

Abstract
Despite the well-documented health benefits of ω-3 polyunsaturated fatty acids (PUFAs), their use in clinical management of hyperglycemia and obesity has shown little success. To better define the mechanisms of ω-3 PUFAs in regulating energy balance and insulin sensitivity, we deployed a transgenic mouse model capable of endogenously producing ω-3 PUFAs while reducing ω-6 PUFAs owing to the expression of a Caenorhabditis elegans fat-1 gene encoding an ω-3 fatty acid desaturase. When challenged with high-fat diets, fat-1 mice strongly resisted obesity, diabetes, hypercholesterolemia, and hepatic steatosis. Endogenous elevation of ω-3 PUFAs and reduction of ω-6 PUFAs did not alter the amount of food intake but led to increased energy expenditure in the fat-1 mice. The requirements for the levels of ω-3 PUFAs as well as the ω-6/ω-3 ratios in controlling blood glucose and obesity are much more stringent than those in lipid metabolism. These metabolic phenotypes were accompanied by attenuation of the inflammatory state because tissue levels of prostaglandin E2, leukotriene B4, monocyte chemoattractant protein-1, and TNF-α were significantly decreased. TNF-α–induced nuclear factor-κB signaling was almost completely abolished. Consistent with the reduction in chronic inflammation and a significant increase in peroxisome proliferator–activated receptor-γ activity in the fat-1 liver tissue, hepatic insulin signaling was sharply elevated. The activities of prolipogenic regulators, such as liver X receptor, stearoyl-CoA desaturase-1, and sterol regulatory element binding protein-1 were sharply decreased, whereas the activity of peroxisome proliferator–activated receptor-α, a nuclear receptor that facilitates lipid β-oxidation, was markedly increased. Thus, endogenous conversion of ω-6 to ω-3 PUFAs via fat-1 strongly protects against obesity, diabetes, inflammation, and dyslipidemia and may represent a novel therapeutic modality to treat these prevalent disorders.
ω-3 polyunsaturated fatty acids (PUFAs) are essential fatty acids that must be derived from the diet because of the lack of endogenous enzymes for ω-3 desaturation in mammals. Modern diets contain excessive levels of ω-6 PUFAs but very low levels of ω-3 PUFAs, leading to an unhealthy ω-6/ω-3 ratio of ∼20:1, instead of the ideal 1:1 for humans (1). Eicosanoid products derived from ω-6 PUFAs (such as prostaglandin [PG] E2 and leukotriene [LT] B4 synthesized from arachidonic acid [AA]) are more potent mediators of inflammation than similar products derived from ω-3 PUFAs (PGE3 and LTB5 synthesized from eicosapentaenoic acid [EPA]). Thus, an imbalanced ω-6/ω-3 ratio in favor of ω-6 PUFAs is highly proinflammatory, which is believed to have contributed to the prevalence of atherosclerosis, obesity, and diabetes (1–5). Indeed, regular consumption of diets rich in ω-3 PUFAs has been associated with low incidences of these diseases, particularly in Icelandic populations, Inuit indigenous people, and Native Americans in Alaska (6–8). However, using fish oil as the primary source of ω-3 PUFAs to treat type 2 diabetes has not met with success (5, 9, 10). Although nutritional studies suggest that high ω-6/ω-3 ratios have contributed significantly to the “obesity epidemic” (11, 12), clinical trials using ω-3 PUFAs as weight-reducing agents have produced conflicting findings of both positive (13–15) and negative effects (16–18). Thus, further systematic studies are needed to fully decipher the functions and underlying mechanisms of ω-3 PUFAs in the regulation of energy balance and insulin sensitivity.
To better define the mechanisms by which ω-3 PUFAs improve cardiometabolic risk, we took advantage of fat-1 transgenic mice that carry a Caenorhabditis elegans fat-1 gene encoding a ω-3 fatty acid desaturase. This enzyme can convert ω-6 to ω-3 PUFAs by adding a double bond at the ω-3 position (19). Consequently, fat-1 not only increases the levels of ω-3 PUFAs but also concomitantly decreases ω-6 PUFAs as well as the ω-6/ω-3 ratio—a goal cumbersome to achieve through dietary means alone but strongly advocated for health benefits. Although not fully equated to a dietary approach, the transgenic method produces the same types of ω-3 PUFAs as those obtained through diets (19). Importantly, the fat-1 model offers numerous advantages in the studies of the health benefits of ω-3 PUFAs. For example, the transgenic model allows elucidation of the mechanisms of actions of ω-3 PUFAs without the compounding issues associated with dietary approaches, such as dose, composition, and duration of treatment applied in different studies (15, 20, 21). The use of fat-1 mice avoid these concerns by feeding exactly the same diet to the transgenic and wild-type (WT) mice. Because FAT-1 is an enzyme, the production of ω-3 PUFAs in mice is limited by the amount of available substrates: ω-6 PUFAs. Indeed, the degree of increase in ω-3 PUFAs (∼3- to 4-fold) required to improve metabolic parameters in fat-1 transgenic mice has been shown to be achievable through dietary means in animals and humans (22). Using fat-1 mice, we can also readily perform genetic studies in different obese and diabetic mice by crossing the transgenic model with these diseased models. Several recent studies have already reported various aspects of metabolic phenotypes related to insulin sensitivity and fatty liver in the fat-1 mice (23–25). However, a systematic investigation is still needed to better define how alterations in the tissue levels of ω-6 and ω-3 PUFAs affect energy balance, lipid and glucose metabolism, chronic inflammation, and the underlying molecular events. Such knowledge will help us determine the therapeutic potential of promoting ω-3 desaturation of fatty acids via FAT-1 activity in the prevention and treatment of obesity and type 2 diabetes.
Materials and Methods
Animals
Fat-1 mice were generated in the C57BL/6 background, and the mice were derived from one of the previously characterized transgenic founders (19). All mice were housed under controlled temperature (22 ± 2°C) and 12 hours of the light-dark cycle with free access to water and food. Mice were fed a regular diet (RD) (5% by weight, 13.8% energy from fat; Prolab Isopro RMH 3000 diet), high-fat diet 1 (HFD1) (21.2% fat by weight, 42% energy from fat; Harlan Teklad), or high-fat diet 2 (HFD2) (safflower oil: HFD1, 1:10 in weight, 52% energy from fat). The majority of the fat content in the 2 high-fat diets is saturated fat based on gas-phase chromatography. High-fat feeding started at 6 weeks of age and continued for 12 weeks. In this study, we have primarily used the fat-1 mice bearing 2 sets of fat-1 transgenes (“homozygous”) in the analysis of metabolic phenotypes. Quantitative PCR assays of tail genomic DNA samples were performed to determine the heterozygotic and homozygotic nature of litters after the mating of heterozygotic fat-1 mice with each other. The mRNA expression levels of the fat-1 transgenes were also determined and found to be consistent with the corresponding genotypes (Supplemental Figure 1, A and B). Analysis of a group of heterozygous fat-1 mice [designated as “fat-1(H)”], revealed that their body weight, fat mass, and glucose intolerance were very similar to those of WT control mice fed a high-fat diet (Supplemental Figure 1, C–F), which was in line with their higher ω-6 PUFA and lower ω-3 PUFA tissue contents than those of the homozygotic fat-1 mice (Supplemental Table 1). In all cases, the WT littermates were used as the control mice in all subsequent metabolic assays. All procedures were approved by the Animal Care and Use Committee of the University of Pittsburgh (Pittsburgh, Pennsylvania) and Nanjing Medical University.
Body composition and energy balance studies
Dual-energy X-ray absorptiometry analysis of body composition was performed on a PIXImus densitometer according to manufacturer's protocol. Oxygen consumption, CO2 production, respiratory quotient, and activity were measured in up to 8 mice simultaneously by using an indirect calorimeter (Oxymax Equal Flow System).
Gas chromatography analysis of fatty acids
Lipids were extracted from food and tail samples according to the method of Bligh and Dyer (26). Gas chromatography was performed on a PerkinElmer Clarus 500 system. Identification of components was by comparison of retention times with those of authentic standards.
Intraperitoneal glucose tolerance test (IPGTT)
The IPGTT was performed by an ip injection of glucose (2 g/kg body weight) after a 16-hour overnight fast. Blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 minutes after injection using a Precision Xtra meter (Abbott Laboratories).
Biochemical analyses of insulin, cholesterol, hepatic triglycerides, LTB4, and PGE2
Serum insulin levels were determined using an insulin ELISA kit (LINCO Research). Serum cholesterol was measured using a cholesterol/cholesteryl ester quantification kit (EMD Biosciences). Hepatic triglycerides were measured with a kit from Sigma-Aldrich. PGE2 and LTB4 were measured by an ELISA (R&D Systems).
Hepatocyte preparation and transfection
Hepatocytes were isolated from 8- to 10-week-old male mice fed the RD. Livers were perfused with collagenase solution as described previously (27). The VP-LXR expression vector and the liver X receptor response element (LXRE)– and peroxisome proliferator–activated receptor response element (PPRE)-driven luciferase reporter gene constructs were provided by Dr. Wen Xie's group (28, 29). VP-LXR was created by fusing the VP16 activation domain of herpes simplex virus to the amino terminus of mouse LXR and driven by a promoter of the rat liver fatty acid–binding protein gene. Peroxisome proliferator–activated receptor (PPAR)-α and PPAR-γ were driven by a cytomegalovirus promoter in the pCMX vector. The LXRE- and the PPRE-driven luciferase vectors were generated with 3 copies of LXRE and PPRE elements, respectively, inserted into the tk-Luc vector. Cells were treated with various agents 24 hours after transfection. After another 24-hour incubation, cellular luciferase activity was measured according to the manufacturer's instructions (Promega), and activities were expressed as a percentage of the corresponding controls. Cotransfected β-galactosidase activity was determined to normalize transfection efficiency.
Analysis of gene expression
Expression levels of monocyte chemoattractant protein-1 (MCP-1), TNF-α, stearoyl-CoA desaturase-1 (SCD-1) were measured using real-time PCR assays. The cDNA was generated from total RNA by reverse transcription using random primers and SuperScript II reverse transcriptase. Mouse 18s-rRNA was used as a reference gene for cDNA input. The probes were labeled with FAM on the 5′ end and with BHQ-1 on the 3′ end. Oligonucleotide sequences are listed in Supplemental Table 2.
Western blot
Phosphorylation of Akt was evaluated via Western blot assays using antibodies against phospho-Akt and total Akt. Analysis of TNF-α–induced IκB-α phosphorylation was performed using an antibody against phospho-IκB-α. A control immunoblot was performed with an antibody against Iκκ-α. Each cellular assay was repeated 3 times as a confirmation. The expression of SCD-1, PPAR-γ, LXR-α, and retinoic acid receptor (RAR)-α was examined using corresponding antibodies with β-actin as the control. All Western blot assays of tissue samples were repeated at least 3 times with 3 to 4 tissue samples per group.
Statistical analysis
Data are presented as means ± SD. Differences between groups were analyzed by a two-tailed Student t test or one-way ANOVA. All values of P < .05 were considered statistically significant.
Results
Resistance to high-fat diet–induced obesity due to increased expenditure of energy
We fed 3 different diets to fat-1 transgenic and WT mice: low-fat RD; HFD1, traditionally used in many studies (30, 31) with 42% of energy derived from lipids; and HFD2, an ω-6 PUFA-enriched high-fat diet, with 52% of energy derived from fat. To approximate the compositions of ω-6 and ω-3 PUFAs in Western diets (1), we added safflower oil to the HFD1 to elevate the ω-6 PUFA content, resulting in a ω-6/ω-3 ratio of ∼20:1 (termed HFD2). Analyses of fatty acid compositions indicated that the total levels of PUFAs (including both ω-6 and ω-3 PUFAs) were very low in the HFD1, representing only ∼0.43% of the total diet content in weight (Tables 1 and 2). This is in contrast to those values for the RD and HFD2, for which the total PUFAs constituted about ∼2% and 2.6% of diet weight, respectively. Interestingly, the ratio of ω-6 to ω-3 in the HFD1 was even less than that in the RD (4.5 vs 6.2 in the RD).
Table 1.
Analyses of Levels of ω-6 and ω-3 PUFAs in Diets
| ω-6, mg/g | ω-3, mg/g | ω-6/ω-3 ratio | |
|---|---|---|---|
| RD | 17.43 ± 0.13 | 2.79 ± 0.34 | 6.22 ± 0.41 |
| HFD1 | 3.54 ± 0.13 | 0.78 ± 0.01 | 4.53 ± 0.08 |
| HFD2 | 25.20 ± 0.08 | 1.26 ± 0.01 | 19.95 ± 0.02 |
The composition of ω-6 and ω-3 PUFAs in the RD, HFD1, and HFD2 was analyzed using standard protocols (see Materials and Methods). Compositions of total ω-6 or ω-3 PUFAs were expressed in milligrams per gram (grams of food). All data presented are means ± SD; n = 6.
Table 2.
Distribution of ω-6 (LA, AA, and DPA) and ω-3 (ALA, EPA, and DHA) Species in Diets
| ω-6 PUFA Species, % |
ω-3 PUFA Species, % |
|||||||
|---|---|---|---|---|---|---|---|---|
| LA | AA | DPA | Total ω-6 | ALA | EPA | DHA | Total ω-3 | |
| RD | 39.95 ± 0.26 | 0.32 ± 0.02 | 0.08 ± 0.00 | 40.36 ± 0.28 | 3.78 ± 0.13 | 1.23 ± 0.08 | 1.24 ± 0.12 | 6.48 ± 0.33 |
| HFD1 | 3.26 ± 0.04 | 0.17 ± 0.01 | 0.01 ± 0.00 | 3.45 ± 0.05 | 0.63 ± 0.00 | 0.04 ± 0.00 | 0.00 ± 0.00 | 0.76 ± 0.00 |
| HFD2 | 21.91 ± 0.09 | 0.04 ± 0.00 | 0.01 ± 0.00 | 21.95 ± 0.09 | 0.92 ± 0.01 | 0.08 ± 0.00 | 0.00 ± 0.00 | 1.10 ± 0.01 |
Abbreviations: ALA, α-lipoic acid; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; LA, linoleic acid. Each species is expressed as a percentage of all fatty acid peaks, ie, the distribution areas of different ω-3 or ω-6 PUFA peaks divided by the total peak areas of all detectable saturated and unsaturated free fatty acids (from the same sample) resolved from the gas chromatography column.
Previous characterizations of fat-1 transgenic mice maintained on ω-6 PUFA-enriched diets or RDs have established that global expression of fat-1 cDNA could increase the levels of ω-3 and lower ω-6 PUFAs in a wide range of tissues: liver, skeletal muscle, brain, and pancreatic islets (19, 32, 33). Our analyses of the tail and liver samples from transgenic mice further confirmed those findings (Tables 3 and 4).
Table 3.
Analysis of ω-6 and ω-3 PUFAs Species in the Tail Samples
| ω-6 PUFA Species, % |
ω-3 PUFA Species, % |
ω-6/ω-3 Ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|
| LA | AA | DPA | Total ω-6 | ALA | EPA | DHA | Total ω-3 | ||
| WT RD | 21.06 ± 0.16 | 2.55 ± 0.05 | 0.08 ± 0.00 | 23.72 ± 0.03 | 0.80 ± 0.01 | 0.17 ± 0.01 | 3.03 ± 0.02 | 4.52 ± 0.01 | 5.25 ± 0.01 |
| fat-1 RD | 19.57 ± 0.63a | 0.19 ± 0.05a | 0.03 ± 0.01 | 19.85 ± 0.63a | 2.47 ± 0.17a | 0.82 ± 0.14a | 3.28 ± 0.09a | 6.74 ± 0.58a | 2.96 ± 0.28a |
| WT HFD1 | 4.47 ± 0.27 | 1.57 ± 0.23 | 0.03 ± 0.01 | 6.14 ± 0.15 | 0.41 ± 0.06 | 0.10 ± 0.01 | 1.57 ± 0.11 | 2.24 ± 0.12 | 2.74 ± 0.24 |
| fat-1 HFD1 | 1.86 ± 0.13a | 0.04 ± 0.01a | 0.01 ± 0.00 | 1.94 ± 0.55a | 0.73 ± 0.11a | 0.70 ± 0.15a | 2.20 ± 0.43a | 3.75 ± 0.45a | 0.52 ± 0.17a |
| WT HFD2 | 21.31 ± 0.60 | 4.07 ± 0.44 | 0.16 ± 0.02 | 25.58 ± 0.55 | 0.25 ± 0.02 | 0.09 ± 0.03 | 1.62 ± 0.13 | 2.32 ± 0.13 | 11.04 ± 0.68 |
| fat-1 HFD2 | 19.01 ± 0.54a | 0.18 ± 0.03a | 0.01 ± 0.00a | 19.20 ± 0.54a | 2.46 ± 0.46a | 0.94 ± 0.03a | 2.13 ± 0.13a | 6.63 ± 0.37a | 2.90 ± 0.20a |
Abbreviations: ALA, α-lipoic acid; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; LA, linoleic acid. Compositions of ω-6 or ω-3 PUFAs were expressed using relative percentages, ie, the distribution areas of ω-3 or ω-6 PUFA peaks divided by the total peak areas of all detectable saturated and unsaturated presented are means ± SD; n = 6.
P < .01 when the fat-1 group was compared with the WT control group.
Table 4.
Analysis of ω-6 and ω-3 PUFA Species in the Liver Samples
| ω-6 PUFA Species, % |
ω-3 PUFA Species, % |
ω-6/ω-3 Ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|
| LA | AA | DPA | Total ω-6 | ALA | EPA | DHA | Total ω-3 | ||
| WT RD | 13.13 ± 0.20 | 3.25 ± 0.12 | 0.04 ± 0.00 | 17.05 ± 0.10 | 0.05 ± 0.01 | 0.27 ± 0.01 | 6.03 ± 0.32 | 6.45 ± 0.30 | 2.65 ± 0.15 |
| fat-1 RD | 9.44 ± 0.67a | 3.04 ± 0.22 | 0.03 ± 0.01 | 13.26 ± 1.50a | 0.06 ± 0.01 | 0.65 ± 0.11a | 9.59 ± 0.09a | 10.30 ± 0.46a | 1.29 ± 0.09a |
| WT HFD1 | 5.09 ± 0.21 | 2.96 ± 0.28 | 0.05 ± 0.01 | 8.18 ± 0.34 | 0.03 ± 0.06 | 0.09 ± 0.01 | 2.14 ± 0.09 | 2.26 ± 0.40 | 2.96 ± 0.05 |
| fat-1 HFD1 | 1.98 ± 0.23a | 0.75 ± 0.09a | 0.01 ± 0.00 | 2.75 ± 0.57a | 0.13 ± 0.03a | 1.85 ± 0.26a | 6.16 ± 1.05a | 8.84 ± 1.87a | 0.32 ± 0.02a |
| WT HFD2 | 20.27 ± 1.98 | 6.78 ± 0.64 | 0.24 ± 0.04 | 29.42 ± 1.70 | 0.16 ± 0.05 | 0.18 ± 0.07 | 2.99 ± 0.42 | 3.45 ± 0.21 | 8.52 ± 0.05 |
| fat-1 HFD2 | 17.09 ± 1.80 | 5.53 ± 0.43b | 0.03 ± 0.01a | 24.12 ± 2.22a | 2.27 ± 0.46a | 0.82 ± 0.12a | 6.49 ± 0.92a | 10.56 ± 2.96a | 2.45 ± 0.31a |
Abbreviations: ALA, α-lipoic acid; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; LA, linoleic acid. Compositions of ω-6 or ω-3 PUFAs are expressed using the relative distribution area of the peaks in a given run of a gas chromatography assay. Data are means ± SD; n = 5.
P < .01, fat-1 group compared with the WT control group.
P < .05.
Fat-1 transgenic mice fed the RD did not differ in body weight from age- and sex-matched WT mice (Figure 1A). When fed the HFD1, WT mice developed severe obesity with the percentage of fat mass reaching nearly 43% of the body weight (Figure 1B). The expression of fat-1 led to a strong suppression of both body weight and fat mass in mice fed the HFD1 (Figure 1, A and B). Consistent with these results, analyses of fatty acid compositions in tail and liver samples suggested that the relative level of ω-6 PUFAs in the fat-1 group was >3-fold lower and that the level of ω-3 PUFAs was considerably higher than those in WT tissues (Tables 3 and 4), resulting in a markedly lower ω-6/ω-3 ratio.
Figure 1.

Body weight and body composition of fat-1 transgenic and WT mice fed regular or high-fat diets. A, Body weights of the mice maintained on a standard chow diet (RD) or a high-fat diet (HFD1 or HFD2) for 12 weeks. Although the body weights for each group at the start were not significantly different from each other, the high-fat diet regimen significantly increased the body weights of WT mice when cross-compared to not only the weight of the mice fed the RD but also the weight of the fat-1 mice fed the same high-fat diet at the same time point (6 or 12 weeks) in a one-way ANOVA. B, Body composition (fat mass and lean mass) using dual-energy X-ray absorptiometry. n = 10 to 12 for different groups. Data are means ± SD. #, P < .01; *, P < .05; HFD1- and HFD2-fed fat-1 mice cross-compared with RD-fed mice and WT mice fed the same diet in a one-way ANOVA. C, Side-by-side comparisons of 2 pairs of male fat-1 transgenic and WT mice fed the HFD1 or HFD2, respectively, for 12 weeks.
To evaluate the impact of a high-fat diet with a high ω-6/ω-3 ratio on body weight, we also used the aforementioned ω-6 PUFA-enriched HFD2. After HFD2 feeding, WT mice rapidly gained body weight and developed severe obesity, with the percentage of fat mass reaching almost 50% of their body composition (Figure 1, A–C). All of the fat-1 mice gained weight as a result of HFD2 feeding, but the gain was significantly below those of WT mice with average fat mass reaching ∼37% of body composition (Figures 1, B and C). The changes in body weight were primarily at the level of fat mass as there was little change in lean mass (Figure 1B). The ratio of ω-6/ω-3 was sharply reduced in HFD2-fed transgenic mouse (∼2.5–2.9) vs WT (∼9–11) tissues (Tables 3 and 4). This was a result of a significant decrease in ω-6 PUFA levels and an almost 3-fold increase in ω-3 PUFA levels. Thus, in the context of high tissue levels of ω-6 PUFAs, the increase in the levels of ω-3 PUFAs and the reduction in ω-6 PUFAs appear to have critical roles in preventing diet-induced obesity. Interestingly, further examination of different ω-6 and ω-3 PUFA species in transgenic tissues revealed a sharp decrease in AA and a corresponding increase in EPA (via addition of a double bond to AA) (Tables 3 and 4), which helps explain the reduced inflammatory state in fat-1 mice (see below).
To further delineate the lean phenotypes of fat-1 transgenic mice, we used an indirect calorimetric approach to evaluate energy balance. We found that energy intake was not significantly different between the transgenic and the WT fed either a regular or high-fat diet (Supplemental Figure 2, A and B). When fed a RD, transgenic and WT mice displayed almost identical levels of oxygen consumption, energy expenditure, and physical activity (Figure 2, A–C). After feeding of a high-fat diet, the values of energy expenditure, adjusted with lean body mass were very similar in the 2 genotypes (Supplemental Figure 3, A and B). With a method used widely in other studies (34–36), differences in energy expenditure were revealed when we put the whole-body weight (including fat tissues) into the consideration of energy balance. Under the HFD1 regimen, oxygen consumption of the fat-1 group was markedly elevated (Figure 2D), and energy expenditure was significantly higher than that of WT mice at most time points of the dark phase (Figure 2E, shaded region) as well as some time points of the light phase (Figure 2E). Consistent with these observations, total physical activity of the fat-1 group was also significantly elevated compared with that of WT mice (Figure 2F). Similar results were observed in HFD2-fed mice (Figure 2, G–I). The differences in energy expenditure observed in high-fat diet–fed mice were probably not an indirect result of drastic differences in body weight between the 2 genotypes at the end of prolonged high-fat feeding. In transgenic mice fed the HFD2 for 3 weeks, we observed elevated oxygen consumption and energy expenditure (adjusted with total or lean body mass) (Supplemental Figure 4, A–C). Interestingly, fat-1–induced production of ω-3 PUFAs also led to a marked increase in the respiratory quotient (Supplemental Figure 4, D and E), which suggested biased usage of fuel toward carbohydrate or protein owing to the low lipid levels in the tissues of transgenic mice (see below). Taken together, these data suggest that endogenous production of ω-3 PUFAs and reduction in the levels of ω-6 PUFAs did not have a significant impact on energy intake but could induce increased energy expenditure.
Figure 2.

Energy expenditure in fat-1 and WT mice fed the RD, HFD1, or HFD2. Oxygen consumption and energy expenditure were assessed using a comprehensive laboratory animal metabolic monitoring system in fat-1 and WT mice fed the RD, HFD1, or HFD2 for 12 weeks. Oxygen consumption, 24-hour profiles of average energy expenditure, and 24-hour total physical activity values during the light phase and dark phase were measured in male fat-1 transgenic and WT mice (A–C, RD; D–F, HFD1; G–I, HFD2). Data are means ± SD of 4 mice per group. #, P < .01; *, P < .05.
Changes in ω-6 and ω-3 compositions via fat-1 expression attenuate high-fat diet–induced insulin resistance
Changes in ω-6 and ω-3 PUFA compositions in fat-1 mice had profound influences on glucose metabolism and insulin sensitivity. The intraperitoneal glucose tolerance test showed that, despite their similarity in body weight while fed the RD, fat-1 mice have much better glucose tolerance than their WT counterparts (Figure 3A). Consistent with such divergence in glucose tolerance curves, activation of Akt by insulin was also much stronger in primary hepatocytes isolated from transgenic mice than in those from WT mice (Figure 3B), suggesting that improved insulin signaling was (at least in part) responsible for the enhanced glucose tolerance.
Figure 3.

Attenuation of diet-induced insulin resistance in fat-1 transgenic mice. A, IPGTT. Male mice, maintained on the RD, HFD1, or HFD2, were injected ip with glucose (2 g/kg body weight). Blood glucose was measured at the indicated time points (means ± SD) (n = 10–12 for different groups). B, Western blot assays and quantitative analysis of insulin (Ins)-induced phosphorylation of Akt (p-Akt) in primary hepatocytes isolated from male fat-1 transgenic or WT mice fed the RD. Cells were stimulated with insulin (0, 0.1, 0.2, 0.4, 1.0, and 5.0 nM) for 15 minutes. The image represents 1 of 3 independent experiments. C, Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated using the formula (fasting plasma insulin × plasma glucose)/22.5. The mean fasting plasma insulin levels for each group are as follows: WT on RD, 0.585 ng/mL; fat-1 on RD, 0.647 ng/mL; WT on HFD1, 1.567 ng/mL; fat-1 on HFD1, 0.683 ng/mL; WT on HFD2, 1.009 ng/mL; and fat-1 on HFD2, 0.655 ng/mL. The mean fasting blood glucose levels for each group are as follows: WT on RD, 138 mg/dL; fat-1 on RD, 98 mg/dL; WT on HFD1, 192 mg/dL; fat-1 on HFD1, 115 mg/dL; WT on HFD2, 198 mg/dL; and fat-1 on HFD2, 143 mg/dL. D, Insulin concentrations measured at the indicated time points during the IPGTT in male fat-1 transgenic or WT mice fed the RD, HFD1, or HFD2. #, P < .01; *, P < .05.
As expected, HFD1 feeding induced strong glucose intolerance in WT mice (Figure 3A). However, transgenic mice that strongly resisted weight gain displayed a glucose disposal curve almost identical to that of the transgenic mice maintained on the RD (Figure 3A). Similarly, HFD2 feeding resulted in overt hyperglycemia (fasting blood glucose of >200 mg/dL) and strong glucose intolerance in WT mice; however, these effects were markedly attenuated in transgenic mice (Figure 3A). Homeostasis model assessment further demonstrated that WT mice fed either high-fat diet were insulin resistant, whereas fat-1 mice maintained their insulin sensitivity despite the challenge of the high-fat diets (Figure 3C). Recently, we reported that endogenous production of ω-3 PUFAs via fat-1 expression in pancreatic islets could augment insulin secretion (37). We measured blood insulin concentrations immediately after ip injection of glucose. With RD feeding, the transgenic mice displayed modestly higher concentrations of blood insulin at 15, 30, and 60 minutes than the WT mice (Figure 3D). In high-fat diet–fed mice, blood concentrations of insulin in the 2 genotypes did not differentiate within the first 15 minutes even though the WT mice had higher blood glucose concentrations than the transgenic mice at this time point (Figure 3A). At later time points (≥30 minutes) when blood glucose levels were still very high, the WT mice had dramatically higher concentrations of insulin than the transgenic mice (Figure 3D), indicative of differential states of insulin resistance in these mice. Taken together, these data suggest that the improved glucose tolerance in fat-1 mice was at least in part a result of enhanced insulin sensitivity. Further studies (such as those using glucose clamps) are needed to evaluate this issue.
Fat-1 expression protects against high-fat diet–induced hypercholesterolemia and hepatic steatosis
Fat-1 expression led to a strong suppression of HFD1- and HFD2-induced hypercholesterolemia (Figure 4A). Remarkably, hepatic triglyceride levels in the transgenic animals were >10-fold lower than those of the WT mice under the RD regimen (RD panel, Figure 4B). Feeding of HFD1 and HFD2 induced severe hepatic steatosis in WT mice but not in fat-1 mice (Figure 4B). Thus, the increase in ω-3 PUFA levels and the concomitant decrease in ω-6 PUFA levels as a result of fat-1 expression could have profound beneficial influence on cholesterol metabolism and hepatic triglyceride levels. Interestingly, when we performed metabolic analysis of heterozygotic fat-1 transgenic mice with less ability to convert ω-6 PUFAs to ω-3 PUFAs than the homozygotic transgenic mice, a striking dichotomy was found in body weight and insulin sensitivity between these 2 genotypes, with the heterozygotic transgenic mice gaining as much body weight and having as much glucose intolerance as the WT mice (Supplemental Figure 1, C–F). However, the blood cholesterol levels of these 2 genotypes were similar and sharply lower than that in the WT mice (Supplemental Figure 1, G and H). This result suggests that lipid metabolism is more sensitive to the elevation of tissue ω-3 PUFAs than energy balance and glucose metabolism.
Figure 4.
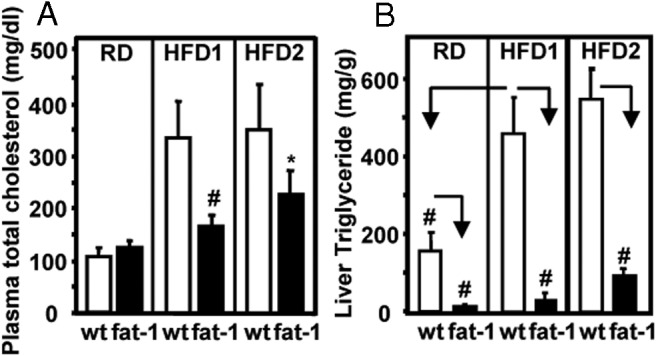
Profiles of plasma total cholesterol and hepatic triglyceride content in fat-1 transgenic and WT mice. Plasma total cholesterol (A) and hepatic triglyceride content (B) in fat-1 transgenic and WT mice fed a regular or high-fat diet for 12 weeks. Data are means ± SD. n = 10 to 12 mice/group. #, P < .01; *, P < .05.
Expression of genes involved in the regulation of lipogenesis and inflammation
To probe the underlying mechanisms, we studied the activities and expression of some of the key regulators of lipid biosynthesis, oxidation, and inflammation. Stimulation of the LXR can lead to activation of SCD-1 expression and SREBP-1 and subsequently lipogenesis in the liver (38–41). After transfecting an LXRE-driven luciferase reporter gene into the primary hepatocytes isolated from RD-fed transgenic or WT mice, we found that, in the absence of cotransfection of LXR, luciferase reporter activity was significantly lower in the cells of transgenic mice than in those of cells of WT mice (Figure 5A). Cotransfection with an LXR-α–expressing construct resulted in an increase in reporter activity (Figure 5A); however, LXR-α–induced luciferase activity was sharply reduced in fat-1 hepatocytes (Figure 5A). To further confirm the specificity of these responses to LXR, we applied an LXR-specific agonist, T0901317, to enhance luciferase activity. Agonist-elicited responses were significantly attenuated in fat-1 primary hepatocytes (Figure 5A). Consistent with the notion of SREBP-1c being a downstream component of the LXR signaling cascade, SREBP-driven luciferase reporter gene activity was significantly decreased in fat-1 transgenic hepatocytes (Figure 5A). In contrast to the reduced activity of these prolipogenic transcription regulators, the activity of PPAR-α, a nuclear receptor that is critical in promoting lipid oxidation, was markedly enhanced, which was further confirmed in the presence of exogenously introduced PPAR-α and PPAR-α agonist, GW7647 (Figure 5B). Similarly, the activity of PPAR-γ was also found to be significantly increased in fat-1 hepatocytes (Figure 5C). A reduction in LXR activity should also predict decreased expression of SCD-1. Indeed, hepatic expression of SCD-1 mRNA and protein was reduced in transgenic mice compared with that in WT mice (Figure 5, D and E). Western blot assays also revealed that hepatic expression levels of nuclear receptors, particularly LXR-α, RAR-α (a heterodimeric partner of LXR), and PPAR-γ, in transgenic mice were similar to those in WT mice (Figure 5, F–H), suggesting that alterations in nuclear receptor activities were unlikely to result in changes in expression.
Figure 5.

Evaluation of the activities of LXR, SREBP-1, PPAR-γ, and SCD-1. A–C, The LXRE- or PPRE-driven luciferase (luc) reporter was expressed in the presence or absence of the corresponding nuclear receptors, LXR (A), PPAR-α (B), and PPAR-γ (C), in primary hepatocytes isolated from fat-1 transgenic and WT mice. Note the difference in scale between the left panel (no cotransfection of LXR) and right panel (with cotransfection of LXR). An SREBP-responsive luciferase reporter (SREBP-luc) was also expressed in primary hepatocytes to assess the activity of SREBP-1. The agents T0901317 (2 μM), GW7647 (1 μM), and 15-PGJ2 (1 μM) are specific agonists for LXR, PPAR-α, and PPAR-γ, respectively. Luciferase activity was expressed as the percentage of control activity (with luciferase activity in wild-type cells in the absence of cotransfection set as 1.0). Each condition was assessed in quintuplet. D, Hepatic expression of SCD-1 mRNA in fat-1 transgenic and WT mice after HFD1 and HFD2 feeding. n = 5 per group. E, Hepatic SCD-1 expression was examined and quantified by immunoblot analyses of 4 samples per group (HFD1) using an antibody to SCD-1. F–H, Western blot assay and quantification of LXRα (F), RARα (G), and PPARγ (H) in livers of male transgenic and wild-type mice fed the RD. Control immunoblots were performed using an antibody against β-actin. Representative images of 3 repeats are shown here. #, P < .01; *, P < .05.
Lipotoxicity and inflammation are implicated in the development of insulin resistance (42). Long-term dietary intake of ω-3 PUFAs has been demonstrated to attenuate inflammation in vivo (43, 44). To evaluate inflammation in fat-1 mice, we measured mRNA levels of TNF-α) and MCP-1 in the liver and/or adipose tissues. After feeding of the HFD1- or HFD2, expression of MCP-1 mRNA was sharply reduced in the liver and adipose tissues of transgenic mice (Figure 6A). Similarly, TNF-α mRNA was decreased in the adipose tissues of fat-1 mice (Figure 6A). Western blot analyses revealed that TNF-α–induced activation of NF-κB was markedly attenuated (Figure 6B). To further evaluate the inflammatory conditions in transgenic mice, we examined the tissue contents of PGE2 and LTB4 (both of which are derived from AA). Consistent with the reduction in ω-6 PUFA levels, hepatic contents of PGE2 and LTB4 were significantly lower in almost all of the fat-1 transgenic groups than in the WT groups (Figure 6, C and D). We also measured serum concentrations of adipocyte-derived cytokines. Serum leptin levels were markedly lower and serum adiponectin levels (adjusted with fat weight) were higher in high-fat diet–fed transgenic mice than in WT mice (Figure 6, E and F). Blood concentrations of TNF-α and interleukin-6 were also significantly lower in high-fat diet–fed transgenic mice than in WT mice (data not shown). Thus, altered lipid metabolism and reduced inflammation could have contributed to the resistance of fat-1 mice to diet-induced obesity, hepatic steatosis, insulin resistance, and hypercholesterolemia.
Figure 6.

Assessment of inflammation with measurement of levels of MCP-1, TNF-α, PGE2, LTB4, leptin, and adiponectin. A, Expression of MCP-1 and TNF-α mRNA in liver and/or adipose tissues of fat-1 transgenic and WT mice fed an HFD1 or HFD2. B, Western blot analyses. Time courses of hepatic IκB-α phosphorylation after treatment with recombinant TNF-α (20 ng/mL). Control immunoblots were undertaken using an antibody against Iκκ-α. This is representative of 3 independent experiments. C and D, Measurements of levels of PGE2 (C) and LTB4 (D) in liver samples of male fat-1 transgenic and WT mice fed a regular of high-fat diet for 12 weeks. E, Plasma leptin concentrations in fat-1 transgenic and WT mice fed a regular or high-fat diet for 12 weeks. F, Plasma adiponectin concentrations expressed per gram of white adipose tissue (WAT) in fat-1 transgenic and WT mice. Data are means ± SD. n = 10 to 12 mice per group. #, P < .01; *, P < .05.
Discussion
Fat-1 transgenic mice provide a highly efficient model to investigate the global impact of varying the species of PUFAs on energy balance and insulin sensitivity in vivo. Although several earlier reports have indicated subsets of metabolic phenotypes in fat-1 mice, such as fatty liver and insulin resistance (23–25), this study represents a first systematic investigation on a wide range of metabolic issues, including energy balance, body composition, glucose and lipid metabolism, insulin sensitivity, inflammatory state, and the expression of many important underlying regulators in metabolism. Consequently, a series of metabolic benefits have been revealed in the fat-1 mice, and such results may guide us to treat major metabolic diseases by adjusting the levels of ω-3- and ω-6 PUFAs in humans.
Numerous clinical and epidemiological studies have shown that excessive levels of ω-6 PUFAs as well as low levels of ω-3 PUFAs in diets contribute to the pathogenesis of obesity and diabetes mellitus (1, 45–47). The metabolic phenotypes of fat-1 transgenic mice have provided novel perspectives about the requirements of ω-6/ω-3 ratios and the relative levels of ω-3 PUFAs in regulating energy balance, insulin sensitivity, and lipid metabolism. We found that transgenic expression of fat-1 did not alter energy intake levels but significantly increased energy expenditure as well as total physical activity in high-rat diet–fed mice. The values of energy expenditure adjusted with lean mass alone did not differ between transgenic and WT mice, but significant elevation of energy expenditure and oxygen consumption was revealed in fat-1 transgenic mice when we adjusted those values with total body weight, a method also adopted in recent studies (34–36). Consistent with such interpretations, transgenic mice also showed elevated energy expenditure even during short-term high-fat feeding when the WT mice have not shown overt obesity. Taken together, our data suggest that fat-1–induced production of ω-3 and a reduction in ω-6 PUFAs levels favor a lean phenotype, primarily through stimulation of energy expenditure.
Previous attempts to use fish oil as the primary source of ω-3 PUFAs to treat type 2 diabetes have not met with success (9). Modern Western diets generally contain low levels of PUFAs and sharply imbalanced ω-6/ω-3 PUFA ratios (1–5). Indeed, our studies revealed that it would take a significant drop in ω-6 (particularly AA) PUFAs and a several fold increase in ω-3 PUFAs (primarily EPA) to fend off obesity and type 2 diabetes (Tables 3 and 4) in the fat-1 mice after feeding of the HFD1 or HFD2. Such a magnitude of alterations in PUFA contents can be hardly achieved clinically through fish-oil compensation without other major nutritional interventions to reduce the intake of ω-6 PUFAs. We want to emphasize that simply reducing the ω-6/ω-3 ratio is not likely to be sufficient to counter obesity and diabetes; the absolute levels of ω-3 PUFAs also appear to be critical. On this note, we observed that heterozygotic fat-1 mice had less ability to convert ω-6 to ω-3 PUFAs than the homozygous fat-1 transgenic mice used in this study (see Material and Methods and Supplemental Table 1). Such transgenic mice, after being fed the HFD1, failed to differentiate themselves from the WT mice in obesity and insulin resistance, even though their ω-6/ω-3 ratio was nearly identical to that of fat-1 mice fed the HFD2 (Tables 3 and 4). The HFD1 contained a very low content of total PUFAs, which was in contrast to that of the HFD2 (Tables 1 and 2). The high levels of dietary ω-6 PUFAs in the HFD2 provided sufficient substrates for the FAT-1 enzyme to produce high levels of ω-3 PUFAs in the tissues (Tables 3 and 4). Interestingly, the heterozygotic transgenic mice still managed to resist the HFD1-induced hypercholesterolemia and hepatic steatosis (Supplemental Figure 1, C–H), suggesting a more stringent requirement of ω-3 PUFAs in managing blood glucose and body weight than in lipid control. Interestingly, an earlier study of fat-1 mice fed a high-fat diet failed to show differences in body weight between the fat-1 and WT mice, which might also be due to the heterozygotic nature of their transgenic mice (23–25). Thus, our findings help explain why the benefits of daily intake of ω-3 PUFAs have been primarily on blood lipid levels and hepatic steatosis (9, 48), not on glucose control and body weight. Any future clinical trials of ω-3 PUFAs as an agent to correct hyperglycemia should couple high (and tolerable) doses of ω-3 PUFAs with reduction in the dietary intake of ω-6 PUFAs.
Molecular analyses of fat-1 mouse tissues have revealed some underlying mechanisms about how ω-3 PUFAs affect metabolism. We probed some of the key regulators of inflammation. The conversion of ω-6 PUFAs to ω-3 PUFAs via fat-1 led to a decrease in the levels of AA and AA-derived lipid mediators of inflammation, such as PGE2 and LTB4, in the livers of fat-1 mice. Similar findings have been reported in islet and colon tissues of fat-1 mice (32, 37), implying a broad down-regulation of inflammation. Chronic low-grade inflammation, particularly via TNF-α-induced NF-κB activation, exerts a strong negative effect on insulin sensitivity and insulin signaling (49–51). The molecular phenotypes of fat-1 mice appear to favor a lessened inflammatory state. Accordingly, TNF-α-induced NF-κB signaling was significantly attenuated. Hepatic and adipose expression of MCP-1 was dramatically lower in high-fat diet–fed fat-1 mice than in WT mice, which by inference indicated reduced infiltration of monocytes and macrophages. PPAR-γ, the activation of which is known to dampen inflammation and sensitize the actions of insulin (52, 53), was stimulated in cells derived from fat-1 mice. Levels of proinflammatory mediators, such as PGE2 and LTB4, were markedly reduced in the tissues of transgenic mice. Taken together, these data suggest that attenuation of inflammation contributes (at least in part) to enhanced glucose metabolism in fat-1 transgenic mice.
By inference, suppression of lipogenesis and elevation of lipid oxidation should lead to reduced adiposity and tissue triglycerides. The exploration of some nuclear receptors helped us delineate the mechanisms underlying the phenotypes in lipid metabolism. In this context, changes in ω-3 and ω-6 PUFAs compositions via fat-1 expression markedly inhibited the activities of prolipogenic LXR and SREBP-1 and elevated the activity of PPAR-α, a nuclear receptor that is critical for promoting lipid oxidation (54). Because we did not see an alteration in protein expression in these nuclear receptors, the sharply increased ω-3 PUFA levels on their activities were probably acting as the ligands of LXR or PPARs and potently out-competed the binding of oxysterol. A reduction in the hepatic expression of SCD-1 in fat-1 transgenic mice might also have contributed to elevated energy expenditure and reduced obesity, both of which were evident in SCD-1 knockout mice (55). Although the mechanisms underlying the antiobesity phenotype in the fat-1 mice remain to be systematically investigated, it is tempting to propose that the effect on energy expenditure is mediated at least in part through a recently identified ω-3 PUFA receptor, GPR120 (56). Deficiency of GPR120 in mice and humans produced symptoms very similar to those of WT mice fed low ω-3 PUFA diets, including reduced energy expenditure, obesity, insulin resistance, and an elevated state of inflammation (57). Alternatively, the drastic reduction in AA (ω-6) by fat-1 expression (Tables 3 and 4) is predicted to reduce AA-derived 2-arachidonoyl glycerol and anadamide, 2 endogenous ligands for endocannabinoid receptor, CB1 (58). Genetic and pharmacological blockade of CB1 has been shown to cause leanness, increased energy expenditure without a concomitant reduction in food intake, and enhanced insulin sensitivity, which are all similar to those in fat-1 mice fed high fat diets (58, 59). Taken together, the interactions of these molecular events should explain at least in part the global impact of changing PUFA species on metabolism, particularly the phenotypes of leanness and resistance to hepatic steatosis after high-fat feeding.
Recently, a wide range of health benefits have been reported in fat-1 transgenic mice, such as suppression of osteoporosis (60) and cancer growth (61, 62), as well as enhancement of insulin secretion and protection of pancreatic islets from inflammatory assault (37, 63). Because nutritional compensation through fish oil can only provide a limited increase in ω-3 PUFAs, converting excessive tissue ω-6 PUFAs to ω-3 PUFAs via fat-1 gene therapy may represent a powerful strategy to treat obesity and type 2 diabetes.
Acknowledgments
We thank Ian Sipula and Rhobert Evans for their technical assistance in measuring energy expenditure and fatty acid compositions, respectively, and Dr. Wen Xie for providing LXR-α, PPAR-α, and PPAR-γ expression constructs as well as LXRE- and PPRE-driven luciferase reporter constructs. We truly appreciate the critical reading of this article by Dr. Erin Kershaw.
This work was supported by the grants from the National Program on Key Basic Research Project of China (973 Program) (Grant 2013CB945202 to A.Z.Z. and F.L.), the National Natural Science Foundation of China (Grant 81170780 to A.Z.Z. and 81372798 to F.L.), the PhD Programs Foundation of the Ministry of Education of China (Grant 20113234110005 to A.Z.Z.), the High-Level Innovative Talents Reward from Jiangsu Province, and the Returned Overseas Chinese Scholar from The First Affiliated Hospital of Nanjing Medical University (to F.L.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the grants from the National Program on Key Basic Research Project of China (973 Program) (Grant 2013CB945202 to A.Z.Z. and F.L.), the National Natural Science Foundation of China (Grant 81170780 to A.Z.Z. and 81372798 to F.L.), the PhD Programs Foundation of the Ministry of Education of China (Grant 20113234110005 to A.Z.Z.), the High-Level Innovative Talents Reward from Jiangsu Province, and the Returned Overseas Chinese Scholar from The First Affiliated Hospital of Nanjing Medical University (to F.L.).
Footnotes
- AA
- arachidonic acid
- EPA
- eicosapentaenoic acid
- HFD1
- high-fat diet 1
- HFD2
- high-fat diet 2
- LT
- leukotriene
- IPGTT
- intraperitoneal glucose tolerance test
- LXR
- liver X receptor
- LXRE
- liver X receptor response element
- MCP-1
- monocyte chemoattractant protein-1
- NF-κB
- nuclear factor-κB
- PG
- prostaglandin
- PPAR
- peroxisome proliferator–activated receptor
- PPRE
- peroxisome proliferator–activated receptor response element
- PUFA
- polyunsaturated fatty acid
- RAR
- retinoic acid receptor
- RD
- regular diet
- SCD-1
- stearoyl-CoA desaturase-1
- SREBP-1
- sterol regulatory element-binding transcription factor 1.
References
- 1. Kang JX. The importance of omega-6/omega-3 fatty acid ratio in cell function. The gene transfer of omega-3 fatty acid desaturase. World Rev Nutr Diet. 2003;92:23–36. [DOI] [PubMed] [Google Scholar]
- 2. Simopoulos AP. Evolutionary aspects of diet and essential fatty acids. World Rev Nutr Diet. 2001;88:18–27. [DOI] [PubMed] [Google Scholar]
- 3. Biarnés M, Montolio M, Nacher V, Raurell M, Soler J, Montanya E. β-Cell death and mass in syngeneically transplanted islets exposed to short- and long-term hyperglycemia. Diabetes. 2002;51:66–72. [DOI] [PubMed] [Google Scholar]
- 4. Coelho DF, Pereira-Lancha LO, Chaves DS, et al. Effect of high-fat diets on body composition, lipid metabolism and insulin sensitivity, and the role of exercise on these parameters. Braz J Med Biol Res. 2011;44:966–972. [DOI] [PubMed] [Google Scholar]
- 5. Kromhout D, de Goede J. Update on cardiometabolic health effects of ω-3 fatty acids. Curr Opin Lipidol. 2014;25:85–90. [DOI] [PubMed] [Google Scholar]
- 6. Kromann N, Green A. Epidemiological studies in the Upernavik district, Greenland. Incidence of some chronic diseases 1950–1974. Acta Med Scand. 1980;208:401–406. [PubMed] [Google Scholar]
- 7. Adler AI, Boyko EJ, Schraer CD, Murphy NJ. Lower prevalence of impaired glucose tolerance and diabetes associated with daily seal oil or salmon consumption among Alaska Natives. Diabetes Care. 1994;17:1498–1501. [DOI] [PubMed] [Google Scholar]
- 8. Schraer CD, Risica PM, Ebbesson SO, Go OT, Howard BV, Mayer AM. Low fasting insulin levels in Eskimos compared to American Indians: are Eskimos less insulin resistant? Int J Circumpolar Health. 1999;58:272–280. [PubMed] [Google Scholar]
- 9. Nettleton JA, Katz R. n-3 long-chain polyunsaturated fatty acids in type 2 diabetes: a review. J Am Diet Assoc. 2005;105:428–440. [DOI] [PubMed] [Google Scholar]
- 10. Mozaffarian D, Rimm EB. Fish intake, contaminants, and human health: evaluating the risks and the benefits. JAMA. 2006;296:1885–1899. [DOI] [PubMed] [Google Scholar]
- 11. Birch EE, Hoffman DR, Castañeda YS, Fawcett SL, Birch DG, Uauy RD. A randomized controlled trial of long-chain polyunsaturated fatty acid supplementation of formula in term infants after weaning at 6 wk of age. Am J Clin Nutr. 2002;75:570–580. [DOI] [PubMed] [Google Scholar]
- 12. Guesnet P, Pugo-Gunsam P, Maurage C, et al. Blood lipid concentrations of docosahexaenoic and arachidonic acids at birth determine their relative postnatal changes in term infants fed breast milk or formula. Am J Clin Nutr. 1999;70:292–298. [DOI] [PubMed] [Google Scholar]
- 13. Couet C, Delarue J, Ritz P, Antoine JM, Lamisse F. Effect of dietary fish oil on body fat mass and basal fat oxidation in healthy adults. Int J Obes Relat Metab Disord. 1997;21:637–643. [DOI] [PubMed] [Google Scholar]
- 14. Fontani G, Corradeschi F, Felici A, et al. Blood profiles, body fat and mood state in healthy subjects on different diets supplemented with omega-3 polyunsaturated fatty acids. Eur J Clin Invest. 2005;35:499–507. [DOI] [PubMed] [Google Scholar]
- 15. Hill AM, Buckley JD, Murphy KJ, Howe PR. Combining fish-oil supplements with regular aerobic exercise improves body composition and cardiovascular disease risk factors. Am J Clin Nutr. 2007;85:1267–1274. [DOI] [PubMed] [Google Scholar]
- 16. Risérus U, Arner P, Brismar K, Vessby B. Treatment with dietary trans10cis12 conjugated linoleic acid causes isomer-specific insulin resistance in obese men with the metabolic syndrome. Diabetes Care. 2002;25:1516–1521. [DOI] [PubMed] [Google Scholar]
- 17. Belury MA, Mahon A, Banni S. The conjugated linoleic acid (CLA) isomer, t10c12-CLA, is inversely associated with changes in body weight and serum leptin in subjects with type 2 diabetes mellitus. J Nutr. 2003;133:257S–260S. [DOI] [PubMed] [Google Scholar]
- 18. Chan DC, Watts GF, Nguyen MN, Barrett PH. Factorial study of the effect of n-3 fatty acid supplementation and atorvastatin on the kinetics of HDL apolipoproteins A-I and A-II in men with abdominal obesity. Am J Clin Nutr. 2006;84:37–43. [DOI] [PubMed] [Google Scholar]
- 19. Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427:504. [DOI] [PubMed] [Google Scholar]
- 20. Jump DB, Clarke SD. Regulation of gene expression by dietary fat. Annu Rev Nutr. 1999;19:63–90. [DOI] [PubMed] [Google Scholar]
- 21. Jump DB. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr Opin Lipidol. 2002;13:155–164. [DOI] [PubMed] [Google Scholar]
- 22. Suzukawa M, Abbey M, Howe PR, Nestel PJ. Effects of fish oil fatty acids on low density lipoprotein size, oxidizability, and uptake by macrophages. J Lipid Res. 1995;36:473–484. [PubMed] [Google Scholar]
- 23. Smith BK, Holloway GP, Reza-Lopez S, Jeram SM, Kang JX, Ma DW. A decreased n-6/n-3 ratio in the fat-1 mouse is associated with improved glucose tolerance. Appl Physiol Nutr Metab. 2010;35:699–706. [DOI] [PubMed] [Google Scholar]
- 24. Kim EH, Bae JS, Hahm KB, Cha JY. Endogenously synthesized n-3 polyunsaturated fatty acids in fat-1 mice ameliorate high-fat diet-induced non-alcoholic fatty liver disease. Biochem Pharmacol. 2012;84:1359–1365. [DOI] [PubMed] [Google Scholar]
- 25. White PJ, Arita M, Taguchi R, Kang JX, Marette A. Transgenic restoration of long-chain n-3 fatty acids in insulin target tissues improves resolution capacity and alleviates obesity-linked inflammation and insulin resistance in high-fat-fed mice. Diabetes. 2010;59:3066–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. [DOI] [PubMed] [Google Scholar]
- 27. Edström S, Ekman L, Ternell M, Lundholm K. Isolation of mouse liver cells: perfusion technique and metabolic evaluation. Eur Surg Res. 1983;15:97–102. [DOI] [PubMed] [Google Scholar]
- 28. Zhou J, Febbraio M, Wada T, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology. 2008;134:556–567. [DOI] [PubMed] [Google Scholar]
- 29. Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hosooka T, Noguchi T, Kotani K, et al. Dok1 mediates high-fat diet-induced adipocyte hypertrophy and obesity through modulation of PPAR-γ phosphorylation. Nat Med. 2008;14:188–193. [DOI] [PubMed] [Google Scholar]
- 31. Choi CS, Fillmore JJ, Kim JK, et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1995–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hudert CA, Weylandt KH, Lu Y, et al. Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proc Natl Acad Sci USA. 2006;103:11276–11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. He C, Qu X, Cui L, Wang J, Kang JX. Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc Natl Acad Sci USA. 2009;106:11370–11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. [DOI] [PubMed] [Google Scholar]
- 35. Ye R, Jung DY, Jun JY, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. 2010;59:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knauf C, Cani PD, Ait-Belgnaoui A, et al. Brain glucagon-like peptide 1 signaling controls the onset of high-fat diet-induced insulin resistance and reduces energy expenditure. Endocrinology. 2008;149:4768–4777. [DOI] [PubMed] [Google Scholar]
- 37. Wei D, Li J, Shen M, et al. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic β-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes. 2010;59:471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Y, Kurdi-Haidar B, Oram JF. LXR-mediated activation of macrophage stearoyl-CoA desaturase generates unsaturated fatty acids that destabilize ABCA1. J Lipid Res. 2004;45:972–980. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Y, Zhang X, Chen L, et al. Liver X receptor agonist TO-901317 upregulates SCD1 expression in renal proximal straight tubule. Am J Physiol Renal Physiol. 2006;290:F1065–F1073. [DOI] [PubMed] [Google Scholar]
- 40. Helleboid-Chapman A, Helleboid S, Jakel H, et al. Glucose regulates LXRα subcellular localization and function in rat pancreatic β-cells. Cell Res. 2006;16:661–670. [DOI] [PubMed] [Google Scholar]
- 41. Mitro N, Mak PA, Vargas L, et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219–223. [DOI] [PubMed] [Google Scholar]
- 42. Ye J. Mechanisms of insulin resistance in obesity. Front Med. 2013;7:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singer P, Shapiro H, Theilla M, Anbar R, Singer J, Cohen J. Anti-inflammatory properties of omega-3 fatty acids in critical illness: novel mechanisms and an integrative perspective. Intensive Care Med. 2008;34:1580–1592. [DOI] [PubMed] [Google Scholar]
- 44. Switzer KC, McMurray DN, Chapkin RS. Effects of dietary n-3 polyunsaturated fatty acids on T-cell membrane composition and function. Lipids. 2004;39:1163–1170. [DOI] [PubMed] [Google Scholar]
- 45. Simopoulos AP. Importance of the ratio of omega-6/omega-3 essential fatty acids: evolutionary aspects. World Rev Nutr Diet. 2003;92:1–22. [DOI] [PubMed] [Google Scholar]
- 46. Leaf A, Kang JX, Xiao YF, Billman GE. Clinical prevention of sudden cardiac death by n-3 polyunsaturated fatty acids and mechanism of prevention of arrhythmias by n-3 fish oils. Circulation. 2003;107:2646–2652. [DOI] [PubMed] [Google Scholar]
- 47. Chang CL, Deckelbaum RJ. Omega-3 fatty acids: mechanisms underlying ‘protective effects’ in atherosclerosis. Curr Opin Lipidol. 2013;24:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis. 2006;189:19–30. [DOI] [PubMed] [Google Scholar]
- 49. Arkan MC, Hevener AL, Greten FR, et al. IKK-β links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. [DOI] [PubMed] [Google Scholar]
- 50. Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-κB: effects on liver homeostasis and beyond. Endocr Rev. 2007;28:365–386. [DOI] [PubMed] [Google Scholar]
- 51. Chiang SH, Bazuine M, Lumeng CN, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138:961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Campbell IW. The clinical significance of PPAR γ agonism. Curr Mol Med. 2005;5:349–363. [DOI] [PubMed] [Google Scholar]
- 53. Ferré P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(suppl 1):S43–S50. [DOI] [PubMed] [Google Scholar]
- 54. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. [DOI] [PubMed] [Google Scholar]
- 55. Ntambi JM, Miyazaki M, Stoehr JP, et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci USA. 2002;99:11482–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oh DY, Olefsky JM. Omega 3 fatty acids and GPR120. Cell Metab. 2012;15:564–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ichimura A, Hirasawa A, Poulain-Godefroy O, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483:350–354. [DOI] [PubMed] [Google Scholar]
- 58. Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nat Neurosci. 2005;8:585–589. [DOI] [PubMed] [Google Scholar]
- 59. Engeli S, Böhnke J, Feldpausch M, et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes. 2005;54:2838–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rahman MM, Bhattacharya A, Banu J, Kang JX, Fernandes G. Endogenous n-3 fatty acids protect ovariectomy induced bone loss by attenuating osteoclastogenesis. J Cell Mol Med. 2009;13:1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu Y, Nie D, Witt WT, et al. Expression of the fat-1 gene diminishes prostate cancer growth in vivo through enhancing apoptosis and inhibiting GSK-3 β phosphorylation. Mol Cancer Ther. 2008;7:3203–3211. [DOI] [PubMed] [Google Scholar]
- 62. Nowak J, Weylandt KH, Habbel P, et al. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis. 2007;28:1991–1995. [DOI] [PubMed] [Google Scholar]
- 63. Weylandt KH, Nadolny A, Kahlke L, et al. Reduction of inflammation and chronic tissue damage by omega-3 fatty acids in fat-1 transgenic mice with pancreatitis. Biochim Biophys Acta. 2008;1782:634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]