

Abstract
We have previously identified a natural occurring, androgen receptor-specific antagonist. Atraric acid (AA) inhibits the transactivation of the androgen receptor (AR) and androgen-mediated growth of AR-expressing human prostate cancer (PCa) cell lines. Here we show that AA treatment of living cells provokes molecular changes of AR signaling. In addition to a deceleration of nuclear translocation a block of the intramolecular amino/carboxy (N/C)-terminal interaction of the AR was observed. Furthermore, using high-resolution confocal fluorescence microscopy, a reduced speckle formation of the AR was observed in line with an increased intranuclear mobility of the receptor. This suggests decreased DNA binding of the AR, which is further indicated by an impaired chromatin recruitment of the AR to the prostate-specific antigen promoter and enhancer shown by chromatin immunoprecipitation experiments. Using inhibitors of the non-receptor tyrosine kinase Src or Akt, known interaction partners of AR, reduced the level of androgen-induced cellular senescence suggesting a partly non-genomic pathway to induce cellular senescence by AA. Using PP2 (4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) pyrimidine or Akt inhibitors, inhibitors of the nonreceptor tyrosine kinase Src or Akt, known interaction partners of AR, reduced the level of androgen-induced cellular senescence, suggesting a partly nongenomic pathway to induce cellular senescence by AA. Treatment of LNCaP cells with AA is associated with hypophosphorylation of the retinoblastoma tumor suppressor and an increase of p16 expression, whereas the p53-p21 signaling pathway seems not be affected by AA treatment. Analyzing human PCa tissue samples treated with AA ex vivo also indicates an induction of cellular senescence associated with an increase of p16 expression but not p21. Taken together, these data indicate that AA exhibits novel features to inhibit AR amino/carboxy-terminal interaction, the AR-mediated nuclear activities and growth of PCa cells.
The androgen receptor (AR) has a major role in normal prostate growth and differentiation but is also the main effector in the development as well as progression of prostate cancer (PCa) (1, 2). PCa as an important age-related disease is one of the most commonly diagnosed cancers and the second leading cause of cancer mortality of men in Western countries (3). Because the activated AR has an important role in the PCa cell proliferation and progression, AR inhibition by androgen ablation and treatment with AR antagonists (antiandrogens) is the main strategy of PCa hormone therapy (2, 4).
The AR as a ligand-dependent transcription factor is a member of the steroid hormone receptor family. In the absence of hormones, the AR is predominantly localized in the cytoplasm in a dynamic chaperone heterocomplex that stabilizes the receptor in a conformation receptive to hormone binding (5). The AR can be activated by AR agonists (androgens) such as T or dihydrotestosterone. Ligand binding induces dissociation of the chaperone complexes, conformational change of the AR and translocation to the nucleus in which the receptor binds to DNA target sequences. Through these androgen response elements (AREs), the AR regulates the transcription of its target genes (6). The AR consists of an amino (N)-terminal domain containing a potent transactivation function, followed by a DNA-binding domain and a carboxy (C)-terminal ligand-binding domain. Ligand-binding induces a conformational change and intra- as well as intermolecular AR N/C-terminal interactions, which play critical roles in both the nuclear shuttling and transcriptional capacity of the receptor (7–9). Furthermore, the AR can act also nongenomically with factors in the cytoplasm that promote cell proliferation involving the nonreceptor tyrosine kinase Src and Akt/protein kinase B (10, 11).
Initially, PCa cells grow androgen dependent but after 1–2 years of treatment, tumors eventually develop therapy resistance. One of several possibilities of therapy resistance is mutations of the human AR that render the used antagonists to potent AR agonists such as bicalutamide (BIC) or hydroxyflutamide. This offers novel opportunities to develop next-generation AR antagonists, which are able to inhibit therapy resistant PCa cells (12, 13).
The natural compound atraric acid (AA), which was isolated from extracts of bark material of the African tree Pygeum africanum, could be identified as a novel and to our knowledge as the first known natural AR antagonist. As a second-generation antiandrogen, AA is able to inhibit the proliferation of both androgen-dependent growing and castration-resistant prostate cancer cells (14, 15). AA competes with androgens for binding to AR and specifically inhibits AR transactivation. It was shown that this compound acts as a highly selective AR antagonist for both wild-type and mutated AR, like the AR T877A mutant that frequently occurs in advanced PCa after treatment with established compounds. In line with this, further results suggest that AA prevents PCa invasiveness and inhibits the expression of endogenous AR target genes like prostate-specific antigen (PSA) in PCa cells (15).
The aim of this study was the further investigation of the underlying molecular mechanisms of AA to act as novel AR antagonist. Understanding how AA inhibits the human AR will be beneficial for the development of new therapeutic strategies against androgen-dependent and castration-resistant PCa. Interestingly, in this study we could observe that AA decelerates the agonist-induced AR nuclear translocation and subsequently reduces DNA binding of AR, resulting in an increased intranuclear mobility. We hypothesize that this compound induces a conformation of the AR distinct from agonists, which is indicated by inhibition of the androgen-mediated intramolecular N/C-terminal interaction. Additionally, AA induces cellular senescence in vitro as well as ex vivo in human PCa cells and tissue. Our findings reveal insight into the molecular mechanisms of AR inhibition by the natural compound AA representing a promising novel AR antagonist.
Materials and Methods
Chemicals and hormones
AA was obtained from Sigma-Aldrich, bicalutamide from Interpharma, and methyltrienolone (R1881) from PerkinElmer. The Src kinase inhibitor, 4-Amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine (PP2), and the Akt kinase inhibitor, 1L6-hydroxymethyl-chiro-insoitol-2-(R)-2-O-methyl-3-O-octadecyl-sn-glycerocarbonat, were obtained from Calbiochem/Merck. All compounds were dissolved in dimethylsulfoxide or ethanol and were added to the culturing medium such that the final concentration of the solvent did not exceed 0.1%. Control incubations (no test compounds) were performed in the presence of only 0.1% dimethylsulfoxide.
Cell lines and culture
The androgen-dependent growing human prostate cancer cell line, LNCaP (lymph node prostate cancer), was cultured in RPMI 1640, and the medium was supplemented with 10% fetal calf serum (Invitrogen), penicillin (100 U/mL), streptomycin (100 U/mL), and 25 mM HEPES (pH 7.8). Hep3B cells stably expressing green fluorescent protein (GFP)-AR or yellow fluorescent protein (YFP)-AR-cyan fluorescent protein (CFP) were cultured in α-MEM supplemented with 5% fetal calf serum, 2 mM L-glutamine, penicillin (100 U/mL), streptomycin (100 U/mL), and G418. Prior to the microscopy analyses, Hep3B cells were cultured for at least 24 hours in medium containing 5% hormone-depleted charcoal-stripped serum (CSS). All cells were cultured in a 5% CO2-95% air, humidified atmosphere at 37°C.
Prostate tissue samples
Specimens were kindly provided by the Institutes of Urology and Pathology (Jena University Hospital, Jena, Germany). Ethical approval was granted (3286–11/11). Gleason scores were between 7 and 9, and the presurgery PSA values were between 4 and 16. Human prostate tissue samples obtained after the prostatectomy of patients were cultured at 5% CO2 and 37°C for 48 hours with the indicated compounds in RPMI 1640. The medium was supplemented with 10% CSS, penicillin (100 U/mL), streptomycin (100 U/mL), and 25 mM HEPES (pH 7.8). Tissue samples were fixed in tissue freezing medium (Sakura) in liquid N2 and using a cryostat sections of 10 μm thickness were prepared for subsequent staining.
Senescence-associated β-galactosidase (SA β-gal) staining
The staining was performed essentially as described earlier (16). Cells or cryosections of PCa tissue were fixed for 5 minutes in 2% glutaraldehyde, washed with PBS, and incubated at 37°C (no CO2) with fresh SA β-gal staining solution [40 mM citric acid/sodium phosphate buffer (pH 6.0) containing 1 mg X-galactosidase (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside)/ml, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2]. The staining solution contains X-galactosidase, a galactopyranosid, which is converted by an active galactosidase into a blue colorant that can be detected in a light microscope after an overnight incubation (16). Three × 200 cells per treatment were counted and the average of the triplets is shown in diagrams.
Senescence-associated heterochromatic foci (SAHF) detection by 4′,6-diamidino-2-phenylindol (DAPI) staining
Cells were washed with PBS and collected by trypsinization. DNA was visualized by DAPI at 1 μg/mL after dropping cells on glass slides and dehydration. Pictures were taken by a fluorescence microscope (17).
Quantitative RT-PCR (qRT-PCR)
RNA was isolated using peqGOLD TriFast (Peqlab) according to the manufacturer's protocol. RNA was used in a one-step qRT-PCR reaction using the SuperScript III Platinum SYBR Green One-Step qRT-PCR kit (Invitrogen) with the following primer sequences (indicated as 5′ → 3′): β-actin, forward, ACAGAGCCTCGCCTTTGCCGA, reverse, CACGATGGAGGGGAAGACG; glyceraldehyde-3-phosphate dehydrogenase, forward, GTGAACCATGAGAAGTATGACAAC, reverse, GAGTCCTTCCACGATACC; cyclin D1, forward, TCAACCTAAGTTCGGTTCCGATG, reverse, GTCAGCCTCCACACTCTTGC; p21, forward, TCGACTTTGTCACCGAGACACCAC, reverse, CAGGTCCACATGGTCTTCCTCTG; E2F1, forward, TGAGACCTCACTGAATCTGACCACC, reverse, CCAGCTCATTGCCAAGAAGTCCAA; p16, forward, CTTGCCTGGAAAGATACCG, reverse, CCCTCCTCTTTCTTCCTCC; and inhibitor of DNA binding 1 (Id1), forward, GGTAAACGTGCTGCTCTACGACATG, reverse, CTCCAGCACGTCATCGACTACATC.
The expression of the housekeeping genes used for normalization is not affected by the treatments.
Protein immunodetection
LNCaP cells were incubated with the indicated compounds for 3 days. Western blot analysis was performed by using the following primary antibodies: AR (BioGenex Laboratories); α-tubulin, β-actin, and pRb (Abcam); p21, p53, acetylated p53 (Lys 379), p16, cyclin D1, and phosphorylated pRb (Ser 807/811; Cell Signaling); and E2F1 (Santa Cruz Biotechnology). As secondary antibody, an antimouse or antirabbit IgG horseradish peroxidase-linked antibody (Santa Cruz Biotechnology) was used and the enhanced chemiluminescence detection method (Amersham Pharmacia Biotech).
Fluorescence-activated cell sorter (FACS) analysis
LNCaP cells were seeded as duplicates in RPMI 1640 with 10% confluence. After 24 hours the cells were treated with different compounds for 3 days. For the FACS analysis, cells were trypsinized, washed two times with PBS, and then fixed for 3 hours with 70% ethanol at −20°C. After that, the cells were incubate for 45 minutes at 4°C while rotating with staining solution (2.5 μg/mL propidium iodide, 0.1 mg/mL ribonuclease A, and 0.05% Triton X-100). Ten thousand cells were analyzed with a CyFlow ML cytometer (Partec), and the cell cycle phases were determined by Cylchred. The percentage of cells in the cell cycle phases is indicated.
Small interfering RNA (siRNA) transfection
For knockdown experiments, stealth RNA interference (Invitrogen) for p16, p21, and Id1 were used. Transient transfections with 10–20 nM siRNA were carried out by electroporation. Scrambled siRNA and one sample without any siRNA, which works as negative control, were applied. The efficiencies of the knockdowns were detected via qRT-PCR to analyze the expression of the specific gene. Furthermore, the transfected cells were treated with different compounds, and the induction of cellular senescence was analyzed.
Chromatin immunoprecipitation (ChIP)
ChIP experiments using LNCaP cells were essentially performed as described earlier (15) with minor changes (Supplemental Data). The following primers were used (indicated as 5′ → 3′): PSA promoter (ARE I), forward, TCTGCCTTTGTCCCCTAGAT, reverse, AACCTTCATTCCCCAGGACT; and PSA enhancer (ARE III), forward, GCAAGCCTGGATCTGAGAGAG, reverse, ACAGCAACACCTTTTTTTTTCTGGATTG.
Fluorescence recovery after photobleaching (FRAP) assays and acceptor photobleaching fluorescence resonance energy transfer (FRET)
FRAP and FRET assays were performed as previously described (17, 18).
Confocal microscopy and live-cell time-laps imaging were performed using a confocal laser-scanning microscope (LSM510; Carl Zeiss MicroImaging, Inc) equipped with a Plan-Neofluar ×40/1.3 NA oil objective (Carl Zeiss MicroImaging, Inc) as described previously (18). Enhanced GFP was excited using a 488 nm argon laser line at moderate laser power, and emission was detected using a 505 to 530 nm bandpass emission filter to obtain images for subcellular localization analysis. Fluorescence intensities were measured in a region of the nucleus and a region just outside the nucleus of the cells, and the relative nuclear intensity was determined using the following equation: relative nuclear intensity (I) = [(Inucleus − Ibackground)/(Icytoplasm − Ibackground) + (Inucleus − Ibackground)].
Results
AA decelerates AR nuclear translocation
Previous results suggest an influence of AA on the translocation of the AR from the cytoplasm to the nucleus (15). To gain a better insight into this process, confocal fluorescence microscopy and time-lapse analysis of AR-negative Hep3B cells stably expressing GFP-tagged AR were applied. Herein the effects of AA on AR nuclear translocation were investigated in comparison with the agonist R1881 and cotreatment of agonist and antagonist in living cells (Figure 1). These experiments indicate that compared with androgens, the cotreatment with AA inhibits the androgen-induced translocation of AR both in a slower and incomplete manner. Androgen treatment leads to a rapid translocation and approximately 90% nuclear localization of the AR within 30 minutes of treatment (Figure 1, A and B). However, after AA treatment the maximal translocation reaches only 50%–60% within 4 hours after the ligand addition (Figure 1B). Cotreatment of the cells with R1881 and AA at the same time induces an intermediate state (Figure 1, A and B). In contrast to androgen alone, the translocation is decelerated and reaches a maximal nuclear localization of 70%–80% after 8–9 hours (Figure 1B). Furthermore, the amount of AR translocated to the nucleus remains constant within at least 24 hours of treatment (data not shown). Thus, the data suggest that in addition to a decelerated nuclear translocation of the AR, AA treatment inhibits the amount of androgen-induced translocated AR, leading to a higher level of cytoplasmatic AR.
Figure 1. AA decelerates AR nuclear translocation in living cells: involvement of HSP90.
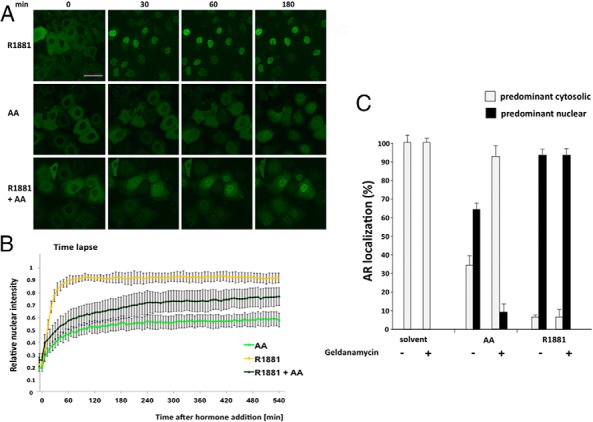
A, Confocal images of Hep3B cells stably expressing GFP-AR at different time points after treatment with R1881 (10−9 M), AA (10−4 M), or cotreatment. Scale, 50 μm. B, Live-cell time lapse imaging of Hep3B cells and quantification of GFP-AR nuclear translocation after addition of the indicated compounds. Calculation of the relative nuclear intensity (I) = [(Inucleus − Ibackground)/(Icytoplasm − Ibackground) + (Inucleus − Ibackground)]. The average of two experiments is shown, and the error bars indicate the variation of the mean of at least 25 cells per time point. C, The HSP90 inhibitor geldanamycin fosters the cytosolic localization of AR in the presence of AA. HeLa cells transfected with GFP-AR were treated for 2 hours with solvent, R1881 (10−10 M), or AA (3 × 10−5 M). Subsequently the HSP90 inhibitor geldanamycin (1 μg/mL) was added for 1 hour, and the intracellular localization of the AR was determined in 2 × 100 cells for each experiment. The deviation of the mean is shown.
Heat shock protein 90 (HSP90) is known to be associated with the AR and to regulate cytosolic retention (19). To reveal whether HSP90 is involved in the cytosolic AA-mediated localization of AR, we used the HSP90 inhibitor geldanamycin, which binds to the ATP binding site of HSP90. Interestingly, the results reveal that the androgen-induced translocation of the AR is not affected by geldanamycin, whereas more AR is located in the cytoplasm with AA treatment similar to cotreatment of agonist with AA (Figure 1C). This suggests that AA-bound AR is associated with HSP90, possibly due to a different AR conformation than induced by agonist.
AA prevents AR N/C-terminal interaction and reduces the agonist-induced intranuclear foci formation and immobilization of AR
A strong correlation between the agonist-induced AR N/C-terminal interaction, DNA binding, and full transcriptional activity of the receptor has been described (20). Therefore, the effect on both the AR N/C-terminal interaction and intranuclear mobility were investigated. The influence of AA on AR N/C-terminal interaction and AR conformation was addressed via FRET measurements of AR tagged with YFP and CFP at the N terminus and C terminus, respectively. The results reveal that AA prevents androgen-mediated relative FRET efficiency (Figure 2A). This indicates that AA induces a different conformation that does not enable AR N/C-terminal interaction, similar to the results observed with the known antagonist BIC (Figure 2A). In addition, it could be shown that this effect is concentration dependent (Supplemental Figure 1).
Figure 2. AA prevents AR N/C-terminal interaction and reduces the agonist-induced intranuclear foci formation and inhibits immobilization of AR.

Hep3B cells stably expressing tagged AR were cultured in CSS medium for 24 hours with R1881 (10−9 M), AA (10−4 M), or BIC (10−6 M) or cotreatment of the antagonists with the androgen R1881. A, FRET assay measurements indicate intramolecular AR N/C interaction inhibited by treatment with AA. The apparent FRET efficiency of YFP-AR-CFP is calculated as the fraction of CFP increase after YFP bleaching (acceptor photobleaching FRET). For normalization cells were transiently transfected with a plasmid for a CFP-YFP fusion construct (equal to 1) and cells cotransfected with CFP and YFP (equal to 0). Values for the treated samples represent the average ± SEM of at least 40 cells measured in three independent experiments. B, High-resolution confocal images of representative nuclei of Hep3B cells expressing GFP-AR at physiological levels after treatment with the indicated compounds. Scale, 5 μm. C, FRAP assays indicate the intranuclear mobility of GFP-AR. In strip-FRAP measurements, fluorescent molecules in a narrow strip spanning the nucleus were bleached for 100 msec at the maximum laser power. Subsequently, fluorescence in the strip was monitored every 100 msec. Fluorescence intensities were plotted against time and normalized to the prebleach value. The mean values of at least 40 cells measured in three independent experiments are plotted. D, AA reduces AR chromatin recruitment to AREs in the PSA promoter and enhancer of human PCa LNCaP cells. ChIP experiments were applied to detect the recruitment of AR at the endogenous PSA promoter and enhancer region in LNCaP cells. Cells were cultured in 10% CSS medium for 3 days and incubated with R1881 (10−10 M) or AA (3 × 10−5 M) or cotreated for 2 hours. The experiment was performed with an AR-specific antibody as well as IgG or without antibody (only beads) as control.
Agonist treatment leads to translocation as well as intranuclear foci formation and speckled distribution of the AR within the nucleus, which is accompanied by reduced mobility, presumably due to transient DNA binding and the subsequent immobilization of the AR (18, 21). In contrast, in the presence of AA, a homogeneous distribution of AR was observed using high-resolution confocal microscopy (Figure 2B). Moreover, AA treatment or cotreatment with the agonist leads to reduced foci formation compared to incubation with R1881 alone (Figure 2B).
A reduced intranuclear foci formation and speckles by AA treatment suggests a reduced AR location at chromatin sites. Therefore, to analyze this further, the FRAP technique was used. In line with the previous results, the FRAP data confirm that AA increases the intranuclear mobility of the AR compared with the transient immobilization by R1881 (Figure 2C). Each of the antagonists, AA as well as BIC alone, lead to a fast redistribution of fluorescence after the bleach pulse, whereas the slower recovery in the presence of androgens indicates transient immobilization, which is suggested to be due to DNA/chromatin binding of the AR. Cotreatment of cells with androgen and either of the antagonists induces an intermediate state and increased mobility (Figure 2C). Similar to the FRET measurement results, this effect is concentration dependent (Supplemental Figures 2 and 3).
Taken together, these data suggest that AA reduces R1881-mediated DNA/chromatin binding, leading to increased nuclear mobility and reduced speckled distribution of AR within the nucleus, therefore contributing to the inhibitory effect on AR function.
To verify the effect of AA on agonist-mediated AR chromatin recruitment, ChIP experiments in the human PCa LNCaP cells were performed to detect the recruitment of the AR to AREs in the promoter and the enhancer of the PSA gene (Figure 2D). Solvent treatment leads to only a weak association of the AR with the PSA enhancer and promoter, whereas AA alone even decreases this. R1881 induces recruitment of the receptor to the investigated AREs and cotreatment of androgens with AA reduces the recruitment of the AR (Figure 2D). Thus, the ChIP data are in accordance with the FRAP results that indicate a reduced chromatin association of the AR due to AA treatment.
AA induces cellular senescence in PCa cells
Cellular senescence displays an irreversible arrest in the G1 phase of the cell cycle and provides a mechanism to inhibit tumor cell growth (22). To investigate whether AA-mediated growth inhibition may be explained by the induction of cellular senescence, androgen-dependent growing LNCaP cells were treated with AA for 72 hours. As markers for the induction of cellular senescence, both staining of the SA β-gal activity and the detection of SAHF via DAPI staining were used (16, 23).
Treatment with AA leads to the induction of SA β-gal activity, indicating the induction of cellular senescence (Figure 3A). These findings are further confirmed by the occurrence of SAHF (Figure 3B). Quantitative analyses revealed that AA induces cellular senescence in LNCaP cells in a concentration-dependent manner, reaching a maximum at 30 μM (Figure 3C). Interestingly, treatment of the cells for 3 days was sufficient for the induction of cellular senescence; a longer incubation with AA did not increase the percentage of SA β-gal-positive cells (Supplemental Figure 4). Furthermore, FACS analyses revealed an increase of the number of cells in the G1 phase of the cell cycle after AA treatment (Supplemental Figure S5). Moreover, the observed proliferation arrest and senescent phenotype seems irreversible because the depletion of AA did not allow the regrowth of cells (Supplemental Figure 6).
Figure 3. The natural AR antagonist AA induces cellular senescence in PCa cells.
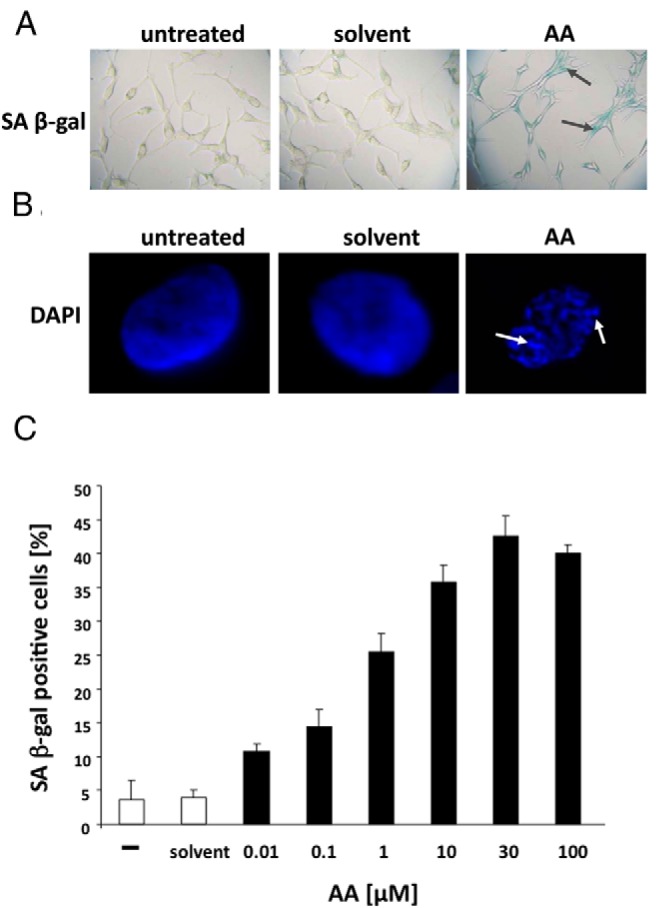
Androgen-dependent growing LNCaP cells were treated with solvent control or the AR antagonist AA (3 × 10−5 M) for 72 hours. A, Phase-contrast microscope pictures of the staining for SA β-gal activity were taken at ×200 magnification. B, Fluorescence microscopy pictures at ×1000 magnification for the detection of SAHF via DAPI staining. SAHFs, as the marker of the induction of cellular senescence, are indicated by arrows. C, Quantification of SA β-gal-positive cells treated with the indicated concentrations. The 3 × 200 cells were counted and plotted as a percentage of positive cells. The error bars show the variation of the mean of triplets.
Thus, these data strongly indicate that AA leads to proliferation arrest in human PCa cells via the induction of cellular senescence.
The Src-Akt pathway is involved in AA-mediated cellular senescence in PCa cell lines
Our results indicate that AA treatment enhances the level of cytosolic localized AR. This suggests that the AA-mediated induction of cellular senescence may be mediated in part through cytosolic AR-interacting factors. Therefore, inhibitors of Src and Akt were used to analyze whether these factors are involved in AA-induced cellular senescence in LNCaP cells (Figure 4, A and B). Interestingly, cotreatment of the Src inhibitor PP2 with AA reduces the level of cellular senescence relative to single treatment with AA (Figure 4A). Similarly, cotreatment of the Akt inhibitor with AA also reduces the level of cellular senescence (Figure 4B), indicating that this nongenomic pathway is involved in the AA-mediated induction of cellular senescence.
Figure 4. Inhibitors of Src and Akt reduce the level of AA-induced cellular senescence.

LNCaP were treated in a similar experimental setup as in Figure 1 with AA (3 × 10−5 M) and cotreated with the indicated inhibitors for 72 hours. Subsequently, SA β-gal staining was performed, and the 3 × 200 cells were counted and plotted as a percentage of positive cells. A, The Src tyrosine kinase inhibitor PP2 was used at 1 μM final concentration. B, The Akt inhibitor (Akti) was used at 1 μM final concentration.
AA-induced cellular senescence in PCa cells is mediated via the Id1-p16-pRb signaling pathway
Two main pathways for the induction of cellular senescence have been described. Activation of the p53-p21 pathway and/or the p16-pRb pathway is frequently involved, either with a combined role or as separate events (24). To gain further insight into the mechanism of AA-mediated induction of cellular senescence, the gene expression from key regulators of both pathways were analyzed (Figure 5A). An induction of p16 but not p21 mRNA was observed after treatment with AA. The mRNA of cyclin D1, a well-known cell cycle factor that is regulated by AR (25), is down-regulated by AA treatment in a similar manner as p21 and Id1 mRNA (Figure 5A).
Figure 5. Treatment with AA modifies the expression of cell cycle regulators.

LNCaP cells were treated for 72 hours with a solvent control or 3 × 10−5 M AA to analyze cell cycle-regulating pathways. A, The mRNA expression of genes of interest was analyzed by qRT-PCR using specific primers for Id1, p21, p16, cyclin D1, and β-actin for normalization. Values of the untreated samples were set as 1. Error bars indicate the SEM of doublets. B, Protein levels were detected via Western blot and quantitatively analyzed using α-tubulin as a loading control. The quantified values are shown below the detected bands and are indicated as fold change compared with the untreated samples, which were set arbitrarily as 1.
Western blot analyses indicate that the p53-p21 signaling pathway is mostly unaffected by AA treatment (Figure 5B). Comparing solvent control with AA treatment, the levels of p53 protein, as an activator of p21, remain unchanged, and an increased acetylation, known to stabilize p53 in senescent cells, could not be observed. On the other hand a dephosphorylation of pRb was detected. Reduced phosphorylation of pRb leads to its activation and hence down-regulation of the transcription factor E2F1 (23). In line with this, a down-regulation of the pRB target E2F1 associated with the up-regulation of p16 are observed at protein level by AA treatment (Figure 5B). Additionally, this is confirmed by the down-regulation of cyclin D1, an inhibitor of pRb. These data indicate that the p16-pRb signaling is addressed by AA treatment, suggesting this to be one underlying mechanism of AA-mediated induction of cellular senescence.
To analyze whether the p16 pathway mediates AA-induced cellular senescence, transient knockdown of p16 and p21 via siRNA transfection were used, and subsequently SA β-gal activity was analyzed (Figure 6). The knockdown was verified by qRT-PCR (Supplemental Figure 7). The data suggest that although p21 has no influence on the AA-mediated induction of cellular senescence compared with control samples, the knockdown of p16 reduces the level of cellular senescence (Figure 6). This indicates that the p21 pathway does not play a major role in the induction of AA-mediated cellular senescence; however, p16 seems to be involved in AA-mediated induction of cellular senescence.
Figure 6. Transient knockdown of cell cycle regulators influence the level of AA-induced cellular senescence.

Transient knockdown of Id1, p16, or p21 were performed via specific siRNA transfection 24 hours prior to AA (3 × 10−5 M) treatment and subsequent SA β-gal staining in LNCaP cells. The 4 × 200 cells were analyzed 3 days after siRNA transfection and treatment in the absence or presence of AA and plotted as a percentage of SA β-gal-positive cells. Scrambled siRNA served as control. Error bars indicate the SD of the mean of triplets.
An inhibitor of p16 gene expression is the cell cycle regulator Id1 via the repression of transcription factors of the E-twenty six family (26). In line with this, a reduction of Id1 gene expression after AA administration was observed (Figure 5A). Additionally, a down-regulation of Id1 expression in LNCaP cells leads to an increase of SA β-gal-positive cells (Figure 6), suggesting that Id1 inhibition is required for the activation of p16 and subsequent induction of cellular senescence triggered by AA treatment.
Thus, the data suggest that the Id1-p16-pRb signaling pathway is involved in the AA-mediated cellular senescence in LNCaP cells.
AA induces cellular senescence in human PCa tissue ex vivo
Because it is known that cellular senescence acts as a natural tumor suppressive mechanism in premalignant tumors and can be induced through different compounds (22), we investigated whether this is also applicable in primary human PCa tissue. The tissue samples obtained from different patients after prostatectomy were cultured with or without AA for 48 hours. Subsequently SA β-gal staining and mRNA expression analysis of the cyclin-dependent kinase inhibitors p16 and p21 were performed. Prostatectomies with nontumor tissues were also derived from different patients and served as control.
Interestingly, AA treatment of human PCa tissue samples ex vivo leads to the induction of SA β-gal activity, visible as increased blue staining in the tissue sections (Figure 7A), which was not observed in this extent in the nontumor tissue used as control (Figure 7A). Quantitative mRNA analysis of the two pathways of cellular senescence revealed an up-regulation of p16 but not of p21 (Figure 7B). The up-regulation of the cell cycle inhibitor p16 ex vivo is in line with the obtained data of LNCaP cells. Hence, AA induces cellular senescence in PCa cells as well as in tumor tissue via the p16 pathway.
Figure 7. AA induces cellular senescence in human PCa tissues ex vivo.

Human PCa tissue (n = 6) and control/nontumor tissue (n = 3) specimens derived from prostatectomy of different patients (∼2 × 2 mm) were treated postoperatively for 48 hours with 10−4 M AA. A, Representative phase-contrast microscope pictures of SA β-gal staining of cryosections (10 μM thickness) of PCa tumor tissue or nontumor tissue. Scale, 300 μm. qRT-PCR was performed for p16 mRNA (B) and p21 mRNA (C). For internal normalization the mRNA expression of β-actin was determined. Box plot of relative p16 and p21 mRNA expression was used to compare the different treatment groups. Boxes show the lower and upper interquartile. Horizontal lines indicate the median and whiskers show a maximum of 1.5 interquartile range. Outliers were excluded.
Taken together, these data suggest that AA induces cellular senescence in human primary tissues ex vivo as well as in LNCaP cells in vitro. Functional analyses in LNCaP cells suggest that the AA-mediated induction of cellular senescence is in part mediated by the p16-pRb pathway. Mechanistically, AA blocks the N/C-terminal interaction of the AR and reduces AR translocation. AR molecules that translocate into the nucleus exhibit higher mobility and less association with DNA and chromatin. These results provide further insights into the mechanism of AR inhibition by the natural compound AA.
Discussion
AA as a natural AR antagonist inhibits the AR-mediated transactivation and proliferation of AR-expressing PCa cell lines. Furthermore, to our knowledge it is the first natural compound described to induce cellular senescence in human PCa cells and tissue specimens ex vivo. Because cellular senescence is a program that arrests cells irreversibly in the G0 phase of the cell cycle, it is an interesting tool to inhibit cancer cell proliferation (22).
Cellular senescence is mediated through exogenous and endogenous stimuli, which cause changes in cell morphology and gene expression profiles (27). Various signaling pathways have been described in oncogene-induced cellular senescence involving the p53-p21 and/or the p16-pRb pathway. Recent publications suggest that cellular senescence occurs naturally during vertebrate embryogenesis as a normal program that plays instructive roles and regulates patterning in development (28, 29). The program of cell cycle arrest mediated through cellular senescence seems to be bypassed in tumor cells (22, 27), and therefore, the exogenous reactivation of the cellular senescence program in malignant cells may represent a potential cancer therapy.
Interestingly, the AR can mediate the induction of cellular senescence in PCa tumor cells. We suggest, based on the observations herein, that AA induces cellular senescence in human PCa cells. This may be an important mechanism of PCa cell growth inhibition mediated by AR antagonists. We observed the AA-mediated induction of cellular senescence in specimens of human PCa tissue as well as in LNCaP cells. These results also indicate that LNCaP cells may be a useful model to analyze the underlying molecular mechanisms of cellular senescence. Our data suggest that the AA-mediated cellular senescence involves the activation of the p16 and pRb signaling pathway, and thereby a hypophosphorylation of pRb occurs.
Moreover, our data suggest that AA administration blocks the N/C-terminal interaction of the AR and reduces the AR translocation into the nucleus in living cells. The block of the N/C-terminal interaction by known antagonists is strongly associated with an inhibition of DNA binding and transcriptional activity of the receptor (20). This is in line with our observations using FRAP assays that indicate a reduced immobilization and a higher mobility of the AR in the presence of AA-bound AR compared with androgens. The data suggest that the AR has a reduced recruitment to chromatin and DNA in the presence of AA. In addition, less AR translocates into the nucleus in the presence of AA, indicating a retention or an increased export of AR into the cytoplasm. This may involve HSP90 because we observed that inhibition of the ATP binding site of HSP90 leads to an increased AR level in the cytoplasm in the presence of AA.
The higher AR levels in the cytoplasm prompted us to analyze known cytoplasmatic signaling pathways in the AA-mediated cellular senescence. Indeed, using inhibitors of Src and Akt, known to interact with the AR (11, 12), the level of cellular senescence is reduced. However, the link between Akt and p16 expression remains unclear. Interestingly, a recent publication also linked p16 expression and Akt activity using several animal and cell models with modulated PTEN (phosphatase and tensin homolog deleted from chromosome 10) expression, which regulates the Akt activity (30). The authors postulate a block of cell cycle reentry through a novel pathway, leading to an increase in p16. This may indicate a novel role of Akt in mediating cellular senescence by induction of the p16-pRb pathway.
Conclusions about a long-term treatment cannot be drawn at this point. Using androgen deprivation or AR antagonist treatment to inactivate the AR may cause resistance to therapy and lead to refractory prostate cancer (4, 31). However, it would be beneficial to analyze pathways to inhibit PCa cell cycle progression to be able to use a combinatorial treatment for improved and long-term inhibition of PCa proliferation.
Thus, the data show that AA inhibits AR function through a reduced nuclear translocation, the inhibition of the N/C-terminal interaction, and accordingly a higher nuclear mobility of the AR associated with lower chromatin/DNA binding. The data further suggest that the AA-mediated induction of cellular senescence is effected by the p16/pRb involving the Src/Akt pathway that may be linked by a yet unknown mechanism. Taken together, these observations provide a molecular pathway of the natural AR antagonist AA, which may be promising in tumor growth inhibition.
Acknowledgments
We are grateful to Dipl. Biochem. Florian Kraft for technical help using FACS.
This work was supported by the German Cancer Aid (to A.B.) and the Boehringer Ingelheim Fonds (to W.H.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the German Cancer Aid (to A.B.) and the Boehringer Ingelheim Fonds (to W.H.).
Footnotes
- AA
- atraric acid
- AR
- androgen receptor
- ARE
- androgen response element
- BIC
- bicalutamide
- C
- carboxy-terminal interaction
- CFP
- cyan fluorescent protein
- ChIP
- chromatin immunoprecipitation
- CSS
- charcoal-stripped serum
- DAPI
- 4′,6-diamidino-2-phenylindol
- FACS
- fluorescence-activated cell sorter
- FRAP
- fluorescence recovery after photobleaching
- FRET
- fluorescence resonance energy transfer
- GFP
- green fluorescent protein
- HSP90
- heat shock protein 90
- Id1
- inhibitor of DNA binding 1
- N
- amino terminal interaction
- PCa
- prostate cancer
- PP2
- 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- PSA
- prostate-specific antigen
- qRT-PCR
- quantitative RT-PCR
- R1881
- methyltrienolone, a nonmetabolized androgen
- SA β-gal
- senescence-associated β-galactosidase
- SAHF
- senescence-associated heterochromatic foci
- siRNA
- small interfering RNA
- YFP
- yellow fluorescent protein.
References
- 1. Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nuclear Receptor Signaling. 2008;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21(12):2855–2863. [DOI] [PubMed] [Google Scholar]
- 3. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. [DOI] [PubMed] [Google Scholar]
- 4. Brinkmann AO, Trapman J. Prostate cancer schemes for androgen escape. Nat Med. 2000;6(6):628–629. [DOI] [PubMed] [Google Scholar]
- 5. Heinlein CA, Chang C. Role of chaperones in nuclear translocation and transactivation of steroid receptors. Endocrine. 2001;14(2):143–149. [DOI] [PubMed] [Google Scholar]
- 6. Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–610. [DOI] [PubMed] [Google Scholar]
- 7. Jones JO, Frank AW, Diamond MI. AR inhibitors identified by high-throughput microscopy detection of conformational change and subcellular localization. ACS Chem Biol. 2009;4(3):199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Royen ME, Cunha SM, Brink MC, et al. . Compartmentalization of androgen receptor protein-protein interactions in living cells. J Cell Biol. 2007;177(1):63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Royen ME, van Cappellen WA, de Vos C, Houtsmuller AB, Trapman J. Stepwise androgen receptor dimerization. J Cell Sci. 2012;125:1970–1979. [DOI] [PubMed] [Google Scholar]
- 10. Migliaccio A, Di Domenico M, Castoria G, et al. . Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res. 15 2005;65(22):10585–10593. [DOI] [PubMed] [Google Scholar]
- 11. Thomas C, Lamoureux F, Crafter C, et al. . Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2013;12(11):2342–2355. [DOI] [PubMed] [Google Scholar]
- 12. Yu J, Akishita M, Eto M, et al. . Src kinase-mediates androgen receptor-dependent non-genomic activation of signaling cascade leading to endothelial nitric oxide synthase. Biochem Biophys Res Commun. 2012;424(3):538–543. [DOI] [PubMed] [Google Scholar]
- 13. Tran C, Ouk S, Clegg NJ, et al. . Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schleich S, Papaioannou M, Baniahmad A, Matusch R. Extracts from Pygeum africanum and other ethnobotanical species with antiandrogenic activity. Planta Med. 2006;72(9):807–813. [DOI] [PubMed] [Google Scholar]
- 15. Papaioannou M, Schleich S, Prade I, et al. . The natural compound atraric acid is an antagonist of the human androgen receptor inhibiting cellular invasiveness and prostate cancer cell growth. J Cell Mol Med. 2009;13(8B):2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dimri GP, Lee X, Basile G, et al. . A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lorenz V, Hessenkemper W, Rödiger J, Kyrylenko S, Kraft F, Baniahmad A. Sodium butyrate induces cellular senescence in neuroblastoma and prostate cancer cells. Horm Mol Biol Clin Invest. 2011;7(1):265–272. [DOI] [PubMed] [Google Scholar]
- 18. Farla P, Hersmus R, Trapman J, Houtsmuller AB. Antiandrogens prevent stable DNA-binding of the androgen receptor. J Cell Sci. 2005;118(Pt 18):4187–4198. [DOI] [PubMed] [Google Scholar]
- 19. Hessenkemper W, Baniahmad A. Targeting heat shock proteins in prostate cancer. Curr Med Chem. 2013;20(22):2731–2740. [DOI] [PubMed] [Google Scholar]
- 20. van Royen ME, van de Wijngaart DJ, Cunha SM, Trapman J, Houtsmuller AB. A multi-parameter imaging assay identifies different stages of ligand-induced androgen receptor activation. Cytometry A. 2013;83(9):806–817. [DOI] [PubMed] [Google Scholar]
- 21. Marcelli M, Stenoien DL, Szafran AT, et al. . Quantifying effects of ligands on androgen receptor nuclear translocation, intranuclear dynamics, and solubility. J Cell Biochem. 2006;98:770–788. [DOI] [PubMed] [Google Scholar]
- 22. Collado M, Gil J, Efeyan A, et al. . Senescence in premalignant tumours. Nature. 2005;436:642. [DOI] [PubMed] [Google Scholar]
- 23. Narita M, Nũnez S, Heard E, et al. . Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2013;113(6):703–716. [DOI] [PubMed] [Google Scholar]
- 24. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. [DOI] [PubMed] [Google Scholar]
- 25. Grosse A, Bartsch S, Baniahmad A. Androgen receptor-mediated gene repression. Mol Cell Endocrinol. 2012;352(1–2):46–56. [DOI] [PubMed] [Google Scholar]
- 26. Ushio K, Hashimoto T, Kitamura N, Tanaka T. Id1 is down-regulated by hepatocyte growth factor via ERK-dependent and ERK-independent signaling pathways, leading to increased expression of p16INK4a in hepatoma cells. Mol Cancer Res. 2009;7(7):1179–1188. [DOI] [PubMed] [Google Scholar]
- 27. Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23(16):2919–2933. [DOI] [PubMed] [Google Scholar]
- 28. Storer M, Mas A, Robert-Moreno A, et al. . Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–1130. [DOI] [PubMed] [Google Scholar]
- 29. Muñoz-Espín D, Cañamero M, Maraver A, et al. . Cell. 2005;155(5):1104–1118. [DOI] [PubMed] [Google Scholar]
- 30. Zeng N, Yang KT, Bayan JA, et al. . PTEN controls β-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell. 2013;12(6):1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burton DG, Giribaldi MG, Munoz A, et al. . Androgen deprivation-induced senescence promotes outgrowth of androgen-refractory prostate cancer cells. PLoS One. 2013;8(6):e68003. [DOI] [PMC free article] [PubMed] [Google Scholar]