

Abstract
Bone morphogenic proteins (BMPs) stimulate bone formation in vivo and osteoblast differentiation in vitro via a Smad signaling pathway. Recent findings revealed that the activation of nuclear factor-κB (NF-κB) inhibits BMP-induced osteoblast differentiation. Here, we show that NF-κB inhibits BMP signaling by directly targeting the Smad pathway. A selective inhibitor of the classic NF-κB pathway, BAY11–770682, enhanced BMP2-induced ectopic bone formation in vivo. In mouse embryonic fibroblasts (MEFs) prepared from mice deficient in p65, the main subunit of NF-κB, BMP2, induced osteoblastic differentiation via the Smad complex to a greater extent than that in wild-type MEFs. In p65−/− MEFs, the BMP2-activated Smad complex bound much more stably to the target element than that in wild-type MEFs without affecting the phosphorylation levels of Smad1/5/8. Overexpression of p65 inhibited BMP2 activity by decreasing the DNA binding of the Smad complex. The C-terminal region, including the TA2 domain, of p65 was essential for inhibiting the BMP-Smad pathway. The C-terminal TA2 domain of p65 associated with the MH1 domain of Smad4 but not Smad1. Taken together, our results suggest that p65 inhibits BMP signaling by blocking the DNA binding of the Smad complex via an interaction with Smad4. Our study also suggests that targeting the association between p65 and Smad4 may help to promote bone regeneration in the treatment of bone diseases.
Bone is a highly dynamic, specialized connective tissue that continuously renews itself throughout life. In adults, old bone is replaced by new bone in an orderly fashion by the interplay between osteoclasts and osteoblasts, a process known as bone remodeling. Osteoclasts resorb bone, and osteoblasts migrate to the resorbed area to form new bone matrix, followed by mineralization. A disturbance in this tight balance between bone resorption and bone formation in favor of resorption underlies the most prevalent bone disease, osteoporosis, which is characterized by reduced bone mass and the deterioration of the bone architecture, resulting in increased bone fragility and fracture risk (1).
Bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily and were originally identified because of their ability to induce ectopic bone formation when implanted into muscle tissue (2, 3). BMPs play critical roles in embryogenesis and tissue homeostasis, and their malfunctions cause human disorders ranging from bone defects to cancer. For this reason, complex mechanisms have evolved to control the access of BMPs to target cells, and BMP signaling is strictly regulated at numerous steps, from ligand availability to the nuclear factors that regulate their transcriptional response. BMP signaling is transduced by 2 types of transmembrane serine-threonine kinase receptors, type I and type II (4, 5). After type II receptors phosphorylate the type I receptors in a ligand-dependent fashion, the activated type I receptors phosphorylate receptor-specific Smads, such as R-Smad1/5/8, in the cytoplasm. The signal is then transduced via the complex formed by R-Smad1/5/8 and Co-Smad4 and is subsequently translocated into the nucleus to regulate BMP-specific target gene expression.
The Smad proteins contain 2 conserved globular domains (the MH1 and MH2 domains) that are connected by a linker region (6). The MH1 domain binds DNA, whereas the MH2 domain binds to membrane receptors for activation, to nucleoporins for nuclear translocation, and to other Smads and nuclear factors to form transcriptional complexes. Smad4 is the product of the tumor suppressor gene deleted in pancreatic carcinoma-4 (DPC-4) (7) and acts as a common mediator of TGF-β, activin, and BMP signaling responses. The MH1 domain of Smad4 directly interacts with DNA (8). The MH2 domain is thought to bind to the phosphorylated C-tail of activated R-Smads (9) and is important for transcriptional activation (10–12) and direct protein interactions with the MH1 domain of R-Smads (13).
Smads play major roles as receptor substrates and transcription factors, but their activity is modulated by multiple regulatory mechanisms (14). Various extracellular antagonists, including noggin, chordin, cerberus, and gremlin, regulate BMP signaling (14, 15). I-Smads, ie, Smad6 and Smad7, function as antagonists of TGF-β/BMP signaling inside cells. In addition to these molecules that primarily act in the cytoplasm, specific molecules act in the nucleus as transcriptional corepressors (14, 15). Importantly, the expression of these regulatory molecules is regulated via negative feedback mechanisms by the stimulation of TGF-β/BMP (14). In addition, Smad7 is induced by various signals other than TGF-β/BMPs, including nuclear factor-κB (NF-κB) (16) and interferon-γ signaling (17). Thus, cells treated with IL-1 (18) or interferon-γ are resistant to the effects of TGF-β and are potentially resistant to the effects of BMPs.
The transcription factor NF-κB plays a key role in immune and inflammatory responses, proliferation, tumorigenesis, and apoptosis (19, 20). To date, 5 proteins with conserved homology in the Rel domain (p65 [RelA], RelB, cRel, NF-κB1 [p50/p105], and NF-κB2 [p52/p100]) have been identified. All 5 proteins share an N-terminal domain of 300 amino acids, designated the Rel homology domain, which is responsible for DNA binding, dimerization, and interactions with the inhibitory IκB proteins. Three proteins, p65, c-Rel, and RelB, contain C-terminal transcriptional activation domains (TADs) that are crucial for their ability to induce target gene expression; however, p50 and p52 homodimers lack TADs and have no intrinsic ability to drive transcription. p65 subunits contain 2 TADs, TA1 (amino acids 521–550), and TA2 (amino acids 428–521) in the C-terminal region (21, 22). In unstimulated cells, NF-κB is predominantly localized in the cytoplasm as part of a complex with inhibitory IκB proteins, including IκBα, IκBβ, IκBϵ, and IκBγ. In response to a variety of stimuli, such as TNF-α or IL-1β, IκBs are phosphorylated (Ser-32 and Ser-36 for IκBα and Ser-19 and Ser-21 for IκBβ) by the activated IκB kinase (IKK) complex. Phosphorylated IκBs are ubiquitinated and then degraded by the 26S proteasome. The IKK complex consists of 2 catalytic kinase subunits, IKKα (IKK1) and IKKβ (IKK2), and a regulatory subunit, NEMO (NF-κB essential modulator), which is also known as IKKγ (19, 20). IKKβ is most critical for the classic (canonical) NF-κB pathway, which is dependent on the degradation of IκB. In this pathway, the p50/p65 heterodimer enters the nucleus and binds to NF-κB-responsive elements to regulate gene expression.
The importance of NF-κB in osteoblast differentiation and bone formation was revealed in a recent study showing that the inhibition of NF-κB in mature osteoblasts in mice by expressing a dominant-negative form of IKKβ increased bone mineral density (BMD) and bone volume due to the increased activity of the osteoblasts (23). Furthermore, the selective inhibition of NF-κB restored the inhibitory effect of TNF-α on BMP2-induced osteoblast differentiation and prevented bone loss in ovariectomized mice (24, 25). We previously reported that activation of the classic NF-κB pathway induced by TNF-α inhibited BMP2-induced osteoblast differentiation by inhibiting the DNA binding of Smad to target genes. Alymphoplasia (aly/aly) mice that have a natural loss-of-function mutation in the Nik gene, which encodes a kinase that is essential for the processing of p100 to p52 in the alternative NF-κB pathway, showed mild osteopetrosis with an increase in several parameters of bone formation (26). In this study, we explored the possible molecular mechanism by which the classic NF-κB pathway regulates BMP-induced bone formation.
Results
Selective NF-κB inhibitor increases the ectopic bone formation induced by BMP2 in vivo
We first examined whether a selective inhibitor of NF-κB, BAY11–77082, enhances the ectopic bone formation induced by BMP2. The ectopic bone formation was not observed with collagen matrix alone (data not shown). Two micrograms of BMP2 was implanted into mice with or without 20 μg of BAY11–77082. BMP2-induced ectopic bones were enlarged and showed enhanced radiopaque areas in the presence of BAY11–77082 compared with those for BMP2 alone (Figure 1A). The mean longest diameter of ectopic bones in the presence of BAY11–77082 was significantly longer than that of BMP2 alone (Figure 1A). A micro-computed tomography (μCT) image of the ectopic bones induced by BMP2 together with BAY11–77082 showed a thick outer bone filled with trabecular bone (Figure 1B). The bone mineral content (BMC) and BMD of these ectopic bones also increased in the presence of BAY11–77082 (Figure 1, C and D). Using hematoxylin and eosin (H&E) staining, we observed thicker cortical bone and a reduced bone marrow area in the presence of BAY11–77082 compared with those for the mice implanted with BMP2 alone (Figure 1E, top panels). The expression of osteocalcin, a marker of mature osteoblasts, in ectopic bones induced by BMP2 together with BAY11–77082 increased compared with that for BMP2 alone (Figure 1F). Although previous studies showed that a selective inhibitor of NF-κB inhibited osteoclastogenesis (27–29), we observed the same number of tartrate-resistant acid phosphatase (TRAP)–positive cells in the ectopic bones formed in the presence and absence of BAY11–77082 (Figure 1E, bottom panels). These results suggest that the inhibition of the NF-κB signaling pathway stimulated BMP2-induced bone formation in vivo.
Figure 1.

BMP2-induced ectopic bone formation is enhanced in the presence of BAY11–77082 in vivo. Two micrograms of BMP2 was implanted subcutaneously to induce ectopic bone formation in the presence or absence of BAY11–77082 in WT mice (n = 8). A, After 4 weeks, the implants were removed and examined using soft X-ray analysis. Scale bar corresponds to 5 mm. The longest diameters of ectopic bones were measured. *, P < .01. B, μCT reconstruction images of ectopic bone in the presence or absence of BAY11–77082 in WT mice. Scale bar corresponds to 1 mm. C and D, BMC (C) and BMD (D) of the ectopic bones in the presence (■) or absence (□) of BAY11–77082 were measured using dual-energy X-ray absorptiometry. *, P < .05; **, P < .01. E, Sections of the implants at 4 weeks were stained with H&E (top panels) and TRAP (bottom panels). Scale bar corresponds to 25 μm. F, Total RNA was isolated, and the expression levels of osteocalcin and GAPDH mRNA in each ectopic bone were measured using RT-PCR analysis. The numbers below the gels represent the intensity of osteocalcin relative to that of GAPDH mRNA. Similar results were obtained in 3 independent experiments.
Absence of p65 but not of p50 enhances BMP2-induced osteoblastic differentiation in MEFs
To further investigate the molecular mechanisms by which the classic NF-κB pathway regulates BMP2-induced bone formation, we examined the effect of NF-κB on BMP2-induced alkaline phosphatase (ALP) activity, a typical marker of osteoblast differentiation, using mouse embryonic fibroblasts (MEFs) prepared from wild-type (WT), p65-deficient (p65−/−), or p50−/− mice because p65−/− mice are embryonic lethal (30). BMP increased ALP activity in MEFs prepared from WT mice, and an even greater increase than occurred in a concentration-dependent manner was observed in MEFs from p65−/− mice (Figure 2, A–C). BMP2-induced expression of type I collagen (ColIa1), osteonectin, and osteocalcin that was associated with osteoblast differentiation was also augmented in MEFs from p65−/− mice (Figure 2D). The negative effect of p65 on the BMP-induced osteoblast differentiation was confirmed in C2C12 cells. The ALP activity induced by the cotransfection of a constitutively activate BMP type I receptor ALK2 [ALK2(Q207D)] and downstream effector, Smad1, was suppressed by the coexpression of p65 in C2C12 cells (Figure 2E).
Figure 2.
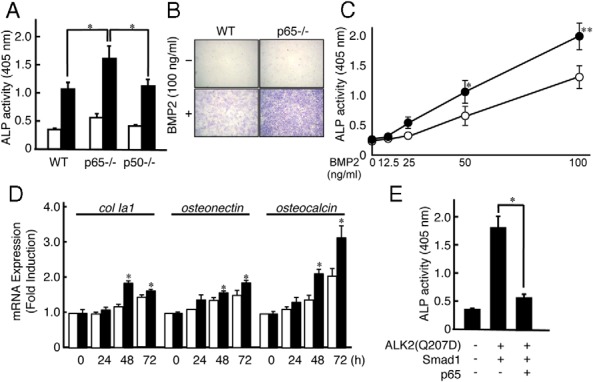
p65 deficiency enhances BMP2-induced osteoblastic differentiation from MEFs. A, MEFs from WT, p65−/−, or p50 −/− mice were treated with (■) or without (□) BMP2 (100 ng/mL) for 72 hours. The cells were then fixed with an acetone-ethanol mixture and incubated with a substrate solution. ALP activity was then determined. Data are expressed as means ± SD (n = 3). *, P < .01. Similar results were obtained in 3 independent experiments. B, Cells were stained for ALP activity. Similar results were obtained in 3 independent experiments. C, MEFs from WT (□) or p65−/− (■) mice were treated with various concentrations of BMP2 for 72 hours. ALP activity was then determined. Data are expressed as means ± SD (n = 3). *, P < .01. Similar results were obtained in 3 independent experiments. D, MEFs from WT (□) or p65−/− (■) mice were treated with BMP2 (100 ng/mL) for the indicated periods. Total RNA was isolated, and type I collagen (ColIa1), osteonectin, osteocalcin, and GAPDH mRNA levels were analyzed using real-time PCR. Data represent means ± SD of the expression levels of osteogenic genes relative to that of GAPDH (n = 3). Similar results were obtained in 3 independent experiments. E, ALP activity was induced by the exogenous expression of empty vector or V5-tagged ALK2(Q207D) with FLAG-tagged Smad1 in the presence or absence of p65 in C2C12 cells. Data are expressed as means ± SD (n = 3). *, P < .01. Similar results were obtained in 3 independent experiments.
p65 inhibits the DNA-binding activities of the Smad proteins induced by BMP2
The expression levels of the BMP receptors and Smad mRNAs were similar between the WT and p65-deficient MEFs (data not shown). We subsequently examined the effects of p65 on the phosphorylation levels of Smad1/5/8 (p-Smad1/5/8), an initial key step in BMP signal transduction (4, 15). Before BMP2 stimulation, the p-Smad1/5/8 level was very low in WT and p65−/− MEFs, as assessed by Western blotting using the anti-p-Smad1/5/8 antibody. BMP stimulation triggered phosphorylation from 10 to 60 minutes which then declined until 240 minutes, and no difference was observed between the WT and p65−/− MEFs (Figure 3A). The expression levels of Smad1 and Smad4 were not changed after the BMP2 stimulation (Figure 3A). The cellular localization of p-Smad1/5/8 that was induced by BMP2 was observed using immunofluorescence. p-Smad1/5/8 was very faint throughout the cytoplasm before stimulation but gradually accumulated in the nucleus and cytoplasm from 30 to 60 minutes in MEFs of both genotypes. The nuclear p-Smad1/5/8 was reduced at 60 to 120 minutes in WT MEFs; however, p-Smad1/5/8 remained in the nucleus at 120 minutes in p65-deficient MEFs (Figure 3B). BMP2-induced Smad1/5/8 phosphorylation in the nucleus was enhanced in MEFs from p65−/− mice compared with that in MEFs from WT mice, even though the histone deacetylase 1 level was constant (Figure 3C).
Figure 3.

p65 inhibits the DNA binding activities of Smad proteins that are induced by BMP2. A, MEFs from WT or p65−/− mice were cultured in the presence of BMP2 (100 ng/mL) for the indicated times. The total cell lysates were immunoblotted with anti-p-Smad1/5/8, Smad1, and p65 antibodies. Anti-β-actin was used as a loading control. Similar results were obtained in 3 independent experiments. B, MEFs from WT or p65−/− mice were treated with or without BMP2 (100 ng/mL) for the indicated periods. The cells were then fixed and incubated with p-Smad1/5/8 antibodies followed by incubation with Alexa Fluor 430–conjugated anti-rabbit IgG. The subcellular localization of Alexa Fluor 430–labeled p-Smad1/5/8 was determined using fluorescence microscopy. To visualize the cell nuclei, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar corresponds to 50 μm. Similar results were obtained in 3 independent experiments. C, MEFs from WT or p65−/− mice were treated with BMP2 (100 ng/mL) for the indicated times. The nuclear fractions were harvested from cultured cells and subjected to immunoblot analysis with p-Smad1/5/8 antibodies. The expression of histone deacetylase 1 (HDAC1) was used as a loading control for the nuclear extracts. Similar results were obtained in 3 independent experiments. D, MEFs from WT or p65−/− mice were cultured in the presence of BMP2 (100 ng/mL) for the indicated times. The chromatin from individual samples was precipitated using p-Smad1/5/8 antibodies. The Id1 promoter was amplified by PCR from the precipitated DNA. The numbers below the gels represent the intensity of the PCR product from the precipitated DNA by anti-p-Smad1/5/8 antibodies relative to the input. Similar results were obtained in 3 independent experiments. E, p65−/− MEFs were transfected with FLAG-tagged Smad1 and HA-tagged Smad4 in the presence of various concentrations (0, 0.5, 1.0, and 2.0 μg) of a p65 expression plasmid for 24 hours. The chromatin from individual samples was precipitated using the FLAG and p65 antibodies. The Id1 promoter was PCR amplified from the precipitated DNA. The numbers below the gels represent the intensity of the PCR product from the precipitated DNA by anti-FLAG and p65 antibodies relative to the input. Similar results were obtained in 3 independent experiments. F, MEFs from WT or p65−/− mice were cultured in the presence of BMP2 (100 ng/mL) for the indicated times. The BMP2-induced Smad1/4 DNA binding activity in the nuclear fractions was measured by EMSA using a BRE probe. Similar results were obtained in 3 independent experiments. n.s., nonspecific binding.
A chromatin immunoprecipitation (ChIP) analysis showed that p-Smad1/5/8 was recruited to the Id1 promoter after 10 minutes of BMP2 stimulation, reached a maximum level at 60 minutes, and then declined until 360 minutes in WT MEFs (Figure 3D). In p65−/− MEFs, however, p-Smad1/5/8 bound to the Id1 promoter longer than in WT cells (see 120 and 360 minutes in Figure 3D). Furthermore, the DNA binding of the FLAG-Smad1 and HA-Smad4 complexes was inhibited by p65 in a dose-dependent manner (Figure 3E). We previously determined that the BMP2-induced DNA binding complex included Smad1/Smad4 as shown by a supershift assay (24, 31). An electrophoresis mobility shift assay (EMSA) also showed that the Smad1/4 complex bound longer to the Id1 promoter in p65-deficient MEFs than in WT MEFs (Figure 3F). These results strongly indicate that p65 inhibits the DNA binding of the Smad1/4 complex to the promoter region of the BMP target genes.
C-terminal TA2 domain of p65 inhibits DNA binding of the Smad1/Smad4 complex
To examine which domain of p65 was involved in the inhibition of the DNA binding of Smad, we generated several p65 deletion mutants and confirmed their expression levels in p65-deficient MEFs (Figure 4, A and B). All of the deletion mutants bound to the DNA of the κB motif as well as to full-length p65 because they had the Rel homology domain (Figure 4C). Although p65 (1–521) was reduced in NF-κB transcriptional activity by 70%, it still inhibited both the BMP2-induced luciferase reporter activity and the DNA binding of the Smad complex with an inhibitory effect similar to that of full-length p65 (Figure 4, D and F). In contrast, neither p65 (1–428) or (1–318), in which both TA1 and TA2 domains have been deleted, showed such inhibitory activity on BMP signaling via the Smad complex (Figure 4, E and F). These findings suggest that the C-terminal TA2 domain, which is present in p65 (1–521), plays an important role in the inhibitory activity of p65.
Figure 4.
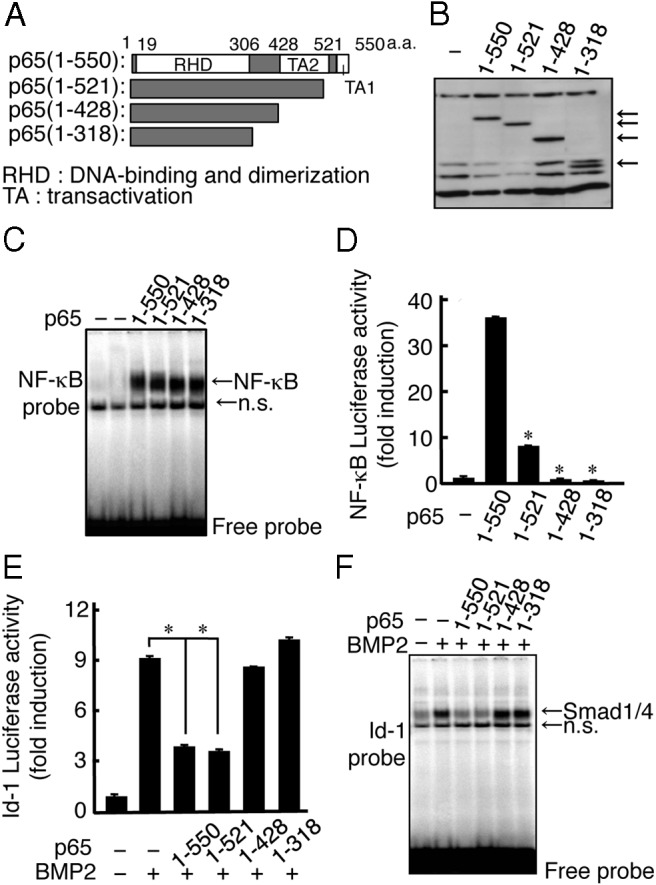
C-terminal TA2 domain of p65 inhibits the DNA binding of the Smad1 and Smad4 complex. A, Schematic outline of the C-terminally truncated forms of the FLAG-tagged p65 constructs used in these experiments. B, p65−/− MEFs were transfected with the C-terminally truncated forms of the FLAG-tagged p65 constructs. The expression levels of the C-terminally truncated forms of FLAG-tagged p65 were examined by Western blotting using the FLAG (M5) antibody. The arrows indicate each of the truncated forms of p65. C, C-terminally truncated forms of FLAG-tagged p65 were transfected in p65−/− MEFs for 24 hours, and the DNA binding activity of NF-κB in the nuclear fractions was measured by EMSA using the κB probe. Similar results were obtained in 3 independent experiments. D, C-terminally truncated forms of FLAG-tagged p65 were transfected with the pNF-κB-luciferase reporter in p65−/− MEFs and were then assayed for luciferase activity after 24 hours. Data are expressed as means ± SD (n = 3). *, P < .01. Similar results were obtained in 3 independent experiments. E, C-terminally truncated forms of FLAG-tagged p65 were transfected with the Id-luciferase reporter in p65−/− MEFs for 24 hours. The cells were treated with BMP2 (100 ng/mL) and then assayed for luciferase activity after 24 hours. Data are expressed as means ± SD (n = 3). *, P < .01. Similar results were obtained in 3 independent experiments. F, C-terminally truncated forms of FLAG-tagged p65 were transfected into p65−/− MEFs for 24 hours. The cells were then treated with BMP2 (100 ng/mL) for 1 hour. The BMP2-induced DNA binding activity in the nuclear fractions was measured by EMSA using a BRE probe. Similar results were obtained in 3 independent experiments. n.s. indicates nonspecific binding.
The C-terminal TA2 domain of p65 interacts with MH1 domain of Smad4 but not Smad1
We next examined the interaction between p65 and Smad using FLAG-tagged p65 and Myc-tagged Smad1 or Smad4 in p65−/− MEFs. The cell extracts were subjected to immunoprecipitation using an anti-FLAG antibody, followed by immunoblotting with an anti-Myc antibody. p65 interacted with Smad4 but not Smad1, and Myc-Smad1 was immunoprecipitated with p65 only in the presence of Myc-Smad4 (Figure 5A). The interaction of endogenous p65 and Smad4 was confirmed in MC3T3-E1 cells in response to BMP2 stimulation (Figure 5B). We further examined the interaction of the p65 deletion mutant and Smad4. Consistent with the inhibitory activity of p65 as shown in Figure 4, not only full-length p65 but also p65 (1–521) associated with Smad4 (Figure 5C). However, the TA2-deficient mutants, p65 (1–428) and (1–318), did not show such an interaction (Figure 5C), suggesting that the TA2 domain of p65 inhibited the DNA binding of the Smad complex by interacting with Smad4. To examine the critical domain of Smad4 in the interaction with p65, we generated 2 Myc-tagged deletion mutants of Smad4 lacking either the MH1 domain (ΔMH1) or the MH2 domain (ΔMH2) (Figure 5D). Full-length FLAG-p65 interacted with full-length Myc-Smad4 and the ΔMH2 mutant but not with the ΔMH1 mutant (Figure 5E). Finally, we examined the role of the TA2 domain of p65 in the interaction with the MH1 domain of Smad4 using a green fluorescent protein (GFP)-TA2 chimeric protein, in which the TA2 domain of p65 was fused to GFP. Figure TA2, but not GFP alone, associated with full-length Smad4 and the ΔMH2 mutant. Consistent with our findings, GFP-TA2 failed to interact with the ΔMH1 mutant of Smad4 (Figure 5F). Furthermore, GFP-TA2, but not GFP alone, inhibited the BMP2-induced luciferase reporter activity (Figure 5G). These results indicate that the TA2 domain of p65 inhibits BMP2 signaling by interacting with the MH1 domain of Smad4.
Figure 5.

The C-terminal TA2 domain of p65 interacts with the MH1 domain of Smad4 but not Smad1. A, p65−/− MEFs were cotransfected with FLAG-tagged p65 and either Myc-tagged Smad1 or Myc-tagged Smad4. The whole-cell lysates were immunoprecipitated (IP) using an anti-FLAG (M5) antibody and immunoblotted (IB) with an anti-Myc antibody. Similar results were obtained in 3 independent experiments. B, MC3T3–E1 cells were treated with or without BMP2 (100 ng/mL) for 1 hour. The whole-cell lysates were immunoprecipitated with anti-p65 (SA171) antibodies and immunoblotted with anti-Smad4 antibodies. Similar results were obtained in 3 independent experiments. C, p65−/− MEFs were cotransfected with the Myc-tagged Smad4 and C-terminally truncated forms of FLAG-tagged p65. Whole-cell lysates were immunoprecipitated with an anti-Myc antibody and immunoblotted with an anti-FLAG (M5) antibody. Similar results were obtained in 3 independent experiments. D, Schematic outlines of the truncated forms of Myc-tagged Smad4 constructs used in these experiments are indicated. E, p65−/− MEFs were cotransfected with FLAG-tagged p65 and truncated forms of Myc-tagged Smad4. The whole-cell lysates were immunoprecipitated with an anti-FLAG (M5) antibody and immunoblotted with an anti-Myc antibody. Similar results were obtained in 3 independent experiments. F, p65−/− MEFs were cotransfected with Myc-tagged forms of truncated Smad4 p65 and GFP or a GFP-tagged TA2 domain of p65. The whole-cell lysates were immunoprecipitated with an anti-Myc antibody and immunoblotted with an anti-GFP antibody. Similar results were obtained in 3 independent experiments. G, GFP (□) or the GFP-tagged TA2 domain of p65 (■) was transfected with the Id-luciferase reporter in p65−/− MEFs for 24 hours. The cells were treated with BMP2 (100 ng/mL) and then were assayed for luciferase activity after 24 hours. Data are expressed as means ± SD (n = 3). *, P < .01. Similar results were obtained in 3 independent experiments.
Discussion
BMP signaling is fine-tuned at multiple levels, including extracellular cues, the interactions of BMP receptors at the plasma membrane, and intercellular events (32). Here, we showed that BMP2-activated NF-κB inhibits BMP signaling at the level of Smad DNA binding via an interaction between Smad4 and p65. The following lines of evidence support our conclusions. First, no differences were observed in the expression levels of the BMP receptors, Smad proteins, including Smad7, or Smurf1 in the presence or absence of BMP2 treatment between WT and p65−/− MEFs (Supplemental Figure 1). Second, although the phosphorylation levels of Smad1/5/8 were nearly comparable between the WT and p65−/− MEFs, the phosphorylation of Smad1/5/8 was present in the nucleus in p65−/− MEFs longer than in the WT MEFs. Third, both the ChIP assay and EMSA analysis revealed that the Smad1/4 complex bound to DNA longer in the p65−/− MEFs than in the WT MEFs. Moreover, the C-terminal TA2 domain of p65 specifically interacted with the MH1 domain of Smad4. Collectively, BMP-activated p65 inhibits BMP-induced bone formation and osteoblast differentiation by inhibiting the DNA binding of the Smad1/Smad4 complex to the promoter region of the BMP/Smad target genes.
Recent findings have indicated that the inhibition of NF-κB in osteoblasts is effective in preventing bone loss induced by an ovariectomy in mice (25). The inhibition of NF-κB in mature osteoblasts of mice expressing a dominant-negative form of IKKβ showed increased BMD and bone volume due to the increased activity of osteoblasts without affecting osteoclast activity. This inhibition also maintained bone formation by preventing the osteoporotic bone loss that was induced by an ovariectomy in mice via the increased expression of Fos-related antigen-1 (Fra-1), which is an essential transcription factor involved in bone matrix formation in vitro and in vivo (23). Furthermore, NF-κB was identified as a crucial factor responsible for inhibiting bone formation in an osteoporosis model (23). These results suggest that the estrogen receptor directly inhibited NF-κB transcription in a ligand-dependent manner by interacting with NF-κB (33). The estrogen receptor is expressed in osteoblasts; therefore, estrogen may negatively regulate NF-κB activities under physiological conditions. However, during the pathogenesis of osteoporosis, this negative regulation may be diminished because of the deficiency of estrogen, resulting in an elevation of basal NF-κB activities in osteoblasts. Another possibility is that proinflammatory cytokines, including TNFα, IL-1β, IL-6, and IL-7, are highly expressed by T cells and other cells during osteoporosis (34). These cytokines potently stimulate NF-κB activities in osteoblasts.
In this study, we showed that BMP2-induced NF-κB activation inhibited osteoblast differentiation as a negative regulator even in the absence of any inflammatory stimuli. The depletion of p65 in MEFs enhanced BMP2-induced osteoblast differentiation, suggesting that NF-κB inhibition, particularly of the p65 subunit, enhanced the bone-forming activity of osteoblasts via a cell autonomous effect. To support this hypothesis, BMP2-induced ectopic bone formation in mice was enhanced in the presence of BAY11–77082. Our study and others have previously reported that selective inhibitors of NF-κB, such as NBD (NEMO-binding domain) peptides or DHMEQ (dehydroxymethylepoxyquinomicin), suppress osteoclastogenesis in vitro and in vivo (35–37). To examine the possibility that the increased BMD in ectopic bone in the presence of BAY11–77082 was due to the inhibition of osteoclastogenesis, we stained sections of BMP2-induced ectopic bones for TRAP to identify osteoclasts. The amount of bone resorption may have increased in the BMP group treated with BAY11–77082 because the total number of TRAP-positive cells was higher in the sections from BMP combined with BAY11–77082 than in the BMP2 group. However, the osteoclast surface ratio, which is known to represent the osteoclast activity in bone remodeling, was similar in both groups, suggesting that BAY11–77082 either did not reduce osteoclast activity or was not fully active at the end of the experiment (data not shown). In addition, it is well known that the degradation rate of the collagen sponge, the carrier of BAY11–77082 used in the current study, is very high (degrades by approximately 70% in the first 7 days after implantation), and BMP2 release from a collagen sponge is lower in the 7-day period from day 7 to day 14 after implantation than in the first 7 days (38–40). Therefore, the amount of BAY11–77082 release from the collagen carrier may have been lower in the later phase of ectopic bone formation than in the early phase of the experiment in the current study, and this fact may explain why the osteoclast number was not reduced in the histological sections of the BAY11–77082–treated group at the end of the experiment. Although we cannot exclude the possibility that osteoclast function was reduced by treatment with BAY11–77082 despite the normal osteoclast number in mice, the treatment in this study might not inhibit osteoclastogenesis in vivo because we previously injected the NF-κB inhibitor daily for 3 weeks to inhibit osteoclastogenesis in vivo. The effect of BAY11–77082 may be dependent on the efficiency of the release of BAY11–77082 from its carrier; therefore, kinetic studies after the local administration of BAY11–77082 need to be performed when a time-course study is conducted.
Previous studies have shown that activated NF-κB inhibits TGF-β signaling at the level of TGF-β type I receptor function via the induction of the synthesis and receptor binding of the inhibitory Smad7 protein. A variety of pathogenic and proinflammatory activators of NF-κB increased Smad7 mRNA in several cell types, including WT MEFs but not p65−/− MEFs (16). The inhibitory function of Smad7 in TGF-β signaling has also been linked to its stable interaction with the ligand-activated type I receptor, thus blocking the association and phosphorylation of its substrates, Smad2 and/or Smad3 (16). These results suggested that the blockade of the activated type I receptor with increased Smad7 that was induced by activated NF-κB was a critical event that resulted in the inhibition of all subsequent steps in the TGF-β/Smad7 signaling cascade. However, in this study, the expression levels of Smad6 and Smad7 were not changed in WT and p65−/− MEFs in the presence or absence of BMP2 (data not shown). Thus, the induction of Smad6 or Smad7 did not contribute to the inhibitory effect of NF-κB on BMP-induced osteoblast differentiation in our system.
The critical elements for the transcription of p65 were mapped between amino acids 435 and 550. Within this region, 2 subdomains, TA1 and TA2, were identified as possessing transactivation activity (21, 22). Although the deletion of the TA1 domain in p65 (1–521) strongly reduced the NF-κB transactivation ability, this p65 mutant inhibited Smad1/Smad4 transactivation ability and DNA binding activity. The deletion of both the TA1 and TA2 domains of p65 (1–428) completely abolished the NF-κB transactivation ability and failed to inhibit BMP/Smad signaling. We also have shown that ΔMH2 but not ΔMH1 interacted with the TA2 domain of p65. Thus, we conclude that the association of the TA2 domain of p65 with the MH1 domain of Smad4 is important to prevent the DNA binding activity of Smad1/Smad4.
We propose that the BMP-induced NF-κB activation negatively regulated BMP signaling and thereby inhibited BMP2-induced osteoblast differentiation in vitro and bone formation in vivo, preventing the overproduction of bone. This regulation of Smad activity occurs during important physiological and pathological processes, such as skeletal development and inflammatory bone diseases, in which the BMP and NF-κB signaling pathways are known to function antagonistically. Mice deficient in p65 are embryonic lethal (30), suggesting that the inhibition of the NF-κB pathway results in life-compromising side effects. Thus, the interaction domain of Smad4 and NF-κB may be a novel therapeutic target for diseases accompanied by bone loss by inducing high bone mass with BMP with less severe side effects.
Materials and Methods
The experimental procedures were approved by the Animal Care and Use Committee of Kyushu Dental University (approval number 10–013).
Reagents
Purified recombinant human BMP2 was purchased from Corefront. Anti-phosphorylated Smad1/5/8 (no. 9511), anti-Smad1 (no. 9743), and anti-Smad4 (no. 9515) antibodies were obtained from Cell Signaling Technology Inc. Anti-IκB (sc-371) was obtained from Santa Cruz Biotechnology. BAY11–77082 and anti-p65 (SA171) antibodies were obtained from BIOMOL. Anti-FLAG M5 and anti-β-actin (AC-15) antibodies were purchased from Sigma-Aldrich. Anti-Myc and anti-GFP antibodies were purchased from Medical and Biological Laboratories and Nakarai Tesque, respectively.
Ectopic bone formation assay
The bone formation effects induced by BMP2 in vivo were examined using an ectopic bone formation assay in the presence or absence of BAY11–77082 in mice. BMP2 (2 μg) and BAY11–77082 (20 μg) were blotted onto a collagen sponge disk (6-mm diameter, 1-mm thickness) made from commercially available bovine collagen sheets (Helistat; Integra Neuro-Sciences), freeze-dried, and maintained at −20°C until being implanted into the mice (38). All procedures were performed under sterile conditions. The mice were anesthetized using diethyl ether gas inhalation, and collagen pellets were surgically implanted into the dorsal muscle pouches (2 pellets/animal) of the mice (8 weeks old). Four weeks after surgery, the mice were euthanized, and the implants were harvested and processed for histological analysis. All of the harvested samples were fixed in PBS-buffered glutaraldehyde (0.25%)-formalin (4%) fixative (pH 7.4) for 2 days at 4°C and washed with PBS for further studies. The longest diameters of the ectopic bones were measured. The BMC and BMD of the ectopic bone were measured using dual-energy X-ray absorptiometry (DCS-600R; Aloka). Three-dimensional reconstruction images of the ectopic bone were obtained by μCT (ScanXmate-E090; Comscan) as described previously (39, 40). The sections of the ectopic bone from each group were stained with H&E and TRAP. The sections were examined using light microscopy.
Cell culture
To generate MEFs from WT, p65-deficient (p65−/−), or p50-deficient (p50−/−) mice, fetuses were harvested at day 13.5 of gestation (41). The embryo head and liver were carefully removed to use for PCR genotyping and Western blot analysis, respectively. The tissue from individual mice was minced and incubated with 0.05% trypsin-EDTA at 37°C for 10 minutes with shaking. After incubation, the fetal tissue was pipetted vigorously to obtain a single-cell suspension. The cells were maintained in DMEM with 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. C2C12 cells, a mouse myoblast cell line, were maintained in DMEM containing 15% FBS with antibiotics. MC3T3-E1 cells, a mouse osteoblast cell line, were maintained in α-MEM containing 5% FBS and antibiotics. All of the cultures were maintained at 37°C in air containing 5% CO2.
Immunofluorescence microscopy
For immunofluorescence analysis, WT MEFs or p65−/− MEFs were seeded onto sterile FBS-coated glass coverslips. The cells were pretreated with BMP2 (100 ng/mL) for the indicated periods. The cells were fixed with 4% paraformaldehyde in PBS for 15 minutes, blocked with 10% goat serum together with 0.5% Triton X-100 in PBS for 15 minutes at room temperature, and incubated with anti-p-Smad 1/5/8 polyclonal antibodies (1:100) for 1 hour at room temperature. After extensive washing, the cells were incubated with Alexa Fluor 430–conjugated anti-rabbit IgG (dilution 1:10 000; Invitrogen) for 1 hour at room temperature. The cells were then washed and mounted in Immunon (Lipshaw). The subcellular localization of Alexa Fluor 430–labeled p65 or p-Smad1/5/8 was determined using fluorescence microscopy (Biorevo BZ-9000; Keyence). To visualize the cell nuclei, the cells were stained with 4′,6-diamidino-2-phenylindole.
Detection of ALP activity and staining
MEFs prepared from WT, p65−/−, and p50−/− mice were seeded at a density of 5.0 × 103 cells/well in 96-well plates with DMEM containing 10% FBS on the day before treatment. The cells were treated with BMP2 (100 ng/mL) for 72 hours. In several experiments, ALP activity was induced by the exogenous expression of V5-tagged ALK2(Q207D) with FLAG-tagged Smad1 in the presence or absence of p65 in C2C12 cells. After 72 hours of treatment, the cells were fixed with an acetone-ethanol mixture (50:50, v/v) and then incubated with a substrate solution (0.1 M diethanolamine, 1 mM MgCl2, and 10 mg/mL p-nitrophenyl phosphate). The reaction was terminated by addition of 5 M NaOH, and then the absorbance was measured at 405 nm using a microplate reader (Bio-Rad). To determine the ALP activity histochemically, the cells were stained for enzymatic activity as described previously (42).
RT-PCR and real-time PCR analysis
Total RNA from ectopic bones or cells was prepared with TRIzol (Invitrogen) and then reverse transcribed into cDNA. The cDNA was amplified by PCR using primers that were specific for osteocalcin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Real-time PCR was performed using SYBR Green Master Mix and a 7300 real-time PCR system (Applied Biosystems) according to the manufacturer's instructions. The samples were matched to a standard curve generated by amplifying serially diluted products using the same PCR parameters. GAPDH expression served as an internal control. The primer sequences have been described previously (43).
ChIP assays
ChIP was performed with a ChIP Assay Kit (Upstate Biotechnology) according to the manufacturer's instructions, using antibodies against p-Smad1/5/8, p65 and FLAG antibodies, or normal IgG. The purified DNA was analyzed by PCR using primers that amplify sequences containing the Id1 promoter, which harbors a BMP2-responsive element (BRE) to which the Smad proteins bind (47). The primer pairs for the Id1 promoter were 5-TAAGTTGACCCTTGGTCAGC-3 (forward) and 5-GACGTCACCCATTCATAAAAC-3 (reverse).
EMSA
The EMSA was performed using WT and p65-deficient cells. The nuclear extracts were prepared from pretreated cells (24). The Smad DNA binding activity in the nuclear fractions was measured using EMSA. The double-stranded BRE probe and the NF-κB probe were radiolabeled using T4 polynucleotide kinase and [γ-32P]ATP (New England Nuclear) using a labeling kit (Takara Shuzo Co). The sequences of the oligo DNAs used were as follows: 5-TCTCCATGGCGACCGCCCGCGCGGCGCCAGCCT-3 (BRE probe) and 5-GATCAGAGGGGACTTTCCGAGG-3 (NF-κB probe). Six micrograms of the nuclear extracts was incubated with the labeled BRE probe. For the supershift experiments, the antibodies were added before the probe, and the mixture was incubated for 15 minutes at room temperature. The reaction mixture was loaded onto a 5% polyacrylamide gel in 0.5× TBE (44.5 mM Tris base, 44.5 mM boric acid, and 1 mM EDTA) and resolved by electrophoresis.
Transfection conditions and the luciferase assay
The p65−/− MEFs were transfected using GeneJuice Transfection Reagent (Merck) according to the manufacturer's instructions. The total amounts of transfected plasmids in each group were equalized by the addition of an empty vector. BMP signaling was monitored using the IdWT4F-luciferase reporter plasmid, which expresses a luciferase protein under the control of a BMP-responsive element in the human ID1 gene, as described previously (31). The NF-κB luciferase plasmid was purchased from Promega. The luciferase activities were measured using a dual-luciferase system (Promega).
Immunoprecipitation and immunoblotting
The cytosolic and nuclear extracts from MEFs were prepared as described previously (24). p65−/− MEFs were transfected with the indicated plasmid using GeneJuice Transfection Reagent. MC3T3-E1 cells were treated with or without BMP2 (100 ng/mL) for 1 hour. The cells were lysed in TNT buffer (20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1% Triton X-100, and 1 mM dithiothreitol) containing protease inhibitors (Roche). For the coprecipitation experiments, whole-cell extracts were incubated for 6 hours at 4°C with anti-FLAG, anti-Myc, anti-GFP, or anti-p65 antibodies coupled to A/G-Sepharose beads. The immune complex was extensively washed with TNT buffer, and the samples were boiled and analyzed by immunoblotting. The protein content was measured with Pierce reagent following the manufacturer's protocol. Then 20 μg of protein was subjected to SDS-PAGE and transferred to a polyvinylidene difluoride membrane at 100 V for 1 hour at 4°C. The membranes were incubated with antibodies at dilutions of 1:500 to 1:1000 in a 5% dry milk solution containing 0.01% azide overnight at 4°C. Subsequently, the blots were washed in a solution containing 10 mM Tris-HCl, 50 mM NaCl, and 0.25% Tween 20 and incubated with a horseradish peroxidase–conjugated secondary antibody. The immunoreactive proteins were visualized using ECL (Amersham Biosciences).
Data analysis
Comparisons were made using an unpaired Student t test. The data are expressed as means ± SD; values of P < .05 were considered significant.
Acknowledgments
S.H.T., H.F., G.S., C.N., and S.K. performed most of the experiments. K.N., K.A., and K.O. performed the radiological assessments and bone histomorphometry. S.H.T. and T.M. performed the histology preparation. T.K., S.O., and M.S. provided the V5-ALK2(Q207D), FLAG-Smad1, Myc-Smad4, Myc-Smad4ΔMH1, Myc-Smad4ΔMH2, and Id-luciferase constructs. T.D. provided the p65-deficient mice. T.K., H.T., M.T., M.H., and C.K. reviewed the intermediate draft. E.J. designed the study, performed the literature review, prepared the initial and final versions of the article, and submitted the document.
This work was supported by a grant obtained from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grants 23390424 [E.J.], 2422009 (to M.H.), and 25293326 [to T.K.]).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by a grant obtained from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grants 23390424 [E.J.], 2422009 (to M.H.), and 25293326 [to T.K.]).
Footnotes
- ALP
- alkaline phosphatase
- BMC
- bone mineral content
- BMD
- bone mineral density
- BMP
- bone morphogenetic protein
- BRE
- BMP2-responsive element
- ChIP
- chromatin immunoprecipitation
- EMSA
- electrophoresis mobility shift assay
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GFP
- green fluorescent protein
- H&E
- hematoxylin and eosin
- IKK
- IκB kinase
- μCT
- micro-computed tomography
- MEF
- mouse embryonic fibroblast
- NF-κB
- nuclear factor-κB
- p-Smad
- phosphorylated Smad
- TAD
- transcriptional activation domain
- TRAP
- tartrate-resistant acid phosphatase
- WT
- wild-type.
References
- 1. Matsuo K, Otaki N. Bone cell interactions through Eph/ephrin: Bone modeling, remodeling and associated diseases. Cell Adh Migr. 2012;6:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. [DOI] [PubMed] [Google Scholar]
- 3. Wozney JM, Rosen V, Celeste AJ, et al. . Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. [DOI] [PubMed] [Google Scholar]
- 4. Katagiri T, Suda T, Miyazono K. The bone morphogenetic proteins. In: Derynck R, Miyazono K, eds. The TGF-β Family. New York, NY: Cold Spring Harbor Publishing; 2008;121–149. [Google Scholar]
- 5. Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. [DOI] [PubMed] [Google Scholar]
- 6. Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 7. Hahn SA, Schutte M, Hoque AT, et al. . DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. [DOI] [PubMed] [Google Scholar]
- 8. Zawel L, Dai JL, Buckhaults P, et al. . Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–7617. [DOI] [PubMed] [Google Scholar]
- 9. Chacko BM, Qin BY, Tiwari A, et al. . Structural basis of heteromeric Smad protein assembly in TGF-β signaling. Mol Cell. 2004;15:813–823. [DOI] [PubMed] [Google Scholar]
- 10. de Caestecker MP, Hemmati P, Larisch-Bloch S, Ajmera R, Roberts AB, Lechleider RJ. Characterization of functional domains within Smad4/DPC4. J Biol Chem. 1997;272:13690–13696. [DOI] [PubMed] [Google Scholar]
- 11. de Caestecker MP, Yahata T, Wang D, et al. . The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. J Biol Chem. 2000;275:2115–2122. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Y, Musci T, Derynck R. The tumor suppressor Smad4/DPC 4 as a central mediator of Smad function. Curr Biol. 1997;7:270–276. [DOI] [PubMed] [Google Scholar]
- 13. Shi Y, Wang YF, Jayaraman L, Yang H, Massagué J., Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell. 1998;94:585–594. [DOI] [PubMed] [Google Scholar]
- 14. Miyazono K. TGF-β signaling by Smad proteins. Cytokine Growth Factor Rev. 2000;11:15–22. [DOI] [PubMed] [Google Scholar]
- 15. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. [DOI] [PubMed] [Google Scholar]
- 16. Bitzer M, von Gersdorff G, Liang D, et al. . A mechanism of suppression of TGF-β/SMAD signaling by NF-κB/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- 17. Weng H., Mertens PR, Gressner AM, Dooley S. IFN-γ abrogates profibrogenic TGF-β signaling in liver by targeting expression of inhibitory and receptor Smads. J Hepatol. 2007;46:295–303. [DOI] [PubMed] [Google Scholar]
- 18. Baugé C, Attia J, Leclercq S, Pujol JP, Galéra P, Boumédiene K. Interleukin-1β up-regulation of Smad7 via NF-κB activation in human chondrocytes. Arthritis Rheum. 2008;58:221–226. [DOI] [PubMed] [Google Scholar]
- 19. Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. [DOI] [PubMed] [Google Scholar]
- 21. Schmitz ML, Stelzer G, Altmann H, Meisterernst M, Baeuerle PA. Interaction of the COOH-terminal transactivation domain of p65 NF-κB with TATA-binding protein, transcription factor IIB, and coactivators. J Biol Chem. 1995;270:7219–7226. [DOI] [PubMed] [Google Scholar]
- 22. Schmitz ML, dos Santos Silva MA, Baeuerle PA. Transactivation domain 2 (TA2) of p65 NF-κB. Similarity to TA1 and phorbol ester-stimulated activity and phosphorylation in intact cells. J Biol Chem. 1995;270:15576–15584. [DOI] [PubMed] [Google Scholar]
- 23. Chang J, Wang Z, Tang E, et al. . Inhibition of osteoblastic bone formation by nuclear factor-κB. Nat Med. 2009;15:682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamazaki M, Fukushima H, Shin M, Katagiri T, Doi T, Takahashi T, Jimi E. Tumor necrosis factor α represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-κB. J Biol Chem. 2009;284:35987–35995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alles N, Soysa NS, Hayashi J, et al. . Suppression of NF-κB increases bone formation and ameliorates osteopenia in ovariectomized mice. Endocrinology. 2010;151:4626–4634. [DOI] [PubMed] [Google Scholar]
- 26. Seo Y, Fukushima H, Maruyama T, et al. . Accumulation of p100, a precursor of NF-κB2, enhances osteoblastic differentiation in vitro and bone formation in vivo in aly/aly mice. Mol Endocrinol. 2010;26:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jimi E, Aoki K, Saito H, et al. . Selective inhibition of NF-κB blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10:617–624. [DOI] [PubMed] [Google Scholar]
- 28. Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J Biol Chem. 2004;279:37219–37222. [DOI] [PubMed] [Google Scholar]
- 29. Takatsuna H, Asagiri M, Kubota T, et al. . Inhibition of RANKL-induced osteoclastogenesis by (−)-DHMEQ, a novel NF-κB inhibitor, through downregulation of NFATc1. J Bone Miner Res. 2005;20:653–662. [DOI] [PubMed] [Google Scholar]
- 30. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. [DOI] [PubMed] [Google Scholar]
- 31. Katagiri T, Imada M, Yanai T, Suda T, Takahashi N, Kamijo R. Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells. 2002;7:949–960. [DOI] [PubMed] [Google Scholar]
- 32. Hartung A, Bitton-Worms K, Rechtman MM, et al. . Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol. 2006;26:7791–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harnish DC. Estrogen receptor ligands in the control of pathogenic inflammation. Curr Opin Investig Drugs. 2006;7:997–1001. [PubMed] [Google Scholar]
- 34. Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 2005;115:3318–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma L, Gao C, Mao Z, et al. . Collagen/chitosan porous scaffolds with improved biostability for skin tissue engineering. Biomaterials. 2003;24:4833–4841. [DOI] [PubMed] [Google Scholar]
- 36. Tangsadthakun C, Kanokpanont S, Sanchavanakit N, et al. . The influence of molecular weight of chitosan on the physical and biological properties of collagen/chitosan scaffolds. J Biomater Sci Polym Ed. 2007;18:147–163. [DOI] [PubMed] [Google Scholar]
- 37. Yamamoto M, Takahashi Y, Tabata Y. Controlled release by biodegradable hydrogels enhances the ectopic bone formation of bone morphogenetic protein. Biomaterials. 2003;24:4375–4383. [DOI] [PubMed] [Google Scholar]
- 38. Zhao B, Katagiri T, Toyoda H, et al. . Heparin potentiates the in vivo ectopic bone formation induced by bone morphogenetic protein-2. J Biol Chem. 2006;281:23246–23253. [DOI] [PubMed] [Google Scholar]
- 39. Soysa NS, Alles N, Weih D, et al. . The pivotal role of the alternative NF-κB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J Bone Miner Res. 2010;25:809–818. [DOI] [PubMed] [Google Scholar]
- 40. Maruyama T, Fukushima H, Nakao K, et al. . Processing of the NF-κB2 precursor p100 to p52 is critical for RANKL-induced osteoclast differentiation. J Bone Miner Res. 2010;25:1058–1067. [DOI] [PubMed] [Google Scholar]
- 41. Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-κB RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Katagiri T, Yamaguchi A, Komaki M, et al. . Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol. 1994;127:1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hirata S, Kitamura C, Fukushima H, et al. . Low-level laser irradiation enhances BMP-induced osteoblast differentiation by stimulating the BMP/Smad signaling pathway. J Cell Biochem. 2010;111:1445–1452. [DOI] [PubMed] [Google Scholar]