

Abstract
The glucocorticoid receptor (GR) was one of the first nuclear hormone receptors cloned and represents one of the most effective drug targets available today for the treatment of severe inflammation. The physiologic consequences of endogenous or exogenous glucocorticoid excess are well established and include hyperglycemia, insulin resistance, fatty liver, obesity, and muscle wasting. However, at the molecular and tissue-specific level, there are still many unknown protein mediators of glucocorticoid response and thus, much remains to be uncovered that will help determine whether activation of the GR can be tailored to improve therapeutic efficacy while minimizing unwanted side effects. This review summarizes recent discoveries of tissue-selective modulators of glucocorticoid signaling that are important in mediating the unwanted side effects of therapeutic glucocorticoid use, emphasizing the downstream molecular effects of GR activation in the liver, adipose tissue, muscle, and pancreas.
Glucocorticoids (GCs) cortisol in humans and corticosterone in rodents are endogenous stress hormones that affect nearly every organ and tissue in the body, regulating diverse physiologic processes including energy homeostasis (metabolism), the immune response, skeletal growth, reproduction, behavior, cell proliferation, and survival (1). In normal physiology, stress-triggered activation of hypothalamic-pituitary-adrenal (HPA) axis induces GC synthesis and secretion from the adrenal cortex, which is tightly regulated by feedback inhibition of the HPA axis (2). The essential role of GCs is to supply enough glucose into the circulation to fuel the brain and ensure survival of the organism under conditions of acute stress or reduced food intake (Figure 1). The mechanisms by which GCs orchestrate this effect include: 1) increased hepatic glucose production (2, 3); 2) decreased peripheral glucose uptake into muscle and adipose tissue (4, 5); 3) increased breakdown of fat and muscle to provide additional substrates for glucose production (6–10); and 4) inhibition of insulin release from β-cells (11).
Figure 1.
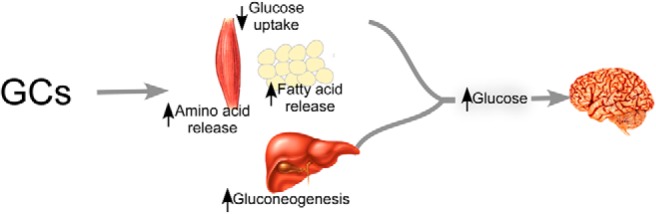
In response to increased GC levels, liver, muscle, and adipose tissue act in concert to increase circulating glucose levels to provide the requisite energy to maintain brain function. GCs increase the transcription of genes involved in de novo gluconeogenesis (liver) while also increasing protein catabolism (muscle) and lipolysis (adipose) to provide substrates for glucose production by the liver.
GCs are elevated basally in obese diabetic mouse models (ob/ob and db/db mice) and in some patients with insulin resistance, and correlate with the occurrence of fatty liver and hyperglycemia (12–15). Likewise, patients with Cushing's syndrome, a disease characterized by chronic elevation of endogenous GCs, have an increased central obesity, hepatosteatosis, and hyperglycemia (16, 17). Adrenalectomy of db/db mice mitigates the diabetic and obese phenotype of this genetic model of disease (18). Signaling through the glucocorticoid receptor (GR) is central to these phenotypes because administration of the GR antagonist RU-486 attenuates hyperglycemia in db/db mice (19, 20).
Synthetic GCs, such as dexamethasone (Dex) and prednisone, have potent antiinflammatory and immunosuppressive effects; and as such, have been widely prescribed over the past 60 years for the treatment of chronic inflammatory diseases, autoimmune diseases, organ transplant rejection, and certain types of leukemia. It is estimated that 1.2% of the US population is currently being treated with a prescription GC drug (21). Chronic use of GCs, however, disturbs normal homeostasis and causes deleterious side effects in humans and rodents including dysregulation of glucose and fat metabolism, hepatosteatosis, insulin resistance, diabetes, osteoporosis, muscle wasting, growth retardation, infertility, cognitive dysfunction, glaucoma, cataracts, and topical skin thinning (1, 3, 5, 22–26). Given the importance of GC drugs in the treatment of chronic inflammatory diseases, the ideal therapeutic would be one that retained the antiinflammatory actions of GCs without inducing the negative side effects. Historically, the pharmaceutical industry tried to tackle this problem by screening for drugs that dissociated GR-mediated transrepression from transactivation, reasoning that the antiinflammatory actions of GCs could be ascribed to transrepression, whereas the unwanted side effects of GCs were a result of combined transactivation and repression (27). This dogma has recently been questioned (28) because dimerization and transactivation of GR are both unequivocally important for its antiinflammatory function (29, 30). For example, GCs directly induce the expression of dual specificity protein phosphatase 1 (Dusp1), a phosphatase that acts to inactivate several transcription factors involved in promoting proinflammatory gene expression (31). We now recognize that tissue, cell, and gene-specific effects of GR activation are orchestrated by many factors including the tissue-specific presence of coregulatory proteins, different GR isoforms, the phosphorylation status of GR, and the sequence of the GR-binding site and flanking DNA on target genes (32, 33). Genome-wide analyses have shown that GR-binding to DNA is highly context specific and relies on the interplay of GR with other proteins (30, 34, 35). Thus, the identification and characterization of molecular factors impinging on GC signaling may provide new insight into mechanisms that can be used to dissociate the positive and negative effects of prescription GC drugs. This review summarizes known mechanisms of GC signal transduction and highlights the more recent findings of novel effectors of GC actions in metabolic tissues.
Overview of GR Biology
Classic mechanisms of GR genomic signaling
At the cellular level, GCs exert both therapeutic and adverse effects through the GR (NR3C1), which belongs to the nuclear receptor superfamily of ligand-activated transcription factors. Upon GC-binding in the cytosol, GR is released from a large multiprotein chaperone complex containing heat shock protein (HSP)-90 and HSP-70. This binding event unmasks the nuclear localization sequence and GR translocates into the nucleus. In the nucleus, it activates or represses transcription of distinct sets of genes either by directly binding to specific DNA sequences (glucocorticoid response elements [GREs]) or through direct protein-protein interactions with other transcription factors and/or coregulators (Figure 2A) (30, 36, 37).
Figure 2.
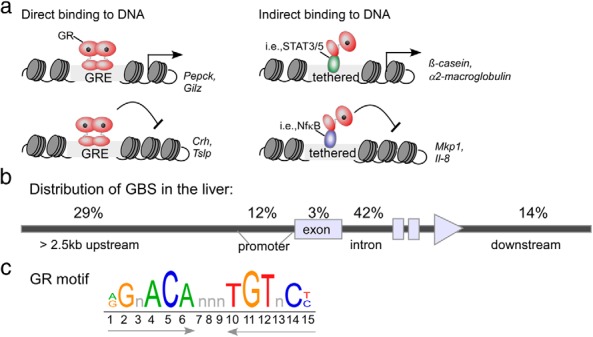
Overview of GR-binding modes. A, GR can bind to DNA as a homodimer to activate or repress gene transcription. Alternatively, GR can tether to other DNA-binding proteins to potentiate activation or repression of genes. The transcription factors STAT3, STAT5, and nuclear factor (NF)-κB have been shown to tether to GR. b, Genomic distribution of GBSs determined from ChIP-seq analysis of mouse liver (adapted from Reference [43]). c, Depiction of the consensus GR-binding motif (IR3) identified by motif-enrichment analysis of ChIP-seq data from mouse liver (43), 3134 cells (34), AtT-20 cells (34), and A549 cells (42) and differentiating 3T3-L1 adipocytes (45). STAT, signal transducer and activator of transcription.
Nongenomic GR signaling
GR has also been shown to participate in nongenomic mechanisms of signal transduction through interactions involving cytosolic GR or specific membrane-bound receptor as reviewed by Stahn et al (38). These nongenomic interactions exert a cellular response within minutes of GC treatment. Although nongenomic mechanisms are not the focus of this review, there are several examples of nongenomic GR interactions that impact the metabolic response to GCs. For example, using kinome analysis, it was found that GC treatment of 3T3 adipocytes hampers insulin signaling by inhibiting insulin receptor (IR) tyrosine kinase activity and phosphorylation of its downstream targets IRS-1 and AKT in a GR-dependent (RU-486 treatment-sensitive) and transcription-independent (actinomycin D-insensitive) mechanism (39).
GR-binding sites revealed from genome-wide chromatin immunoprecipitation (ChIP) studies
Genome-wide studies using ChIP combined with next-generation sequencing (ChIP-seq) have shown that most GR-binding sequences (GBSs) are not present in the typical promoter regions near the transcription start site (TSS) of genes (Figure 2B). A significant number of GBSs are actually located in intragenic regions and at distances greater than 25 kb from the TSS (40). The role that distal binding sites have on transcriptional regulation is not yet clear, and it is difficult to assign a specific binding event to changes in a potential gene target on a genomic scale. In fact, only 50% of the genes that have a GR-binding site within 100 kb of a TSS demonstrate a Dex-dependent transcriptional response (34, 35, 41). Moreover, comparing different GBSs to the DNase accessibility of those same sites demonstrated that approximately half of the GBS motifs had an absolute requirement for accessible chromatin in A549 lung carcinoma cells (34, 42).
ChIP-seq studies performed in mouse liver found that genes activated and repressed by GR are actively remodeled upon GC treatment (ie, have altered DNase hypersensitivity sites [43]). CCAAT/enhancer-binding protein β (C/EBPβ) appears to direct GR-binding to the liver genome through a highly cooperative mechanism whereby 62% of GR-binding sites are pre-occupied by C/EBPβ prior to GR-binding (43). Importantly, by performing an extensive comparison of genome-wide GR-binding sites from mouse liver to previously published GR ChIP-seq data derived from cell lines (ie, mouse mammary adenocarcinoma [3134] [34], pituitary gland tumor [AtT20] [44], preadipocyte embryonic fibroblast [3T3-L1] [45], and myoblasts [C2C12] [40]), the authors found that only 0.5% of the 11 000 binding sites in liver are shared between the 4 other cell types, and 83% of sites are unique to liver tissue (43). Despite the unique locations of the GR-binding sites in different tissues, the consensus GR response element that was enriched in each of the studies was very similar and consisted of an inverted repeat with 3-nucleotide spacer (Figure 2C). The above mentioned studies further reinforce the idea that cell/tissue-specific expression of proteins dictates GR-binding sites and transcriptional output.
Molecular Mediators of GR Activity
GC signaling can be viewed as a series of essential steps starting with 1) the availability of active GC in the target tissue; 2) binding of the GC/GR complex to DNA and/or accessory factors important for signal activation; and 3) the increase or decrease of GR gene products that contribute to physiologic responses. This review focuses exclusively on the events occurring at the level of GR or downstream of GR and does not report on the important role of prereceptor metabolism in GC/GR signaling. In this regard, the enzyme 11β-hydroxysteroid dehydrogenase 1 has been shown to be critical for generating active GC within tissues and is currently being investigated as a drug target for the treatment of metabolic disease and has been summarized in a recent review (46). Below, we describe molecular mediators of GR activation in individual metabolic tissues.
Liver
Role of GCs in the regulation of hepatic gluconeogenesis
The activation of the HPA axis is a physiologic response to stress that is intended to be of short duration, allowing the body to acutely elevate glucose levels to provide the energy needed to overcome the stressful stimuli. Prolonged GC activation, as in exogenous GC administration or Cushing's syndrome, results in hyperglycemia and insulin resistance. The phosphoenolpyruvate carboxykinase (Pepck) and glucose-6-phosphatase (G6Pc) enzymes catalyze, respectively, the rate-limiting step and final step in hepatic glucose production (47). Pepck is highly regulated at a transcriptional level by complex hormonal and dietary stimuli. Functional GR-binding sites have been characterized in the promoter regions of Pepck and G6Pc (48, 49). GC-mediated up-regulation of Pepck requires binding of GR along with several accessory factors including hepatocyte nuclear factor 4 (HNF-4α), hepatocyte nuclear factor 3 (HNF-3), C/EBPβ, forkhead transcription factor (FOXO1), retinoic acid receptor, retinoid X receptor, COUP-TF (chicken ovalbumin upstream promoter transcription factor), and peroxisome proliferator-activated receptor γ (PPARγ2) to the Pepck promoter (44, 49–52). Several coactivators including, PPAR coactivator 1α (PGC1α), glucocorticoid receptor interacting protein 1, p160 transcriptional cofactor steroid receptor coactivator 1 (SRC-1), and the acetyltransferase p300/cAMP response element binding protein (CREB)-binding protein (CBP) are also involved in Pepck transactivation (20, 53–56). In fact, GCs directly induce Pgc1α in hepatocytes (57). PGC1α has a marked effect on the expression of gluconeogenic genes because of its ability to interact with multiple transcription factors (ie, GR, HNF-4α, CREB, and FOXO1) (58, 59). Elegant Pepck promoter mutation and ChIP studies by a number of groups have shown that the binding of accessory factors facilitates the recruitment of GR to the nonconsensus GRE, which is necessary for full GC-induced Pepck expression (44, 49, 58, 60–62).
Chromatin-modifying enzymes are responsible for maintaining the Pepck promoter in an open conformation, thereby allowing the transcription factors to access chromatin. The histone acetyltransferases p300 and CBP are recruited to the Pepck promoter in response to GCs (55, 62). Insulin strongly represses the induction of Pepck, in part, through the loss of p300/CBP, resulting in chromatin condensation (62). SMAD6 is a transcription factor that belongs to the TGFβ family. SMAD6 suppresses GR-mediated transactivation by attracting histone deacetylase 3 (HDAC3) to DNA-bound GR and subsequently, antagonizing acetylation of histone H3 and H4 induced by SRC-1 (63). In another unique mechanism, Nadar et al (64) recently found that phosphorylation of GR at serine 232 by MAPK p38 can discharge p300 and the SWF/SNF chromatin-remodeling complex component SNF2 from gluconeogenic GR-bound promoters in rat liver. Because AMP-activated kinase (AMPK) activates MAPK p38, this provides a mechanism by which the organism “senses” its low-energy status and turns off ATP-consuming metabolic programs including gluconeogenesis (65). Indeed, when rats were treated with the AMPK activator AICAR, Dex-induced hepatic steatosis and gluconeogenesis were attenuated (64).
Liver × receptors (LXRs) are nuclear hormone receptors that are activated by endogenous cholesterol metabolites. Recently, we have shown that compared with wild-type (Wt) mice, Lxrβ-/- mice are protected from GC-induced hyperglycemia, hyperinsulinemia, and hepatic steatosis despite being similarly sensitive to immunosuppression. Following Dex-treatment, Lxrβ-/- mice remain insulin sensitive and do not show increased liver expression of Pepck. Moreover, neither GR nor LXRβ regulate each other's expression; however, LXRβ is required for GR recruitment to the GRE of the Pepck gene following GC induction (26).
LXRα has been demonstrated to repress the expression of the GR target genes Pepck and G6pc (66–69). In vivo studies in rodents have shown that activation of LXRα decreases liver levels of these genes along with hepatic glucose production (68, 69). It was recently shown that treatment of hepatoma cells with LXR ligands also suppressed GC-induced Pepck and G6pc expression (70). In agreement, dosing the rats for 3 days with an LXR ligand (GW3965) attenuated the increase in plasma glucose that is observed after a single dose of Dex (70). Using gel shift and ChIP experiments, the authors demonstrated that these effects were mediated by LXRα, because LXRα/ retinoid X receptor α heterodimers competed with GR to bind to the GRE of the rat G6Pc gene (70). Taken together, the studies mentioned above suggest isoform-specific roles for LXRα and LXRβ in GC-induced gluconeogenesis. Whereas endogenous expression of LXRβ is required for a maximal GR occupancy on the GRE of gluconeogenic genes in the liver, activation of LXRα by a synthetic ligand appears to suppress GC-activated gluconeogenic gene transcription by inhibiting the recruitment of GR to the GREs of these genes.
Several transcription factors have been shown to regulate GR levels directly. Farnesoid X receptor (FXR) is a ligand-activated transcription factor that responds to endogenous bile acids. Recently, Renga et al (71) found that FXR activation up-regulates GR, Pepck, and G6Pc expression, only when the mice were in the fasted state. In the fed state, FXR activation results in a decrease of GR, Pepck, and G6Pc expression, likely through the mechanism of induction of the small heterodimer partner, a nuclear receptor repressor regulated by FXR (71, 72). The authors further describe a distal FXR-binding site (ER8) on the human GR promoter and show that activation of FXR is necessary to up-regulate GR transcription in response to bile acids. They have also shown, following fasting, Fxr-/- mice exhibit hypoglycemia and have decreased expression of GR, Pepck, and G6Pc compared with Wt mice. Moreover, decreased expression of GR in Fxr-/- mice protected them from hyperglycemia caused by 4 days of Dex treatment (71). Similarly, Lu et al (73) found that Yin Yang 1 (YY-1), a transcription factor involved in cell proliferation and differentiation, also regulates hepatic gluconeogenesis. YY-1 expression is induced in mice during fasting by cAMP/protein kinase A/CREB signaling and in the state of insulin resistance (in db/db mice). Under these conditions, YY-1 along with the SRC-1 coactivator complex is recruited to the promoter of GR to induce its expression, resulting in increased GR signaling and increased hepatic gluconeogenesis (73).
Altering the nuclear translocation of GR is another mechanism of changing the GC/GR liver response. Inhibition of HDAC6 activity results in hyperacetylation of Hsp90 and impaired chaperone-dependent activation of GR (74, 75). Recently, Winkler et al (76) found that Hdac6-/- mice are protected from Dex-induced glucose intolerance, insulin resistance, and hyperglycemia due to decreased nuclear translocation of GR. They further showed that HDAC6 inhibition by tubacin had no effect on Dex-mediated suppression of inflammatory gene expression in lipopolysaccharide-induced human THP-1 monocytes, suggesting that this may be a new mechanism to dissociate the positive and negative actions of GR (76).
It is well recognized that catabolic hormones such as GCs oppose the actions of insulin in the liver. It is therefore not surprising that GCs modulate the expression of proteins and lipids that inhibit the insulin-signaling pathway and contribute to GC-induced hepatic glucose production. For example, GCs have been shown to increase the expression of Trb3, a mammalian homolog of Drosophila tribbles, in hepatocytes (77, 78). Du et al (78) found that TRB3 functions as a negative modulator of insulin signaling where it binds directly to AKT, preventing its phosphorylation and activation in diabetic db/db mouse livers, in turn promoting hyperglycemia and glucose intolerance (78). In a second example, Holland et al (79) showed that Dex treatment increases ceramide synthesis and accumulation in the liver by inducing the expression of ceramide synthetic genes including serine palmitoyltransferase, the rate-limiting enzyme in ceramide synthesis. Reducing ceramide levels by myriocin (serine palmitoyltransferase inhibitor) pretreatment protects mice from developing Dex-induced insulin resistance and glucose intolerance. In addition, mice heterozygous for dihydroceramide desaturase-1 (Des1), an enzyme that converts inactive dihydroceramide into active ceramide, are protected from Dex-induced insulin resistance (79). Together these studies highlight new mechanisms regulated by GCs that impact hepatic gluconeogenesis and insulin resistance.
Role of GCs in hepatic lipogenesis
Elevated GC levels modulate genes involved in lipolysis and triglyceride (TG) synthesis in a tissue-specific manner. GC effects on the liver are thought to be partially responsible for hypertriglyceridemia and hepatic steatosis seen in Cushing's patients (17, 80, 81). Dolinsky et al (84) observed that treatment of mice with Dex caused accumulation of TGs in the liver by reducing TG lipolysis and increasing TG synthesis (82) (Figure 3). In the same study, Dex had no effect on hepatic very low density lipoprotein (VLDL) secretion rates in vivo or in isolated primary hepatocytes but did find a 50% decrease in TG turnover in hepatocytes with Dex treatment. They also observed that the proportion of secreted TG derived from de novo sources is increased with Dex, whereas utilization of stored TG for secretion was reduced (82). Triacylglycerol hydrolase (TGH, also known as Ces3) is a lipase that hydrolyzes intracellular TG within hepatocytes prior to incorporation into VLDL. Although GR does not regulate Tgh transcriptionally, Tgh mRNA stability is decreased by Dex treatment (82).
Figure 3.
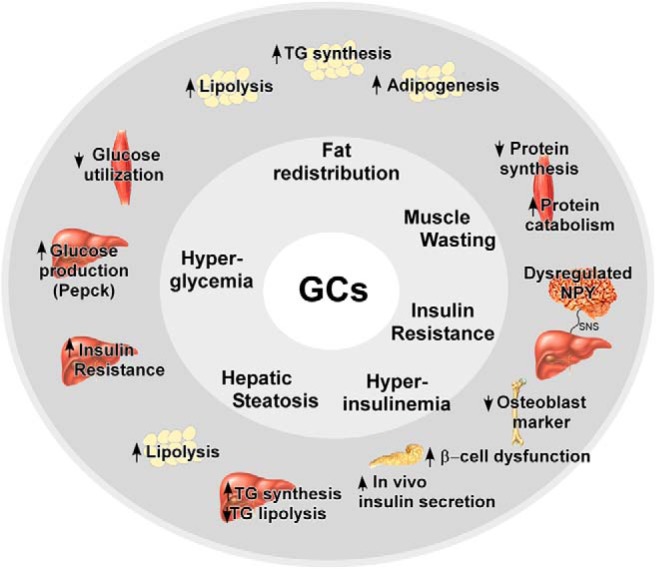
A summary of selected GC-responsive tissues and GC-induced pathways discussed in this review. TG, triglyceride; SNS, sympathetic nervous system.
We have shown that Lxrβ-/- mice are resistant to developing GC-induced hyperinsulinemia and hepatosteatosis (26). LXRβ appears to affect GC-regulated lipid metabolism by modulating glucose metabolism and insulin sensitivity. It has previously been shown in normal or diabetic rats, and in isolated primary hepatocytes, that the potent lipogenic actions of GCs in the liver are dependent on insulin (83–86). Compared with Wt mice, Dex-treated Lxrβ-/- mice are more insulin sensitive. This observation could explain the decreased GC-induced TG accumulation in Lxrβ-/- mice compared with Wt mice (26).
A study by Lemke et al (87) has shown that hepatic overexpression of Hes1 (hairy inducer of split-1) completely protects mice from liver steatosis caused by GR activation. GC treatment represses the expression of Hes1, a transcriptional repressor, in the liver. Adenovirus-mediated hepatic overexpression of Hes1 increases the expression of pancreatic lipase (Pnl) and pancreatic lipase-related protein (Pnlrp 2) in mice treated with Dex for 3 weeks (87). Hepatic Pnl and Pnlrp2 contribute to TG hydrolysis and the subsequent stimulation of fatty acid oxidation and ketogenesis (88). Moreover, liver-specific knockdown of GR in db/db mice induces Hes1 expression and improves hepatic steatosis. These data suggest that GC-mediated inhibition of the transcription of Hes1 results in a decreased expression of Pnl and Pnlrp2, which in turn elevates hepatic TG accumulation. Furthermore, the authors showed that Hes1 is a direct target of GR using both ChIP and promoter reporter studies (87). This study indicates that Hes1 is likely a GR primary target gene that controls hepatic TG storage by regulating genes involved in TG hydrolysis in the liver. A recent study using Dex-treated Hes1 liver-specific knockout mice found that Hes1 plays a more global role as a master repressor that must first be down-regulated for GR to have its effects on carbohydrate metabolism (89).
MED1, a component of mediator cofactor complex, has been shown to coactivate several nuclear receptors including GR (90, 91). Gene expression analysis in Med1-/- mouse embryonic fibroblast cells found that Med1 was selectively required for the regulation of GR target genes (90). Moreover, Dex-induced hepatic steatosis was attenuated in liver-specific Med1-/- mice compared with Wt mice. The expression of medium- and short-chain acyl-coenzyme A dehydrogenases (Mcad and Scad), which encode enzymes involved in FA oxidation, are repressed by 3 days of Dex treatment in Wt mice. In Med1-liver specific knockout mice, the repression of Mcad and Scad by Dex is reduced, and the mice are protected from hepatosteatosis (92). It is not clear whether this is a direct or indirect effect of GR because functional GR-binding sites in the promoters of Mcad or Scad have not yet been identified.
Adipose tissue
Role of GCs in lipogenesis, adipogenesis, and lipolysis
GCs have complex effects on adipose tissue, causing both increased lipolysis and increased adiposity (Figure 3). One of the clinical features of GC excess in Cushing's syndrome is abdominal obesity with limited sc fat on the extremities. Abdominal obesity is strongly associated with insulin resistance, cardiovascular disease, and hypertension and thus is a significant contributor to morbidity and mortality (93–95). The complexity of GC signaling in adipose tissue depends not only on the specific adipose depot in question but also the intracellular concentration of GC. Multiple mechanisms contributing to the complicated effects of GCs on adipose tissue have been recently summarized by Peckett et al (96) and we highlight a few studies below.
Using stable isotope-labeling techniques, it has been shown that TG synthesis and lipolysis are increased concurrently in inguinal fat pads of mice treated with Dex and in sc and visceral fat from Cushingoid (CRH-Tg) mice (97, 98). Compared with mice treated with vehicle, Dex treatment increases the expression of numerous TG synthesis genes (Scd2, Gpat3, Gpat4, Agpat2, and Lpin1); lipolytic genes (Lipe and Mgll); lipid transport genes (Cd36, Lrp1, Slc27a2, and Vldlr); and lipid storage genes (S3–S12) in inguinal fat (98). At least one functional GBS has been found within the genomic sequence of the genes mentioned above with the exception of Agpat2 (98). Moreover, it has been shown that ex vivo cultures of visceral adipocytes but not sc adipocytes from GC-treated rats showed higher lipolytic rates than the control cultures, suggesting GCs may promote the flux of TGs from the visceral white adipose tissue (WAT) to liver (99). Campbell et al (99) found that treatment of rats with corticosterone for 10 days also increased visceral adiposity due to increased preadipocyte differentiation as opposed to adipocyte hypertrophy. In agreement with this observation, Dex is routinely used as a component of the in vitro adipogenic differentiation cocktail (100).
Angiopoietin-like4 (ANGPTL4, fasting-induced adipose factor) is a protein inhibitor of lipoprotein lipase. Angptl4 is synthesized and secreted from the liver and white adipose tissue (WAT), in response to fasting and elevated GC levels. Increased expression of Angptl4 in WAT inhibits extracellular lipoprotein lipase and promotes intracellular lipolysis; together, these result in higher circulating TGs and increased mobilization of lipids from WAT to the plasma (101). Accordingly, Koliwad et al (102) found that Angptl4-/- mice are protected from hypertriglyceridemia and hepatosteatosis caused by 4 days of Dex treatment. The authors showed that Angptl4 is a direct target of GR using both ChIP and promoter reporter studies (102). Subsequent studies by Gray et al (101) showed that 24 hours of fasting induced Angptl4 expression in the liver and WAT in a GR-dependent manner (the effect was attenuated by RU-486 treatment). In addition, lipolysis induced by fasting or Dex treatment was severely impaired in WAT from Angptl4-/- mice (101). These studies suggest that Angptl4 is a primary target of GR in adipocytes that promotes lipolysis and redistribution of lipids from adipocytes to the liver.
Skeletal muscle
The muscle is the largest insulin-sensitive organ in the body and thus plays a major role in maintaining glucose homeostasis (103). In response to stress, GCs act on the muscle and oppose the actions of insulin, resulting in enhanced protein degradation, decreased protein synthesis, and supressed insulin-stimulated glucose uptake (Figure 3). Prolonged GC exposure can lead to muscle wasting (atrophy) and insulin resistance (104). Muscle-specific deletion of GR found that GR expression was necessary to promote muscle atrophy in response to Dex treatment (105). Interestingly, both genomic and nongenomic mechanisms have been found to be at play in skeletal muscle to regulate protein and glucose metabolism through modulation of the insulin/IGF-1 pathway. The role of GR in muscle has been recently reviewed in detail by Kuo et al (104). Although the detailed molecular mechanisms are still being elucidated, a few of the key components will be discussed below.
Effect of GCs on muscle wasting
GCs promote skeletal muscle atrophy through increased catabolic (protein breakdown) and decreased anabolic (protein synthesis) pathways. On the catabolic side, 2 important genes, muscle RING-finger protein-1 (MuRF1) and muscle atrophy F-box (MAFbx) play a key role in promoting muscle atrophy and are induced with increasing concentrations of exogenous or endogenous glucocorticoids (105).
MuRF1 and MAFbx are ubiquitin ligases that mark proteins for degradation by the proteasome (106). MuRF1 has been shown to physically interact with the myosin heavy chain of skeletal muscle to target it for degradation, and the loss of myosin heavy chain in response to GCs is attenuated in Murf1-/- mice (106). MAFbx was found to bind and antagonize MyoD, a transcription factor responsible for muscle differentiation and repair (107). MAFbx was also found to target eIF3-f (a protein involved in the initiation phase of translation) for degradation (108). Using Dex to induce muscle atrophy, Baehr et al (109) found that Murf1-/- mice exhibited muscle sparing compared with Wt mice; however, this was not observed in Mafbx-/- mice, indicating that MuRF1 and MAFbx are differentially sensitive to various atrophy models (109).
Mechanistically, GCs induce the up-regulation of MuRF1 and MAFbx by different means. MuRF1 possesses a GRE in its proximal promoter, and its regulation by GCs depends on GR homodimerization (110). In contrast, MAFbx is activated through an indirect mechanism, in which competitive binding of GR with AKT leads to the downstream activation of FOXO1/3, transcriptional regulators of MAFbx, thus, indirectly inducing MAFbx expression (111). MuRF1 also has a FOXO1-binding site near its GRE that has been shown to act synergistically with GR to increase its expression (110).
Myostatin plays an important role in the anti-anabolic effects of GCs. The myostatin gene contains a GRE in its promoter, and GC treatment increases its expression. Myostatin negatively regulates skeletal muscle mass by inhibiting muscle cell proliferation and protein synthesis (112). Dex treatment of myostatin -/- mice resulted in a sparing of skeletal muscle compared with Wt mice, and a decrease in the expression of Murf1 and Mafbx, suggesting that myostatin may also have a role in their regulation (113). Several studies have shown that myostatin inhibits AKT (114–116), which would lead to increased expression of the atrophy genes.
Downstream of AKT, mammalian target of rapamycin (mTOR) plays an important role in regulating protein synthesis by mediating the phosphorylation of S6K1 and 4E-BP1 (117). GCs inhibit mTOR, which leads to a decrease in protein synthesis. As outlined by a recent review, there are several GR primary target genes (ie, Klf-15, Sesn1, Depdc6, Ddit4, and Mknk2) that are known to interact with mTOR and may contribute to GC-inhibited mTOR signaling (104). Two of these primary GR target genes, Ddit4 (REDD1) and KLF15, have been shown to inhibit mTOR via distinct mechanisms. Ddit4 inhibits mTOR by increasing activity of TSC1/2 (proteins which form complexes and help to regulate mTOR) (118). KLF15 inhibits mTOR by activating transcription of the mitrochondrial enzyme, Bcat2, which reduces mTOR activity (118). Furthermore, it was shown that KLF15 up-regulates the atrophy genes MuRF1 and MAFbx. Interestingly, mTOR activation blocks GR transcription, thus attenuating GR-induced atrophy (118). Therefore, the cross talk between GR and mTOR is an important factor for skeletal muscle regulation.
Effect of GCs on muscle glucose homeostasis
Impaired insulin action in skeletal muscle is considered to be a key defect in insulin-stimulated glucose disposal in insulin-resistant, obese, and diabetic individuals (119). GCs inhibit glucose uptake and utilization in muscles. Glucose uptake is suppressed mainly through inhibition of GLUT4 translocation to the membrane (5, 120, 121). Moreover, glycogen synthase activity is decreased following GC treatment (5, 121, 122). Under normal conditions, when insulin is present, glycogen synthase kinase 3 (downstream of phosphatidylinositol 3-kinase [PI3K]) is phosphorylated and inactive, leaving glycogen synthase derepressed. However, following GC treatment, glycogen synthase kinase 3 phosphorylation is decreased, which activates it, leading to repression of glycogen synthase (123). Notably, the above genes are downstream of AKT in the insulin-signaling pathway.
GCs further decrease glucose utilization by inhibiting glucose oxidation in the muscle. Pyruvate dehydrogenase kinase 4 (PDK4) is a negative regulator of glucose oxidation, and GCs strongly up-regulate Pdk4 expression in skeletal muscle via GRE binding (and binding of FOXO1) thereby decreasing overall glucose utilization in muscle cells (124). This correlates with earlier findings that starvation and diabetes increase the expression of PDK4 in muscle leading to inactivation of pyruvate dehydrogenase (125). GR mRNA expression in skeletal muscle has also been shown to be increased in obese patients with type 2 diabetes when compared with healthy controls, and these levels decreased during treatment in concert with improved insulin sensitivity (126).
GR directly impairs insulin signaling
Dysregulation of protein and glucose metabolism in skeletal muscle both involve impaired insulin signaling. In the absence of GCs, insulin binds and activates the insulin receptor, which phosphorylates IRS, activating PI3K, leading to AKT activation. Activated AKT causes inactivation of FOXO1, thereby limiting atrophy gene expression. Many groups have found that muscle atrophy relates to impaired IRS-1-associated PI3K/AKT activity (127–129); however, until recently it was not clear how this was occurring. In 2009, Hu et al discovered that activated GR competitively binds to p85α, the regulatory subunit of PI3K (130, 131) and decreases its ability to associate with IRS-1. Because of this nongenomic GR interaction, AKT is not phosphorylated and FOXO1/3 are active, which contributes to increased MuRF1 and MAFbx expression (131). GR expression in muscle is necessary for Dex-mediated impairment in AKT phosphorylation (111). Using ChIP-seq and microarray analyses, Kuo et al (40) found GR-binding regions in 8 genes involved in regulating the insulin/IGF-1 pathway. Among these, p85α was regulated at the transcriptional level through a GRE. Furthermore, short hairpin RNA studies of p85α in myotubes found that the ability of GCs to inhibit AKT was compromised and atrophy gene expression diminished (40). Conversely, adenoviral overexpression of p85α reduced myotube diameters and decreased protein synthesis, mimicking the effects of GCs (40). These data suggest that p85α may be responsible for suppression of insulin signaling in response to GCs through genomic and nongenomic mechanisms.
Pancreas
The pancreas elicits a complex response to elevations in GC levels as it secretes insulin and glucagon in response to changes in circulating blood glucose. Studies in mice and humans have shown that following Dex administration both glucagon and insulin levels are elevated concurrently (23, 132). With GC treatment it is expected that insulin secretion would be elevated to counteract increased circulating blood glucose; however, the importance of elevated glucagon during chronic GC therapy is not well studied. Further complicating our understanding of the actions of GCs in the pancreas, conflicting reports have emerged from in vivo and in vitro studies.
Perfusion of isolated pancreas from Dex-treated rats found that compared with vehicle treatment, the first phase of glucose-induced insulin release was unaffected, but the priming effect of glucose (ie, augmented secretion compared with first phase) was lost in the second phase of glucose-induced insulin release (133). The authors suggest that the loss of priming by glucose was due to increased demands on β-cells mediated by the GC treatment. In contrast, a recent study in rat islets isolated from Dex-treated animals demonstrated that more docked secretory granules are present in the β-cells, resulting in enhanced insulin secretion in response to glucose (134).
GC treatment causes increased circulating insulin levels both in mice and humans (23, 24, 132, 135, 136). It is generally accepted that during GC therapy, hyperinsulinemia observed in vivo is due to the body's attempt to counteract the hyperglycemia due to insulin resistance in the liver (Figure 3). In agreement, Nicod et al (135) demonstrated that after 2 days of Dex administration to healthy individuals, heightened insulin secretion fully compensated for Dex-mediated insulin resistance in skeletal muscle and adipose but not in liver because endogenous hepatic glucose production remained elevated during a hyperglycemic clamp (135). Moreover, there appears to be an acute effect of GCs on insulin secretion. When oral glucose tolerance tests were performed immediately (5 minutes) after receiving a single iv bolus of hydrocortisone in healthy humans, it was found that the rise in plasma glucose was attenuated during the initial 15 minutes, which led to lower glucose levels during the first 2 hours in hydrocortisone group. This was accompanied by enhanced circulating insulin and C-peptide levels during the initial 15 minutes, and a 35% increase in the first-phase β-cell function. These studies suggest that both direct (GC action at the pancreas) and indirect (hyperglycemia) cues signal together to orchestrate whole-body insulin secretion in response to GCs.
In contrast to the above experiments in which GCs were administered subchronically in vivo, mechanistic studies performed in tissue culture using isolated islets and insulin-producing cells show that GC treatment inhibits insulin secretion and increases β-cell apoptosis (11, 137, 138). Ullrich et al (139) found that GC treatment causes induction of serum/GC-regulated kinase 1 (Sgk-1) in insulin-secreting INS-1 cells, which, in turn, up-regulates the activity of voltage-gated K(+) channels. Increased K(+) channel activity reduces Ca(2+) entry through voltage-gated Ca(2+) channels and insulin release. In agreement, Dex significantly blunted glucose-induced insulin release in islets isolated from Wt but not Sgk1-/- mice (139). These data suggest that the GR target gene Sgk-1 is a key protein that causes GC-mediated dysregulation in insulin secretion. Reich et al (138) recently found that Dex induced the expression of Txnip, a negative regulator of the antioxidant thioredoxin in β-cells of mice and human islets, resulting in apoptosis. These effects were reversed by cotreatment of Dex with RU-486. In agreement, Txnip-/- mice have more functional β-cells compared with control littermates (140). In summary, pancreatic GC actions are not completely understood and render opposing responses in isolated cell systems compared with an intact animal.
Interorgan Interplay Influencing GC-Induced Metabolic Adaptations
The interplay between organ systems plays an important role in mediating the metabolic consequences of glucocorticoid excess. For example, GC-induced lipolysis of adipose tissue enhances free fatty acid release into the bloodstream, enhancing fatty acid uptake into the liver, which results in hepatic steatosis. Other examples of this interorgan communication have been shown recently in different contexts involving nontraditional metabolic organs, a few examples of which will be discussed below.
GC-mediated GR activation has been shown to increase PPARα expression (57). Bernal-Mizrachi et al (141) determined that Pparα-/- mice are protected from Dex-induced hyperglycemia, hyperinsulinemia, and hypertension and that PPARα reconstitution in nondiabetic Dex-treated Pparα-/- mice liver is sufficient to increase gluconeogenic gene expression, hyperglycemia, hyperinsulinemia, and hypertension. A subsequent study found that selective hepatic afferent vagotomy decreased Dex-mediated hyperglycemia, hyperinsulinemia, insulin resistance, Pepck and PPARα expression, and hypertension in Wt mice. Moreover, PPARα reconstitution in nondiabetic Dex-treated Pparα-/- mice increased blood glucose, insulin, Pepck activity, and blood pressure in sham-operated animals but not hepatic vagotomized mice (142). These studies suggest that a complex interaction between hepatic PPARα expression and vagal afferent pathway is necessary for GC-induced metabolic effects.
GR is highly expressed in the hypothalamic paraventricular nucleus (PVN) and arcuate nucleus (ARC), where GCs have been shown to increase the expression of neuropeptide Y (NPY). NPY neurons projecting from the ARC to the PVN are essential for balancing feeding behavior and glucose metabolism. Recently, Yi et al (143) found that local administration of Dex into the ARC, but not into the PVN, during a hyperinsulinemic-euglycemic clamp induced severe hepatic insulin resistance in mice. This effect was prevented by either intracerebroventricular coadministration of the NPY1 receptor antagonist BIBP3226 or by hepatic sympathetic denervation (143). This study suggests that GC signaling in the ARC neurons modulates hepatic insulin responsiveness via NPY and the sympathetic nervous system (Figure 3).
A recent study suggests an interesting new role for osteoblasts (“bone forming cells”) in mediating the effects of GCs on fuel metabolism. Transgenic mice expressing the GC degrading enzyme 11β-hydroxysteroid dehydrogenase 2, under the control of the rat collagen type 1 promoter, produced osteoblasts with disrupted intracellular GC signaling at the prereceptor level. Using these mice, the authors found that disruption of GC signaling in the osteoblast attenuates GC-induced obesity and hyperglycemia and partially prevents GC-induced insulin resistance and glucose intolerance (24). Osteocalcin is a key protein secreted by osteoblasts the expression of which is decreased by GC treatment. Hepatic overexpression of osteocalcin in Wt mice prevented GC-induced increases in hepatic lipid deposition and partially protected against insulin resistance (24). These data point to a novel GC-mediated cross talk between the skeleton and whole-body glucose metabolism (Figure 3).
Concluding Remarks
Although the mechanisms of GR action have been studied for more than 30 years, we continue to uncover new and remarkable ways in which GR coordinates genomic and nongenomic actions to achieve a specific physiologic response. The development of new GC therapeutics with distinct activity profiles are showing promise at separating a subset of side effects from the desired antiinflammatory effects, although their exact mechanisms of achieving this dissociation are still not completely clear (144, 145). Several proteins described in this review such as, LXR, FXR, AMPK, and HDAC6, are amenable to small-molecule targeting and may prove to be effective at limiting GR-mediated side effects when dosed in combination with currently prescribed GCs. We anticipate that the recent technological advances in genome-wide sequencing will allow researchers to probe deeper into the mechanisms of GR gene regulation, exploring mRNA transcription and splicing, micro RNA, and long non-coding RNA, to uncover additional tissue-specific or gene-specific regulatory pathways that contribute to GR function.
Acknowledgments
We apologize to our colleagues whose work we accidentally overlooked or were unable to cite due to space constraints.
This work was supported by Canadian Institutes of Health Research (MOP-125900) and the Natural Sciences and Engineering Research Council of Canada (RGPIN 356873–08).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by Canadian Institutes of Health Research (MOP-125900) and the Natural Sciences and Engineering Research Council of Canada (RGPIN 356873–08).
Footnotes
- AMPK
- AMP-activated kinase
- ANGPTL4
- angiopoietin-like 4
- ARC
- arcuate nucleus
- CBP
- CREB binding protein
- C/EBPβ
- CCAAT/enhancer-binding protein β
- ChIP
- chromatin immunoprecipitation
- ChIP-seq
- ChIP combined with next-generation sequencing
- CREB
- cAMP response element-binding protein
- Dex
- dexamethasone
- FOX
- forkhead transcription factor
- FXR
- farnesoid X receptor
- G6Pc
- glucose-6-phosphatase
- GBS
- GR-binding sequence
- GC
- glucocoticoid
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HDAC
- histone deacetylase
- HNF
- hepatocyte nuclear factor
- HPA
- hypothalamic-pituitary-adrenal
- HSP
- heat shock protein
- LXR
- liver X receptor
- MAFbx
- muscle atrophy F-box
- mTOR
- mammalian target of rapamycin
- MuRF1
- muscle RING-finger protein-1
- NPY
- neuropeptide Y
- PDK
- pyruvate dehydrogenase kinase
- Pepck
- phosphoenolpyruvate carboxykinase
- PGC
- PPAR coactivator 1α
- PI3K
- phosphatidylinositol 3-kinase
- Pnl
- pancreatic lipase
- Pnlrp
- Pnl-related protein
- PPAR
- peroxisome proliferator-activated receptor
- PVN
- paraventricular nucleus
- SRC-1
- steroid receptor coactivator 1
- TGH
- triacylglycerol hydrolase
- TSS
- transcription start site
- VLDL
- very low density lipoprotein
- WAT
- white adipose tissue
- Wt
- wild type
- YY-1
- Yin Yang 1.
References
- 1. Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem. 2011;286(5):3177–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275(1–2):43–61. [DOI] [PubMed] [Google Scholar]
- 3. Lewis RL, Roger A. The effect of the adrenal cortex on carbohydrate metabolism. Endocrinology. 1940(27):971. [Google Scholar]
- 4. Sakoda H, Ogihara T, Anai M, et al. . Dexamethasone-induced insulin resistance in 3T3-L1 adipocytes is due to inhibition of glucose transport rather than insulin signal transduction. Diabetes. 2000;49(10):1700–1708. [DOI] [PubMed] [Google Scholar]
- 5. Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism. 1998;47(1):3–6. [DOI] [PubMed] [Google Scholar]
- 6. Hasselgren PO. Glucocorticoids and muscle catabolism. Curr Opin Clin Nutr Metab Care. 1999;2(3):201–205. [DOI] [PubMed] [Google Scholar]
- 7. Divertie GD, Jensen MD, Miles JM. Stimulation of lipolysis in humans by physiological hypercortisolemia. Diabetes. 1991;40(10):1228–1232. [DOI] [PubMed] [Google Scholar]
- 8. Guillaume-Gentil C, Assimacopoulos-Jeannet F, Jeanrenaud B. Involvement of non-esterified fatty acid oxidation in glucocorticoid-induced peripheral insulin resistance in vivo in rats. Diabetologia. 1993;36(10):899–906. [DOI] [PubMed] [Google Scholar]
- 9. Odedra BR, Bates PC, Millward DJ. Time course of the effect of catabolic doses of corticosterone on protein turnover in rat skeletal muscle and liver. Biochem J. 1983;214(2):617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munck A. Glucocorticoid inhibition of glucose uptake by peripheral tissues: old and new evidence, molecular mechanisms, and physiological significance. Perspect Biol Med. 1971;14(2):265–269. [DOI] [PubMed] [Google Scholar]
- 11. Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99(3):414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rose AJ, Herzig S. Metabolic control through glucocorticoid hormones: an update. Mol Cell Endocrinol. 2013;380(1–2):65–78. [DOI] [PubMed] [Google Scholar]
- 13. Reynolds RM, Walker BR, Syddall HE, Whorwood CB, Wood PJ, Phillips DI. Elevated plasma cortisol in glucose-intolerant men: differences in responses to glucose and habituation to venepuncture. J Clin Endocrinol Metab. 2001;86(3):1149–1153. [DOI] [PubMed] [Google Scholar]
- 14. Ohshima K, Shargill NS, Chan TM, Bray GA. Adrenalectomy reverses insulin resistance in muscle from obese (ob/ob) mice. Am J Physiol. 1984;246(2 Pt 1):E193–E197. [DOI] [PubMed] [Google Scholar]
- 15. Livingstone DE, Grassick SL, Currie GL, Walker BR, Andrew R. Dysregulation of glucocorticoid metabolism in murine obesity: comparable effects of leptin resistance and deficiency. J Endocrinol. 2009;201(2):211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Plotz CM, Knowlton AI, Ragan C. The natural history of Cushing's syndrome. Am J Med. 1952;13(5):597–614. [DOI] [PubMed] [Google Scholar]
- 17. Rockall AG, Sohaib SA, Evans D, et al. . Hepatic steatosis in Cushing's syndrome: a radiological assessment using computed tomography. Eur J Endocrinol. 2003;149(6):543–548. [DOI] [PubMed] [Google Scholar]
- 18. Shimomura Y, Bray GA, Lee M. Adrenalectomy and steroid treatment in obese (ob/ob) and diabetic (db/db) mice. Horm Metab Res. 1987;19(7):295–299. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Nakagawa Y, Wang Y, et al. . Increased glucocorticoid receptor and 11β-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to the phenotype of type 2 diabetes in db/db mice. Diabetes. 2005;54(1):32–40. [DOI] [PubMed] [Google Scholar]
- 20. Friedman JE, Sun Y, Ishizuka T, et al. . Phosphoenolpyruvate carboxykinase (GTP) gene transcription and hyperglycemia are regulated by glucocorticoids in genetically obese db/db transgenic mice. J Biol Chem. 1997;272(50):31475–31481. [DOI] [PubMed] [Google Scholar]
- 21. Overman RA, Yeh JY, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken). 2013;65(2):294–298. [DOI] [PubMed] [Google Scholar]
- 22. Scerif M, Fuzesi T, Thomas JD, et al. . CB1 receptor mediates the effects of glucocorticoids on AMPK activity in the hypothalamus. J Endocrinol. 2013;219(1):79–88. [DOI] [PubMed] [Google Scholar]
- 23. Wise JK, Hendler R, Felig P. Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man. J Clin Invest. 1973;52(11):2774–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brennan-Speranza TC, Henneicke H, Gasparini SJ, et al. . Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J Clin Invest. 2012;122(11):4172–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab. 1982;54(1):131–138. [DOI] [PubMed] [Google Scholar]
- 26. Patel R, Patel M, Tsai R, et al. . LXRβ is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J Clin Invest. 2011;121(1):431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. [DOI] [PubMed] [Google Scholar]
- 28. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013;154(3):993–1007. [DOI] [PubMed] [Google Scholar]
- 29. Nixon M, Andrew R, Chapman KE. It takes two to tango: dimerisation of glucocorticoid receptor and its anti-inflammatory functions. Steroids. 2013;78(1):59–68. [DOI] [PubMed] [Google Scholar]
- 30. Uhlenhaut NH, Barish GD, Yu RT, et al. . Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49(1):158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shipp LE, Lee JV, Yu CY, et al. . Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS One. 2010;5(10):e13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324(5925):407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007;3(6):e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. John S, Sabo PJ, Thurman RE, et al. . Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43(3):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Biddie SC, John S, Sabo PJ, et al. . Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 2011;43(1):145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007;275(1–2):13–29. [DOI] [PubMed] [Google Scholar]
- 37. Phuc Le P, Friedman JR, Schug J, et al. . Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet. 2005;1(2):e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stahn C, Lowenberg M, Hommes DW, et al. . Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol. 2007;275(1–2):71–78. [DOI] [PubMed] [Google Scholar]
- 39. Lowenberg M, Tuynman J, Scheffer M, et al. . Kinome analysis reveals nongenomic glucocorticoid receptor-dependent inhibition of insulin signaling. Endocrinology. 2006;147(7):3555–3562. [DOI] [PubMed] [Google Scholar]
- 40. Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci USA. 2012;109(28):11160–11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reddy TE, Pauli F, Sprouse RO, et al. . Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19(12):2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reddy TE, Gertz J, Crawford GE, Garabedian MJ, Myers RM. The hypersensitive glucocorticoid response specifically regulates period 1 and expression of circadian genes. Mol Cell Biol. 2012;32(18):3756–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grøntved L, John S, Baek S, et al. . C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013;32(11):1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chakravarty K, Cassuto H, Reshef L, Hanson RW. Factors that control the tissue-specific transcription of the gene for phosphoenolpyruvate carboxykinase-C. Crit Rev Biochem Mol Biol. 2005;40(3):129–154. [DOI] [PubMed] [Google Scholar]
- 45. Steger DJ, Grant GR, Schupp M, et al. . Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010;24(10):1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gathercole LL, Lavery GG, Morgan SA, et al. . 11β-Hydroxysteroid dehydrogenase 1: translational and therapeutic aspects. Endocr Rev. 2013;34(4):525–555. [DOI] [PubMed] [Google Scholar]
- 47. Hers HG, Hue L. Gluconeogenesis and related aspects of glycolysis. Annu Rev Biochem. 1983;52:617–653. [DOI] [PubMed] [Google Scholar]
- 48. Vander Kooi BT, Onuma H, Oeser JK, et al. . The glucose-6-phosphatase catalytic subunit gene promoter contains both positive and negative glucocorticoid response elements. Mol Endocrinol. 2005;19(12):3001–3022. [DOI] [PubMed] [Google Scholar]
- 49. Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK, et al. . Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem. 1993;268(8):5353–5356. [PubMed] [Google Scholar]
- 50. Hall RK, Sladek FM, Granner DK. The orphan receptors COUP-TF and HNF-4 serve as accessory factors required for induction of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Proc Natl Acad Sci USA. 1995;92(2):412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hall RK, Yamasaki T, Kucera T, Waltner-Law M, O'Brien R, Granner DK. Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein-1 gene expression by insulin. The role of winged helix/forkhead proteins. J Biol Chem. 2000;275(39):30169–30175. [DOI] [PubMed] [Google Scholar]
- 52. Wang JC, Stromstedt PE, O'Brien RM, Granner DK. Hepatic nuclear factor 3 is an accessory factor required for the stimulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Mol Endocrinol. 1996;10(7):794–800. [DOI] [PubMed] [Google Scholar]
- 53. Herzog B, Hall RK, Wang XL, Waltner-Law M, Granner DK. Peroxisome proliferator-activated receptor γ coactivator-1α, as a transcription amplifier, is not essential for basal and hormone-induced phosphoenolpyruvate carboxykinase gene expression. Mol Endocrinol. 2004;18(4):807–819. [DOI] [PubMed] [Google Scholar]
- 54. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1 α): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24(1):78–90. [DOI] [PubMed] [Google Scholar]
- 55. Wang XL, Herzog B, Waltner-Law M, Hall RK, Shiota M, Granner DK. The synergistic effect of dexamethasone and all-trans-retinoic acid on hepatic phosphoenolpyruvate carboxykinase gene expression involves the coactivator p300. J Biol Chem. 2004;279(33):34191–34200. [DOI] [PubMed] [Google Scholar]
- 56. Yoon JC, Puigserver P, Chen G, et al. . Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413(6852):131–138. [DOI] [PubMed] [Google Scholar]
- 57. Lemberger T, Staels B, Saladin R, Desvergne B, Auwerx J, Wahli W. Regulation of the peroxisome proliferator-activated receptor α gene by glucocorticoids. J Biol Chem. 1994;269(40):24527–24530. [PubMed] [Google Scholar]
- 58. Rhee J, Inoue Y, Yoon JC, et al. . Regulation of hepatic fasting response by PPARγ coactivator-1α (PGC-1): requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc Natl Acad Sci USA. 2003;100(7):4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Puigserver P, Rhee J, Donovan J, et al. . Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature. 2003;423(6939):550–555. [DOI] [PubMed] [Google Scholar]
- 60. Scott DK, O'Doherty RM, Stafford JM, Newgard CB, Granner DK. The repression of hormone-activated PEPCK gene expression by glucose is insulin-independent but requires glucose metabolism. J Biol Chem. 1998;273(37):24145–24151. [DOI] [PubMed] [Google Scholar]
- 61. Wang JC, Stafford JM, Scott DK, Sutherland C, Granner DK. The molecular physiology of hepatic nuclear factor 3 in the regulation of gluconeogenesis. J Biol Chem. 2000;275(19):14717–14121. [DOI] [PubMed] [Google Scholar]
- 62. Hall RK, Wang XL, George L, Koch SR, Granner DK. Insulin represses phosphoenolpyruvate carboxykinase gene transcription by causing the rapid disruption of an active transcription complex: a potential epigenetic effect. Mol Endocrinol. 2007;21(2):550–563. [DOI] [PubMed] [Google Scholar]
- 63. Ichijo T, Voutetakis A, Cotrim AP, et al. . The Smad6-histone deacetylase 3 complex silences the transcriptional activity of the glucocorticoid receptor: potential clinical implications. J Biol Chem. 2005;280(51):42067–42077. [DOI] [PubMed] [Google Scholar]
- 64. Nader N, Ng SS, Lambrou GI, et al. . AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK. Mol Endocrinol. 2010;24(9):1748–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25. [DOI] [PubMed] [Google Scholar]
- 66. Cao G, Liang Y, Broderick CL, et al. . Antidiabetic action of a liver x receptor agonist mediated by inhibition of hepatic gluconeogenesis. J Biol Chem. 2003;278(2):1131–1136. [DOI] [PubMed] [Google Scholar]
- 67. Grefhorst A, van Dijk TH, Hammer A, et al. . Differential effects of pharmacological liver X receptor activation on hepatic and peripheral insulin sensitivity in lean and ob/ob mice. Am J Physiol Endocrinol Metab. 2005;289(5):E829–E838. [DOI] [PubMed] [Google Scholar]
- 68. Laffitte BA, Chao LC, Li J, et al. . Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci USA. 2003;100(9):5419–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Commerford SR, Vargas L, Dorfman SE, et al. . Dissection of the insulin-sensitizing effect of liver X receptor ligands. Mol Endocrinol. 2007;21(12):3002–3012. [DOI] [PubMed] [Google Scholar]
- 70. Nader N, Ng SS, Wang Y, Abel BS, Chrousos GP, Kino T. Liver x receptors regulate the transcriptional activity of the glucocorticoid receptor: implications for the carbohydrate metabolism. PLoS One. 2012;7(3):e26751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Renga B, Mencarelli A, D'Amore C, et al. . Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. FASEB J. 2012;26(7):3021–3031. [DOI] [PubMed] [Google Scholar]
- 72. Lu TT, Makishima M, Repa JJ, et al. . Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6(3):507–515. [DOI] [PubMed] [Google Scholar]
- 73. Lu Y, Xiong X, Wang X, et al. . Yin Yang 1 promotes hepatic gluconeogenesis through upregulation of glucocorticoid receptor. Diabetes. 2013;62(4):1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kovacs JJ, Murphy PJ, Gaillard S, et al. . HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–607. [DOI] [PubMed] [Google Scholar]
- 75. Murphy PJ, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J Biol Chem. 2005;280(40):33792–33799. [DOI] [PubMed] [Google Scholar]
- 76. Winkler R, Benz V, Clemenz M, et al. . Histone deacetylase 6 (HDAC6) is an essential modifier of glucocorticoid-induced hepatic gluconeogenesis. Diabetes. 2012;61(2):513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stayrook KR, Bramlett KS, Savkur RS, et al. . Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146(3):984–991. [DOI] [PubMed] [Google Scholar]
- 78. Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300(5625):1574–1577. [DOI] [PubMed] [Google Scholar]
- 79. Holland WL, Brozinick JT, Wang LP, et al. . Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5(3):167–179. [DOI] [PubMed] [Google Scholar]
- 80. Shibli-Rahhal A, Van Beek M, Schlechte JA. Cushing's syndrome. Clin Dermatol. 2006;24(4):260–265. [DOI] [PubMed] [Google Scholar]
- 81. Taskinen MR, Nikkila EA, Pelkonen R, Sane T. Plasma lipoproteins, lipolytic enzymes, and very low density lipoprotein triglyceride turnover in Cushing's syndrome. J Clin Endocrinol Metab. 1983;57(3):619–626. [DOI] [PubMed] [Google Scholar]
- 82. Dolinsky VW, Douglas DN, Lehner R, Vance DE. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. Biochem J. 2004;378(Pt 3):967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kirk CJ, Verrinder TR, Hems DA. Fatty acid synthesis in the perfused liver of adrenalectomized rats. Biochem J. 1976;156(3):593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mangiapane EH, Brindley DN. Effects of dexamethasone and insulin on the synthesis of triacylglycerols and phosphatidylcholine and the secretion of very-low-density lipoproteins and lysophosphatidylcholine by monolayer cultures of rat hepatocytes. Biochem J. 1986;233(1):151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cai Y, Song Z, Wang X, Jiao H, Lin H. Dexamethasone-induced hepatic lipogenesis is insulin dependent in chickens (Gallus gallus domesticus). Stress. 2011;14(3):273–281. [DOI] [PubMed] [Google Scholar]
- 86. Diamant S, Shafrir E. Modulation of the activity of insulin-dependent enzymes of lipogenesis by glucocorticoids. Eur J Biochem. 1975;53(2):541–546. [DOI] [PubMed] [Google Scholar]
- 87. Lemke U, Krones-Herzig A, Berriel Diaz M, et al. . The glucocorticoid receptor controls hepatic dyslipidemia through Hes1. Cell Metab. 2008;8(3):212–223. [DOI] [PubMed] [Google Scholar]
- 88. Inagaki T, Dutchak P, Zhao G, et al. . Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5(6):415–425. [DOI] [PubMed] [Google Scholar]
- 89. Revollo JR, Oakley RH, Lu NZ, et al. . HES1 is a master regulator of glucocorticoid receptor-dependent gene expression. Sci Signal. 2013;6(304):ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen W, Roeder RG. The Mediator subunit MED1/TRAP220 is required for optimal glucocorticoid receptor-mediated transcription activation. Nucleic Acids Res. 2007;35(18):6161–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen W, Roeder RG. Mediator-dependent nuclear receptor function. Semin Cell Dev Biol. 2011;22(7):749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jia Y, Viswakarma N, Fu T, Yu S, Rao MS, Borensztajn J, Reddy JK. Conditional ablation of mediator subunit MED1 (MED1/PPARBP) gene in mouse liver attenuates glucocorticoid receptor agonist dexamethasone-induced hepatic steatosis. Gene Expr. 2009;14(5):291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Després JP. Abdominal obesity as important component of insulin-resistance syndrome. Nutrition. 1993;9(5):452–459. [PubMed] [Google Scholar]
- 94. Arnaldi G, Mancini T, Polenta B, Boscaro M. Cardiovascular risk in Cushing's syndrome. Pituitary. 2004;7(4):253–256. [DOI] [PubMed] [Google Scholar]
- 95. Mancini T, Kola B, Mantero F, Boscaro M, Arnaldi G. High cardiovascular risk in patients with Cushing's syndrome according to 1999 WHO/ISH guidelines. Clin Endocrinol (Oxf). 2004;61(6):768–777. [DOI] [PubMed] [Google Scholar]
- 96. Peckett AJ, Wright DC, Riddell MC., The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism. 2011;60(11):1500–1510. [DOI] [PubMed] [Google Scholar]
- 97. Harris C, Roohk DJ, Fitch M, Boudignon BM, Halloran BP, Hellerstein MK. Large increases in adipose triacylglycerol flux in Cushingoid CRH-Tg mice are explained by futile cycling. Am J Physiol Endocrinol Metab. 2013;304(3):E282–E293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yu CY, Mayba O, Lee JV, Tran J, Harris C, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One. 2010;5(12):e15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Campbell JE, Peckett AJ, D'Souza AM, Hawke TJ, Riddell MC. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am J Physiol Cell Physiol. 2011;300(1):C198–C209. [DOI] [PubMed] [Google Scholar]
- 100. Pantoja C, Huff JT, Yamamoto KR. Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro. Mol Biol Cell. 2008;19(10):4032–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gray NE, Lam LN, Yang K, Zhou AY, Koliwad S, Wang JC. Angiopoietin-like 4 (Angptl4) protein is a physiological mediator of intracellular lipolysis in murine adipocytes. J Biol Chem. 2012;287(11):8444–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Koliwad SK, Kuo T, Shipp LE, et al. . Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism. J Biol Chem. 2009;284(38):25593–25601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zierath JR, Wallberg-Henriksson H. From receptor to effector: insulin signal transduction in skeletal muscle from type II diabetic patients. Ann NY Acad Sci. 2002;967:120–134. [DOI] [PubMed] [Google Scholar]
- 104. Kuo T, Harris CA, Wang JC. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol. 2013;380(1–2):79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab. 2012;302(10):E1210–E1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Clarke BA, Drujan D, Willis MS, et al. . The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6(5):376–385. [DOI] [PubMed] [Google Scholar]
- 107. Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem. 2005;280(4):2847–2856. [DOI] [PubMed] [Google Scholar]
- 108. Lagirand-Cantaloube J, Offner N, Csibi A, et al. . The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27(8):1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Baehr LM, Furlow JD, Bodine SC. Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J Physiol. 2011;589(Pt 19):4759–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, Bodine SC. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab. 2008;295(4):E785–E797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhao W, Qin W, Pan J, Wu Y, Bauman WA, Cardozo C. Dependence of dexamethasone-induced Akt/FOXO1 signaling, upregulation of MAFbx, and protein catabolism upon the glucocorticoid receptor. Biochem Biophys Res Commun. 2009;378(3):668–672. [DOI] [PubMed] [Google Scholar]
- 112. Ma K, Mallidis C, Bhasin S, et al. . Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab. 2003;285(2):E363–E371. [DOI] [PubMed] [Google Scholar]
- 113. Gilson H, Schakman O, Combaret L, et al. . Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology. 2007;148(1):452–460. [DOI] [PubMed] [Google Scholar]
- 114. Amirouche A, Durieux A-C, Banzet S, et al. . Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150(1):286–294. [DOI] [PubMed] [Google Scholar]
- 115. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296(6):C1258–C1270. [DOI] [PubMed] [Google Scholar]
- 116. Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297(5):C1124–C1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. J Endocrinol. 2008;197(1):1–10. [DOI] [PubMed] [Google Scholar]
- 118. Shimizu N, Yoshikawa N, Ito N, et al. . Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011;13(2):170–182. [DOI] [PubMed] [Google Scholar]
- 119. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–S163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Weinstein SP, Paquin T, Pritsker A, Haber RS. Glucocorticoid-induced insulin resistance: dexamethasone inhibits the activation of glucose transport in rat skeletal muscle by both insulin- and non-insulin-related stimuli. Diabetes. 1995;44(4):441–445. [DOI] [PubMed] [Google Scholar]
- 121. Morgan SA, Sherlock M, Gathercole LL, et al. . 11β-Hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes. 2009;58(11):2506–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Coderre L, Srivastava AK, Chiasson JL. Effect of hypercorticism on regulation of skeletal muscle glycogen metabolism by insulin. Am J Physiol. 1992;262(4 Pt 1):E427–E433. [DOI] [PubMed] [Google Scholar]
- 123. Burén J, Lai Y, Lundgren M, Eriksson JW, Jensen J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch Biochem Biophys. 2008;474(1):91–101. [DOI] [PubMed] [Google Scholar]
- 124. Connaughton S, Chowdhury F, Attia RR, et al. . Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol. 2010;315(1):159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wu P, Inskeep K, Bowker-Kinley M, Popov K, Harris R. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes. 1999;48(8):1593–1599. [DOI] [PubMed] [Google Scholar]
- 126. Vestergaard H, Bratholm P, Christensen NJ. Increments in insulin sensitivity during intensive treatment are closely correlated with decrements in glucocorticoid receptor mRNA in skeletal muscle from patients with Type II diabetes. Clinical Science. 2001;101(5):533–540. [DOI] [PubMed] [Google Scholar]
- 127. Bodine SC, Stitt TN, Gonzalez M, et al. . Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3(11):1014–1019. [DOI] [PubMed] [Google Scholar]
- 128. Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004;15(6):1537–1545. [DOI] [PubMed] [Google Scholar]
- 129. Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology. 2006;147(9):4160–4168. [DOI] [PubMed] [Google Scholar]
- 130. Leis H, Page A, Ramirez A, et al. . Glucocorticoid receptor counteracts tumorigenic activity of Akt in skin through interference with the phosphatidylinositol 3-kinase signaling pathway. Mol Endocrinol. 2004;18(2):303–311. [DOI] [PubMed] [Google Scholar]
- 131. Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest. 2009;119(10):3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lee SN, Peng B, Desjardins R, Pintar JE, Day R, Lindberg I. Strain-specific steroidal control of pituitary function. J Endocrinol. 2007;192(3):515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Grill V, Rundfeldt M. Abnormalities of insulin responses after ambient and previous exposure to glucose in streptozocin-diabetic and dexamethasone-treated rats. Role of hyperglycemia and increased B-cell demands. Diabetes. 1986;35(1):44–51. [DOI] [PubMed] [Google Scholar]
- 134. Rafacho A, Marroqui L, Taboga SR, et al. . Glucocorticoids in vivo induce both insulin hypersecretion and enhanced glucose sensitivity of stimulus-secretion coupling in isolated rat islets. Endocrinology. 2010;151(1):85–95. [DOI] [PubMed] [Google Scholar]
- 135. Nicod N, Giusti V, Besse C, Tappy L. Metabolic adaptations to dexamethasone-induced insulin resistance in healthy volunteers. Obes Res. 2003;11(5):625–631. [DOI] [PubMed] [Google Scholar]
- 136. Viguerie N, Picard F, Hul G, et al. . Multiple effects of a short-term dexamethasone treatment in human skeletal muscle and adipose tissue. Physiol Genomics. 2012;44(2):141–51. [DOI] [PubMed] [Google Scholar]
- 137. Ranta F, Avram D, Berchtold S, et al. . Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. 2006;55(5):1380–1390. [DOI] [PubMed] [Google Scholar]
- 138. Reich E, Tamary A, Sionov RV, Melloul D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated β cell death. Diabetologia. 2012;55(4):1048–1057. [DOI] [PubMed] [Google Scholar]
- 139. Ullrich S, Berchtold S, Ranta F, et al. . Serum- and glucocorticoid-inducible kinase 1 (SGK1) mediates glucocorticoid-induced inhibition of insulin secretion. Diabetes. 2005;54(4):1090–1099. [DOI] [PubMed] [Google Scholar]
- 140. Chen J, Hui ST, Couto FM, et al. . Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic β-cell mass and protects against diabetes. FASEB J. 2008;22(10):3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bernal-Mizrachi C, Weng S, Feng C, et al. . Dexamethasone induction of hypertension and diabetes is PPAR-α dependent in LDL receptor-null mice. Nat Med. 2003;9(8):1069–1075. [DOI] [PubMed] [Google Scholar]
- 142. Bernal-Mizrachi C, Xiaozhong L, Yin L, et al. . An afferent vagal nerve pathway links hepatic PPARα activation to glucocorticoid-induced insulin resistance and hypertension. Cell Metab. 2007;5(2):91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yi CX, Foppen E, Abplanalp W, et al. . Glucocorticoid signaling in the arcuate nucleus modulates hepatic insulin sensitivity. Diabetes. 2012;61(2):339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Brandish PE, Anderson K, Baltus GA, et al. . The preclinical efficacy, selectivity and pharmacologic profile of MK-5932, an insulin-sparing selective glucocorticoid receptor modulator. Eur J Pharmacol. 2014;724:102–111. [DOI] [PubMed] [Google Scholar]
- 145. De Bosscher K. Selective glucocorticoid receptor modulators. J Steroid Biochem Mol Biol. 2010;120(2–3):96–104. [DOI] [PubMed] [Google Scholar]