for 6 to 48 hours with 100nM T3 or TA3. No difference in cell appearance was seen after 48 hours of incubation with T3 or TA3 (Supplemental Figure 2). Expression values were corrected for the housekeeping gene PPIA, and the fold change with untreated cells was calculated. T3 and TA3 significantly induced the expression of the T3-responsive genes Hairless (HR), ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) and Kruppel-like factor 9 (KLF9) after both 6 (Figure 4A) and 48 (Figure 4B) hours. No significant induction of neurogranin (RC3) expression was seen by T3 or TA3 after 6 or 48 hours. This may be related to the brain region-specific regulation of RC3 expression by T3 (36, 37). The response to TA3 was slightly lower than to T3 for HR after 6 hours and for HR and ENPP2 after 48 hours. The significant results after 6 hours of incubation with T3 or TA3 suggest that they were produced by a direct genomic effect. These findings indicate that equal concentrations of T3 and TA3 have similar effects on the expression of TH-responsive genes (criterion 4). Similar results were obtained using 10nM instead of 100nM T3 or TA3 (data not shown).
Figure 4. Changes in expression of TH-responsive genes after incubation of SH-SY5Y cells for 6 (A) or 48 (B) hours with 0.1μM T3 or TA3.
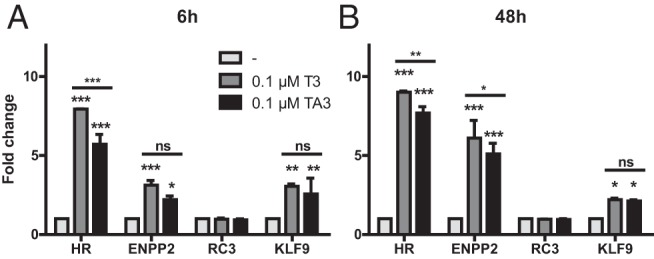
Expression values were corrected for the housekeeping gene cyclophilin A, and expressed as fold change vs control incubation without TH. Results are presented as mean ± SEM of 2 experiments performed in triplicate. Significance represents T3 or TA3 vs no treatment using matched 2-way ANOVA with Bonferroni posttest. *, P < .05; **, P < .01; ***, P < .001.
TA3 restores neural differentiation in the hypothyroid brain
To evaluate the action of TA3 in vivo, we took advantage of athyroid Pax8-KO mice that do not produce endogenous THs and therefore show a severely retarded brain development (32, 38). We treated newborn WT and Pax8-KO mice once a day by sc injections of TA3 (200 ng/g BW) or saline and analyzed brain morphology of these animals at P12. Cerebellar development was monitored by visualizing PC morphology with an antibody against calbindin. Compared with WT littermates, saline-treated Pax8-KO mice showed a grossly retarded PC dendritic outgrowth consistent with the hypothyroid state of these animals (Figure 5A). TA3 treatment was sufficient to normalize PC dendrite formation in Pax8-KO mice without affecting PC development in WT animals.
Figure 5. Thyromimetic actions of TA3 in the developing mouse brain.

Pax8-KO mice and WT littermates were injected daily with TA3 (200 ng/g BW) starting at P1 and analyzed at P12. Control animals received saline injections. A, PCs in sagittal cerebellar sections through the vermis region were visualized by calbindin immunostaining, and the thickness of the molecular layer (ML) that comprises PC dendrites was determined on 3 consecutive sections of each animal (males; n = 4 per genotype). Whereas in saline-treated Pax8-KO mice, PCs showed a strongly reduced dendritic outgrowth and therefore a thinner ML, TA3 treatment of Pax8-KO mice restored normal PC outgrowth and consequently ML thickness. B, Coronal forebrain sections of the same animals were immunostained with an antibody against MBP and revealed a significantly reduced immunoreactivity in the cerebral cortex of saline-treated Pax8-KO mice. In contrast, TA3 treatment of Pax8-KO mice resulted in normal MBP expression. Scale bar, 50 μm. **, P < .01; ***, P < .001. Abbreviation: AU, arbitrary unit.
Another hallmark of a congenital hypothyroid central nervous system is a reduced myelination because the expression of myelin components such as MBP is positively controlled by TH (39). We therefore analyzed MBP expression in the cerebral cortex by immunohistochemistry and indeed found a strongly reduced MBP immunoreactivity in saline-treated Pax8-KO mice compared with WT littermates (Figure 5B). TA3 treatment did not affect MBP expression in the cerebral cortex of WT animals but restored normal MBP immunoreactivity in Pax8-KO mice.
Finally, we determined the number of PV-immunopositive GABA-interneurons in the cerebral cortex where hypothyroidism leads to a strongly reduced PV immunoreactivity (40, 41). As depicted in Figure 6 for the retrosplenial as well as the somatosensory cortex, PV-positive neurons were found in Pax8-KO mice only upon TA3 treatment. Altogether, our findings indicate that during early postnatal periods, TA3 can indeed prevent neural abnormalities that are caused by insufficient TH supply (criterion 5).
Figure 6. TA3 stimulates the maturation of PV-immunoreactive neurons in the somatosensory and retrosplenial cortex.

Coronal forebrain sections of WT and Pax8-KO mice at P12 were immunostained with an antibody against PV that labels a subset of GABAergic interneurons. PV immunoreactivity was quantified on 3 consecutive sections from each animal using 4 animals per genotype and normalized to the respective cortical area. In saline-treated Pax8-KO mice, PV immunoreactivity was strongly reduced but could be partially restored upon TA3 treatment. Scale bar, 50 μm. **, P < .01; ***, P < .001. Abbreviation: AU, arbitrary unit.
TA3 promotes neural development in Mct8/Oatp1c1-dKO mice
With the generation of Mct8/Oatp1c1-dKO mice, we recently established a mouse model that exhibits the same abnormal serum TH profile as the patients with AHDS mutations, whereas the brain TH content in these animals is strongly reduced due to a highly compromised transport of TH into the brain (34). To elucidate whether TA3 treatment promotes neural differentiation in these animals as well, we treated Mct8/Oatp1c1-dKO mice with either 50 or 400 ng/g BW TA3 daily between P1 and P12 and analyzed cerebellar Purkinje cell morphology and cerebral myelination as described above. In agreement with previous findings (34), saline-injected Mct8/Oatp1c1-dKO mice exhibited a strongly reduced thickness of the molecular layer and decreased MBP immunoreactivity compared with saline-treated WT animals (Figure 7). TA3 treatment in turn led to a dose-dependent stimulation of cerebellar Purkinje cell dendritogenesis as well as cortical myelination in Mct8/Oatp1c1-dKO mice. Normalization, however, was achieved only by applying the higher TA3 dose (400 g/g BW). These data indicate that TA3 is also able to exert thyromimetic actions in the central nervous system of Mct8/Oatp1c1-dKO mice, an animal model for human MCT8 deficiency.
Figure 7. TA3 promotes cerebellar development and cortical myelination in Mct8/Oatp1c1-dKO mice.

Mct8/Oatp1c1-dKO (M/O dKO) mice were daily injected with TA3 at a dose of either 50 ng/g BW (50) or 400 ng/g BW (400) between P1 and P12. As controls, WT and M/O-dKO mice were treated with saline. Visualization of PC dendrites on sagittal cerebellar sections by calbindin immunostaining (A, upper row) revealed a reduced PC arborization in M/O-dKO mice, whereas TA3 treatment visibly stimulated dendritogenesis. Coronal forebrain sections of the same animals were immunostained with an antibody against MBP and revealed a significantly reduced immunoreactivity (IR) in the cerebral cortex of saline-treated M/O-dKO mice, whereas animals receiving TA3 exhibited elevated MBP IR. However, quantification of the thickness of the molecular layer (ML) as well as of the cortical MBP IR revealed that only the application of a high dose of TA3 was sufficient in normalizing both parameters in Mct8/Oatp1c1-dKO animals (B). Scale bar in A, 40 μm (upper row) and 80 μm (lower row). *, P < .05; **, P < .01; ***, P < .001. Abbreviation: AU, arbitrary unit.
Discussion
The aim of our study was to obtain evidence that TA3 is a valuable compound for the treatment of patients with MCT8 mutations, in particular when it is administered to young children where TH-dependent brain development is still an ongoing process. TA3 fulfills all 5 criteria required for an effective therapy of these patient: 1) TA3 has a high affinity for the T3 receptor (27), 2) TA3 is transported into brain cells by a transporter other than MCT8, 3) TA3 is converted by D3 in the same manner as T3, 4) TA3 produces similar neuronal gene responses as T3, and 5) TA3 restores neural differentiation in the hypothyroid brain.
Cellular transport of TA3 has been studied before in rat anterior pituitary cells (42) and cardiomyocytes (43). Evidence was provided that uptake of TA3 by these cells is mediated by plasma membrane transporters, but the identity of TA3 transporters remains to be determined.
Our in vitro studies in neuronal cell lines suggest that TA3 is almost as potent as T3 in the regulation of gene responses. These data fit well with our previous in vitro findings of a robust T3-like effect of TA3 on dendrite formation in cerebellar Purkinje cells (44). The T3-like effects of TA3 are further supported by our in vivo data showing that treatment of athyroid Pax8-KO mice as well as of Mct8/Oatp1c1-dKO mice with TA3 results in normal cerebellar PC dendritogenesis, indicating that TA3 can replace T3 in developing neurons. Moreover, TA3 treatment restores normal MBP expression, suggesting that TA3 can replace T3 in promoting myelination. Using 3 different test systems, 1) a neuroblastoma cell line, 2) in vitro cultured mouse PCs (5), and 3) TH-dependent brain areas, our data demonstrate that TA3 has potent T3-like effects in the regulation of neuronal gene expression.
A major advantage of TA3 is the extensive clinical experience with this compound that has been administered already for many years to patients with TH resistance caused by TRβ receptor mutations to reduce thyrotoxicosis (26, 45). It has been administered to thyroid cancer patients to suppress TSH (24). It is known as a safe drug to administer for long periods to suppress TSH in humans. All these features make it an attractive therapeutic option for AHDS patients.
A disadvantage of TA3 is its short half-life of ∼6 hours in humans, which makes it necessary to administer it 3 to 4 times a day (46). Therefore, we recently studied also the effects of TA4 as a natural metabolite of T4 (44). We could demonstrate that TA4 is a good therapeutic option because it is endogenously converted by D1 and D2 to bioactive TA3 and does not have such a short half-life as TA3. The major drawback is the limited experience with TA4 application in humans (47, 48). Consequently, extensive clinical studies would be needed before TA4 may be available for the treatment of AHDS patients. Therefore, TA3 administration appears more suitable for therapy of AHDS patients despite its short half-life.
What can we expect from administration of TA3 to AHDS patients? Based on the animal studies and the in vitro findings, TA3 administration will reduce serum T3 levels and, consequently, diminish symptoms of peripheral thyrotoxicosis such as muscle wasting, tachycardia, and weight loss. Of course, this assumes that the effect of the decrease in serum T3 outweighs the effects of administered TA3 on peripheral tissues. With respect to psychomotor development, we anticipate beneficial effects if TA3 is administered in particular to young children, because TH-dependent brain development still occurs after birth (49). If the treatment is initiated later in life, brain damage is likely to be largely irreversible as is known from other causes of TH insufficiency, such as congenital hypothyroidism (50).
To conclude, our in vitro and in vivo studies indicate that TA3 is a promising compound for the treatment of patients with MCT8 mutations. It fulfills all criteria required for such a therapeutic approach, ie, 1) high T3 receptor affinity, 2) MCT8-independent cellular transport, 3) T3-like metabolism by D3, 4) T3-like effects in neuronal cells, and 5) T3-like responses in the hypothyroid brain. These properties combined with the extensive clinical experience with this compound make TA3 an attractive drug to treat the peripheral thyrotoxicosis of MCT8 patients with the possibility to improve psychomotor development when administered to young children.
Acknowledgments
We thank Wim Klootwijk for the technical assistance. We thank the Sherman family, the Smile Foundation, and the Jérôme Leujeune Foundation as well as the BMBF (E-RARE project Thyronerve) for financial support of this work.
This work was supported by Smile Foundation with support from the Sherman family and by the Jérôme Lejeune Foundation and BMBF (E-RARE project Thyronerve).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by Smile Foundation with support from the Sherman family and by the Jérôme Lejeune Foundation and BMBF (E-RARE project Thyronerve).
Footnotes
- AHDS
- Allan-Herndon-Dudley syndrome
- BW
- body weight
- DITPA
- 3,5-diiodothyropropionic acid
- dKO
- double knockout
- FBS
- fetal bovine serum
- GABA
- γ-aminobutyric acid
- KO
- knockout
- MBP
- myelin basic protein
- MCT8
- monocarboxylate transporter 8
- P1
- postnatal day 1
- PC
- Purkinje cell
- PMA
- phorbol 12-myristate 13-acetate
- PV
- parvalbumin
- TA3
- 3,3′,5-triiodothyroacetic acid
- TH
- thyroid hormone
- TRβ
- TH receptor-β
- WT
- wild-type.
References
- 1. Friesema EC, Ganguly S, Abdalla A, et al. . Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–40135. [DOI] [PubMed] [Google Scholar]
- 2. Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20:2761–2772. [DOI] [PubMed] [Google Scholar]
- 3. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Friesema EC, Grueters A, Biebermann H, et al. . Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. [DOI] [PubMed] [Google Scholar]
- 5. Allan W, Herndon C, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Ment Defic. 1944;48:325–334. [Google Scholar]
- 6. Stevenson RE, Goodman HO, Schwartz CE, Simensen RJ, McLean WT Jr, Herndon CN. Allan-Herndon syndrome. I. Clinical studies. Am J Hum Genet. 1990;47:446–453. [PMC free article] [PubMed] [Google Scholar]
- 7. Bialer MG, Lawrence L, Stevenson RE, et al. . Allan-Herndon-Dudley syndrome: clinical and linkage studies on a second family. Am J Med Genet. 1992;43:491–497. [DOI] [PubMed] [Google Scholar]
- 8. Zorick TS, Kleimann S, Sertié A, Zatz M, Rosenberg S, Passos-Bueno MR. Fine mapping and clinical reevaluation of a Brazilian pedigree with a severe form of X-linked mental retardation associated with other neurological dysfunction. Am J Med Genet A. 2004;127A:321–323. [DOI] [PubMed] [Google Scholar]
- 9. Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252:663–666. [DOI] [PubMed] [Google Scholar]
- 10. Friesema EC, Visser WE, Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol. 2010;322:107–113. [DOI] [PubMed] [Google Scholar]
- 11. Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005;153:359–366. [DOI] [PubMed] [Google Scholar]
- 12. Zung A, Visser TJ, Uitterlinden AG, Rivadeneira F, Friesema EC. A child with a deletion in the monocarboxylate transporter 8 gene: 7-year follow-up and effects of thyroid hormone treatment. Eur J Endocrinol. 2011;165:823–830. [DOI] [PubMed] [Google Scholar]
- 13. Visser WE, Vrijmoeth P, Visser FE, Arts WF, van Toor H, Visser TJ. Identification, functional analysis, prevalence and treatment of monocarboxylate transporter 8 (MCT8) mutations in a cohort of adult patients with mental retardation. Clin Endocrinol (Oxf). 2013;78:310–315. [DOI] [PubMed] [Google Scholar]
- 14. Wémeau JL, Pigeyre M, Proust-Lemoine E, et al. . Beneficial effects of propylthiouracil plus l-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008;93:2084–2088. [DOI] [PubMed] [Google Scholar]
- 15. Bernal J. Thyroid hormones and brain development. Vitam Horm. 2005;71:95–122. [DOI] [PubMed] [Google Scholar]
- 16. Ceballos A, Belinchon MM, Sanchez-Mendoza E, et al. . Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3′-triiodo-l-thyronine. Endocrinology. 2009;150:2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006;147:4036–4043. [DOI] [PubMed] [Google Scholar]
- 18. Trajkovic M, Visser TJ, Mittag J, et al. . Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jansen J, Friesema EC, Kester MH, et al. . Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007;92:2378–2381. [DOI] [PubMed] [Google Scholar]
- 20. Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology. 2008;149:2184–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Visser WE, Jansen J, Friesema EC, et al. . Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat. 2009;30:29–38. [DOI] [PubMed] [Google Scholar]
- 22. Di Cosmo C, Liao XH, Dumitrescu AM, Weiss RE, Refetoff S. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology. 2009;150:4450–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verge CF, Konrad D, Cohen M, et al. . Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. J Clin Endocrinol Metab. 2012;97:4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mueller-Gaertner HW, Schneider C. 3,5,3′-Triiodothyroacetic acid minimizes the pituitary thyrotrophin secretion in patients on levo-thyroxine therapy after ablative therapy for differentiated thyroid carcinoma. Clin Endocrinol (Oxf). 1988;28:345–351. [DOI] [PubMed] [Google Scholar]
- 25. Anzai R, Adachi M, Sho N, Muroya K, Asakura Y, Onigata K. Long-term 3,5,3′-triiodothyroacetic acid therapy in a child with hyperthyroidism caused by thyroid hormone resistance: pharmacological study and therapeutic recommendations. Thyroid. 2012;22:1069–1075. [DOI] [PubMed] [Google Scholar]
- 26. Radetti G, Persani L, Molinaro G, et al. . Clinical and hormonal outcome after two years of triiodothyroacetic acid treatment in a child with thyroid hormone resistance. Thyroid. 1997;7:775–778. [DOI] [PubMed] [Google Scholar]
- 27. Messier N, Langlois MF. Triac regulation of transcription is T(3) receptor isoform- and response element-specific. Mol Cell Endocrinol. 2000;165:57–66. [DOI] [PubMed] [Google Scholar]
- 28. Klootwijk W, Friesema EC, Visser TJ. A nonselenoprotein from amphioxus deiodinates triac but not T3: is triac the primordial bioactive thyroid hormone? Endocrinology. 2011;152:3259–3267. [DOI] [PubMed] [Google Scholar]
- 29. Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008;22:1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buntinx M, Vanderlocht J, Hellings N, et al. . Characterization of three human oligodendroglial cell lines as a model to study oligodendrocyte injury: morphology and oligodendrocyte-specific gene expression. J Neurocytol. 2003;32:25–38. [DOI] [PubMed] [Google Scholar]
- 31. Kester MH, Kuiper GG, Versteeg R, Visser TJ. Regulation of type III iodothyronine deiodinase expression in human cell lines. Endocrinology. 2006;147:5845–5854. [DOI] [PubMed] [Google Scholar]
- 32. Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. 1998;19:87–90. [DOI] [PubMed] [Google Scholar]
- 33. Flamant F, Poguet AL, Plateroti M, et al. . Congenital hypothyroid Pax8(−/−) mutant mice can be rescued by inactivating the TRalpha gene. Mol Endocrinol. 2002;16:24–32. [DOI] [PubMed] [Google Scholar]
- 34. Mayerl S, Müller J, Bauer R, et al. . Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest. 2014;124:1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martínez de Arrieta C, Morte B, Coloma A, Bernal J. The human RC3 gene homolog, NRGN contains a thyroid hormone-responsive element located in the first intron. Endocrinology. 1999;140:335–343. [DOI] [PubMed] [Google Scholar]
- 37. Morte B, Iñiguez MA, Lorenzo PI, Bernal J. Thyroid hormone-regulated expression of RC3/neurogranin in the immortalized hypothalamic cell line GT1–7. J Neurochem. 1997;69:902–909. [DOI] [PubMed] [Google Scholar]
- 38. Horn S, Heuer H. Thyroid hormone action during brain development: more questions than answers. Mol Cell Endocrinol. 2010;315:19–26. [DOI] [PubMed] [Google Scholar]
- 39. Barradas PC, Vieira RS, De Freitas MS. Selective effect of hypothyroidism on expression of myelin markers during development. J Neurosci Res. 2001;66:254–261. [DOI] [PubMed] [Google Scholar]
- 40. Berbel P, Marco P, Cerezo JR, DeFelipe J. Distribution of parvalbumin immunoreactivity in the neocortex of hypothyroid adult rats. Neurosci Lett. 1996;204:65–68. [DOI] [PubMed] [Google Scholar]
- 41. Gilbert ME, Sui L, Walker MJ, et al. . Thyroid hormone insufficiency during brain development reduces parvalbumin immunoreactivity and inhibitory function in the hippocampus. Endocrinology. 2007;148:92–102. [DOI] [PubMed] [Google Scholar]
- 42. Everts ME, Visser TJ, Moerings EP, et al. . Uptake of triiodothyroacetic acid and its effect on thyrotropin secretion in cultured anterior pituitary cells. Endocrinology. 1994;135:2700–2707. [DOI] [PubMed] [Google Scholar]
- 43. Verhoeven FA, Van der Putten HH, Hennemann G, Lamers JM, Visser TJ, Everts ME. Uptake of triiodothyronine and triiodothyroacetic acid in neonatal rat cardiomyocytes: effects of metabolites and analogs. J Endocrinol. 2002;173:247–255. [DOI] [PubMed] [Google Scholar]
- 44. Horn S, Kersseboom S, Mayerl S, et al. . Tetrac can replace thyroid hormone during brain development in mouse mutants deficient in the thyroid hormone transporter mct8. Endocrinology. 2013;154:968–979. [DOI] [PubMed] [Google Scholar]
- 45. Anzai R, Adachi M, Sho N, Muroya K, Asakura Y, Onigata K. Long-term 3,5,3′-triiodothyroacetic acid therapy in a child with hyperthyroidism caused by thyroid hormone resistance: pharmacological study and therapeutic recommendations. Thyroid. 2012;22:1069–1075. [DOI] [PubMed] [Google Scholar]
- 46. Menegay C, Juge C, Burger AG. Pharmacokinetics of 3,5,3′-triiodothyroacetic acid and its effects on serum TSH levels. Acta Endocrinol (Copenh). 1989;121:651–658. [DOI] [PubMed] [Google Scholar]
- 47. Burger AG, Engler D, Sakoloff C, Staeheli V. The effects of tetraiodothyroacetic and triiodothyroacetic acids on thyroid function in euthyroid and hyperthyroid subjects. Acta Endocrinol (Copenh). 1979;92:455–467. [DOI] [PubMed] [Google Scholar]
- 48. Lerman J, Pitt-Rivers R. Physiologic activity of triiodo-and tetraiodo-thyroacetic acid in human myxedema. J Clin Endocrinol Metab. 1956;16:1470–1479. [DOI] [PubMed] [Google Scholar]
- 49. Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab. 2007;3:249–259. [DOI] [PubMed] [Google Scholar]
- 50. Gilbert ME, Lasley SM. Developmental thyroid hormone insufficiency and brain development: A role for brain-derived neurotrophic factor (BDNF)? Neuroscience. 2013;239:253–270. [DOI] [PubMed] [Google Scholar]